
Abstract
c-Met receptor tyrosine kinase is a proto-oncogene whose aberrant activation is attributed to a lower rate of survival in most cancers. Natural product-derived inhibitors known as “fourth generation inhibitors” constitute more than 60% of anticancer drugs. Furthermore, consensus docking approach has recently been introduced to augment docking accuracy and reduce false positives during a virtual screening. In order to obtain novel small-molecule Met inhibitors, consensus docking approach was performed using Autodock Vina and Autodock 4.2 to virtual screen Naturally Occurring Plant-based Anti-cancer Compound–Activity–Target database against active and inactive conformation of c-Met kinase domain structure. Two hit molecules that were in line with drug-likeness criteria, desired docking score, and binding pose were subjected to molecular dynamics simulations to elucidate intermolecular contacts in protein–ligand complexes. Analysis of molecular dynamics simulations and molecular mechanics Poisson–Boltzmann surface area studies showed that ZINC08234189 is a plausible inhibitor for the active state of c-Met, whereas ZINC03871891 may be more effective toward active c-Met kinase domain compared to the inactive form due to higher binding energy. Our analysis showed that both the hit molecules formed hydrogen bonds with key residues of the hinge region (P1158, M1160) in the active form, which is a hallmark of kinase domain inhibitors. Considering the pivotal role of HGF/c-Met signaling in carcinogenesis, our results propose ZINC08234189 and ZINC03871891 as the therapeutic options to surmount Met-dependent cancers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During recent years, a paradigm shift has occurred in cancer research. Identification of the molecular pathways involved in cancer initiation and progression will aid scientists to develop new therapies according to the molecular profile of patients [1]. Receptor tyrosine kinases (RTKs) are pharmaceutically attractive targets for this purpose because they play a critical role in tumor formation. They regulate a variety of signal-transduction pathways within a cell [2], of which c-Met and its ligand, hepatocyte growth factor (HGF), mediate epithelial tissue remodeling, migration, morphogenesis, cell growth, differentiation, and angiogenesis [2, 3]. Deregulated activation of c-Met-signaling pathway through multiple mechanisms like activation, mutation, gene amplification, and heterodimerization has been reported to correlate with high tumor grade and lower rate of survival in most cancers [3, 4]. Moreover, in 2011, c-Met was found as a marker of pancreatic cancer stem cell, which made it a suitable therapeutic target for cancer treatment [5].
The Met receptor that belongs to the family of RTK is a heterodimer of an extracellular α-chain and a transmembrane β-chain. Upon binding of HGF, receptor dimerization and autophosphorylation of tyrosine residues of kinase domain in β sub-unit occurs, leading to recruitment of multiple effector proteins and promotes the activation of MAPK-signaling, PI3K/Akt-signaling, STAT-signaling, FAK-signaling pathways etc. Over the last decade, small-molecule inhibitors targeting ATP-binding site in kinase domain have been developed and tested in clinical trials [1, 6]. Two distinct binding patterns have been proposed for these ATP-competitive inhibitors: type I compounds (e.g., PF-023410661) bind to activated DFG-in conformation and interact with Met 1160 in the hinge region and Tyr 1230 in the activation loop. Although they are specific, they display limited activity toward Tyr 1230 mutation, while type II compounds (e.g., BMS-777607) are less selective and bind in the inactive DFG-out conformation formed by conformational changes of the Asp1222 side chain [2, 3, 7].
As evidenced by scientific literature, there is a great interest in the screening of natural products for drug development. It has been reported that natural products have great diversity and novel chemical structure which render them useful as new inhibitors. Accordingly, inhibitors derived from natural products have sometimes been called “fourth generation inhibitors” [8]. Actually, natural compounds and their derivatives constitute more than 60% of anticancer drugs [9]. In addition, phytochemicals have been used increasingly for many reasons including low cost, fewer adverse effects, and well-tolerated in the body. Thus, phytochemicals have been discovered as promising compounds for prevention, treatment, or reversal of drug resistance in cancer [10]. Moreover, previous investigations have revealed some flavonoids modulated HGF/c-Met axis in cancer cells; but the intermolecular contacts are unknown [11, 12].
Nowadays, molecular docking is a well-accepted computational method in structure-based drug design for identifying novel hit molecules, which employs a scoring function to evaluate ligand binding orientation and affinity [13,14,15]. In conventional molecular docking, if there is a difference in pose prediction identifying the correct pose will be impossible, while agreement in docked pose will increase accuracy and reduce false positive notably during a virtual screening. This is the objective of consensus docking, which enhances ligand pose prediction by more than one scoring algorithm [16]. Houston et al. reported that a significant improvement in pose prediction accuracy was achieved using Autodock and Vina in 82% of cases. Interestingly, this percentage increased up to 92% when three docking programs; Autodock, Vina, and Dock 6.5, were used simultaneously [16]. It is worthy of note that a Lamarckian Genetic Algorithm and an Amber-force-field-based scoring function have been implemented in Autodock and Vina, respectively [16, 17]. Berry et al. carried out a multi-step Autodock Vina-Glide-GOLD consensus docking procedure to identify novel inhibitors against human coronavirus validated by MD simulation [18]. However, a recent study by Tuccinardi et al focused on the reliability of the consensus docking results in binding pose prediction using ten docking procedures [19].
In this study, we used consensus docking procedure to screen Naturally Occurring Plant-based Anti-cancer Compound–Activity–Target (NPACT) database. For this purpose, information regarding NPACT entries were obtained from the ZINC database. Following consensus docking, MD simulation and molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) binding energy calculation were performed to elucidate the binding mode of the hit compounds as novel c-Met kinase inhibitors.
Materials and Methods
Selection of Data Set
The three-dimensional, ready-to-dock format of 1574 ligands available in NPACT database was retrieved from the ZINC database (http://zinc.docking.org/) on January 18, 2015. ZINC has emerged as a widely-used database of purchasable compounds suitable for virtual screening [20]. Using physicochemical properties, 738 ligands were selected, which conformed to Lipinski’s rule of five [21] and Veber’s rules for drug-like molecules and oral bio-availability [22].
Structure Preparation
The crystal structures for active conformation of the c-Met kinase domain (PDB ID: 2RFS) [23] and inactive conformation (PDB ID: 1R0P) [24, 25] were retrieved from the Protein Data Bank (http://www.rcsb.org/pdb/home/home.do) and prepared for docking calculation by eliminating hetero atoms and water molecules. Thereafter, missing residues in protein structures were modified using MODELLER v.9.11 [26] based on the original FASTA sequence. For the representative structures of c-Met, the Autodock Tools (ADT) 1.5.7 was used to merge non-polar hydrogens to carbon atoms and compute Gasteiger charges. Rotatable bonds and Gasteiger charges were also assigned by the prepare_ligand4.py utility of ADT for the selected ligands.
Consensus Docking
Autodock Vina 1.1.2 was used for protein–ligand docking against 2RFS and 1R0P structures with the default parameters [27]. The 500 top ranked ligands were further docked using Autodock 4.2.6 [28]. A grid box of 126 × 126 × 126 points with 0.153 Å spacing was defined by centering on the ligand in the active site. When running a docking using the Lamarckian GA, there were a number of parameters to set and, except the number of Lamarckian genetic algorithm runs was set to 100; other parameters were considered as their default values. In all docking studies, residues F1089, V1092, K1110, Y1159, M1160, M1211, D1222, M1229, and Y1230 in the c-Met ATP-binding site were allowed to be flexible. The lowest energy conformation in the Autodock Vina result and the largest cluster of Autodock 4.2 were selected as the resultant docked structure. The root mean square deviation (RMSD) calculation was used to assess the docking poses of Autodock Vina and Autodock 4.2 as described by Houston et al. [16]. Consequently, poses with RMSD more than 2.0 Å were rejected. To generate a molecular representation of each protein-ligand complex, PyMOL Molecular Graphics System v. 1.7.2 was used. ZINCPharmer (http://zincpharmer.csb.pitt.edu/) was also used to identify the pharmacophore features of the hit molecules [29].
Molecular Dynamics (MD) Simulation
Gromacs 4.5.3 [30] was used to perform MD simulation with the Amber03 force field parameters. To perform MD simulations, the best Autodock Vina result was considered as the initial conformation. Before the simulation, electrostatic potential (ESP) charges for drug candidates were computed employing Gaussian03 (B3LYP/6-31G* to optimize the geometry and #P HF/6-31G* SCF = Tight Pop = MK IOp (6/33 = 2) to calculate ESP); then, the restrained electrostatic potential (RESP) method was employed to assign atomic charges. To obtain ligand topology files based on generalized Amber force field, antechamber was used. A distance of 1 nm was set between a cubic box of TIP3P water molecules and the solute. Steepest descent algorithm was applied until the maximum force of the whole system was below 1000 kJ/mol. During 500 ps of NVT and NPT ensembles, all complexes were simulated to equilibrate at 300 K temperature and 1 bar pressure, respectively. Final simulation runs were carried out for 20 ns with a time step of 2 fs under V-rescale temperature and Parrinello–Rahman pressure coupling algorithms. The particle–mesh Ewald (PME) algorithm was utilized to calculate long-range electrostatics, while Linear Constraint Solver (LINCS) method was used to constrain all covalent bond lengths. A 1.2 nm cutoff was set for van der Waals interactions. VMD 1.9.2 [31] and PyMOL Molecular Graphics System v. 1.7.2 were used to visualize trajectories and generate images, respectively.
MM-PBSA Binding Energy Calculation
Binding-free energy calculations based on MM-PBSA method were estimated using the g_mmpbsa tool [32]. Snapshots saved at every 10 ps from the last 5 ns of each MD trajectory were considered for calculating each energy term. The solute, solvent, and vacuum dielectric constants were set at 2, 80, and 1, respectively, for polar solvation calculation. The solvent-accessible surface area (SASA) model was also used to compute non-polar solvation energy.
Results and Discussion
Database Screening using Consensus Docking
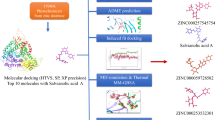
A schematic procedure of consensus docking is shown in Fig. 1. A compilation of 1574 plant-derived compounds available in ZINC database, a currently widely-used database for virtual screening, was filtered according to Lipinski and Veber rules. These physico-chemical criteria can be employed to select desirable compounds having membrane permeability as well as to be easily absorbed in the human body. The processed ligand set was then docked through consensus docking method [16]. As mentioned earlier, this method is reasonable particularly when funds for purchasing compounds are limited. Calculation of RMSD between binding modes predicted by Autodock 4.2 and Vina for each complex produced a small set of 98 molecules. Of these 98 ligands, two hits were in line with good-docking score and estimated IC50, and the presence of essential interactions with key residues in the ATP-binding site were considered for further evaluation.

Flowchart of the consensus docking procedure followed to choose hit compounds with c-Met kinase inhibitory activity
The 2D structures of two hit compounds alongside their molecular weight are shown in Table 1. ZINC08234189, a steroid lactone with a molecular weight of 470.61 g/mol, chemically consists of the 4-hydroxy-5,6-epoxy-2-en-1-one moiety and unsaturated lactone side chain which play crucial roles in biological activity [33].
With regard to ZINC03871891, it is an ortho-diphenolic diterpene which has 330.42 g/mol molecular weight. The structure of ZINC03871891 has an abietane carbon skeleton with a lactone moiety oriented across the central ring [34].
Docked Structures of the Hit Compounds
In order to reject false positives, it is crucial to evaluate the binding mode and affinity of the preliminary hit compounds. Table 2 shows the docking results with associated interaction profile in details. Interestingly, ZINC08234189 exhibited higher affinity in comparison to ZINC03871891 with the inactive conformation of c-Met as evidenced by Autodock binding energy of −12.91 kcal/mol. Analysis of its interaction pattern showed that the phenolic hydroxyl group of ZINC08234189 engaged in two hydrogen bonds with the carboxyl group of P1158 and the amino group of M1160 in the hinge region while hydrophobic contacts were present with V1092 and M1211 in the central hydrophobic region. Other residues like I1084, A1108, L1157, and F1089 in the two hydrophobic subpockets, and M1229 and Y1230 in the activation loop are involved in the hydrophobic interactions (Fig. 2a).

Binding poses of the two hit molecules: a 1R0P in complex with ZINC08234189, and b 2RFS in complex with ZINC03871891. The N and C lobes of tyrosine kinase domain are shown in cyan and green, respectively. The predicted binding mode using Autodock 4.2 and Vina are displayed in blue and yellow stick representation, respectively. Key residues involved in interaction are represented in the stick format. The length of Hydrogen bonds colored as yellow dotted lines is indicated in Angstrom (color figure online)
In the case of ZINC03871891 bound to the active conformation, it did not form hydrogen bond with any residues in the hinge region, a typical characteristic of all kinase inhibitors targeting the ATP-binding site [25, 35]. Instead, hydrophobic interactions were found with M1160, suggesting a lower binding affinity with an IC50 of 18.76 nM. Alternatively, residue V1092, L1140, L1157, and M1211 comprising the hydrophobic subpockets as well as D1222 (activation loop) are involved in hydrophobic interactions. The side chain of N1209 along with the carboxyl group of R1208 in the catalytic loop also formed a bidentate H-bond with this potent inhibitor as illustrated in Fig. 2b.
Furthermore, Fig. 3 shows the pharmacophoric features of the hit molecules. It revealed that O4 on compound ZINC08234189 had hydrogen donor (HD) and acceptor (HA) features, leading to hydrogen bonds with P1158 (O) and M1160 (N), which resulted in a strong interaction, while O6 on hydroxyl group had HD property. In addition, the scaffold of ZINC08234189 had five hydrophobic features. With regard to ZINC03871891, its backbone had four hydrophobic and one aromatic features, whereas O4 had HA property that tended to approach Y1230. Besides, O1 and O2 with HD and HA features formed hydrogen bonds with HD of R1208 (O) and N1209 (OD1) in the active site.

Pharmacophore features of the two hit molecules. a ZINC08234189 b ZINC03871891. Aromatic, hydrogen donor, hydrogen acceptor, and hydrophobic features are shown in violet, white, orange, and green spheres, respectively (color figure online)
MD Simulations
Although we selected two complexes according to consensus docking: 1R0P bound with ZINC08234189 and 2RFS bound with ZINC03871891 to compare the stability and binding affinity of the aforementioned compounds computationally, the apo form of c-Met and all protein-ligand complexes were subjected to MD simulations and MM/PBSA binding energy calculation method.
Given that Autodock Vina is believed to be superior in ligand pose prediction than Autodock 4.2 [36, 37], docking results of Vina having lowest binding energy were chosen for MD simulation. It should be noted that docking simulation provides a single snapshot of interactions between ligand and protein both in rigid and semi-flexible docking. Thus, in order to gain a deeper insight into the effects of protein structural changes and flexibility on the complex interaction profile, a 20-ns MD simulation was performed. The Cα RMSD in comparison with the initial structure indicated that 1R0P in the apo state experienced continuous fluctuation rising to 0.32 nm at the end of the simulation period. Upon binding of ZINC08234189 at the active site, the average values of conformational changes increased from 0.2 to 0.28 nm, while these changes were negligible for ZINC03871891 (Fig. 4a). In consideration of 2RFS without any ligand, the RMSD value increased to 0.26 nm for the first 4 ns. Thereafter, the receptor reached a plateau for the last 16 ns. Binding stability of 2RFS-ZINC03871891 as a function of time was also analyzed and it was shown that the system converged well with the range of 0.15–0.25 nm compared to ZINC08234189 (Fig. 4b). However, it is obvious that the RMSD value of 1R0P-ZINC08234189 was smaller than 2RFS-ZINC08234189. It was observed that around 3 and 5 ns RMSD value of ZINC08234189 underwent noticeable fluctuation due to the lactone ring rotation for 1R0P and 2RFS, respectively, and remained stable till the end (Fig. 4c, d). However, ZINC03871891 achieved relatively stable coordination throughout the entire simulation process. The same results were also observed after the initial and MD-averaged structures were superimposed (Fig. 5).

Root mean square deviation (RMSD) of the Cα atoms of the inactive (1R0P) and active (2RFS) conformation and ligand heavy atoms against the initial structure over 20 ns MD simulation. a 1R0P, b 2RFS, c RMSD of ligand backbone in complex with 1R0P, d RMSD of ligand backbone in complex with 2RFS

Superimposed structures of c-Met inhibitors using Autodock Vina (green with a blue ligand) and MD-averaged structure of the last 5 ns (cyan with a yellow ligand) (color figure online)
To investigate the residual mobility in ligand-bound and unbound forms, root mean square fluctuations (RMSF) of individual residues were also monitored in each case. As shown in Fig. 6, binding of hit molecules did not induce any remarkable changes in flexibility of ATP-binding site residues relative to the protein in the apo state. In particular, these fluctuations were minimal in any state of 2RFS. Thereafter, the protein structure compactness was analyzed using the g_gyrate utility of the Gromacs 4.5.3 software (Fig. 7, Table 3). It was shown that in its apo state, the protein was stable with no obvious structural expansion and contraction, whereas ZINC08234189 led to lower Rg value especially in complex with 1R0P within the range of 2.02–2.10 nm. In addition, an obvious expansion has occurred for ZINC03871891 bound to 2RFS after 6 ns.

Root mean square fluctuations (RMSF) analysis of c-Met kinase domain residues in the presence and absence of hit molecules over 20 ns MD simulation period. a 1R0P, b 2RFS

Radius of gyration of c-Met kinase domain in the presence and absence of hit molecules over 20 ns MD simulation period. a 1R0P, b 2RFS
Table 3 presents information regarding hydrogen bonds formed throughout the trajectory for all protein-ligand complexes. The percentage of H-bonds presence during the last 5 ns of MD simulation was measured to examine their stability, though only contacts populated with over 40% of the simulation time are listed.
As summarized in Table 3, ZINC08234189 formed hydrogen bonds with ε-amino group of K1110 (79.12% occupancy) in 1R0P (inactive form), while ZINC08234189-2RFS complex formed two hydrogen bonds with main chain nitrogen and oxygen atoms of M1160 and P1158 in the hinge region, respectively, being the main determinant for the overall stability of the complex. For ZINC03871891-1R0P, two hydrogen bonds with the main chain oxygen atom of R1227 were detected with occupancy of about 95 and 65%, while only an amino group of M1160 participated in H-bond interaction between ZINC03871891 and 2RFS.
Binding-Free Energy Analysis by MM-PBSA Calculation
Recent studies have reported MM/PBSA method as a simulation technique to estimate biomolecular interaction-free energy. Although it increases computational cost, it provides more accurate information than energy-scoring functions that have been used in docking softwares for rescoring top-ranked hits [17]. Five hundred snapshots extracted between 15–20 ns with equal intervals were selected as input for analysis as reported in Table 4. The results showed that both selected molecules had higher binding affinity of −128.27 ± 0.48 and −113.19 ± 0.59 kJ/mol for ZINC03871891 and ZINC08234189, respectively toward 2RFS than 1R0P, whereas the latter structure showed −113.71 ± 0.51 and −107.60 ± 0.81 kJ/mol for ZINC03871891 and ZINC08234189, respectively, though this difference was not considerable for ZINC08234189. In addition, ZINC03871891 has less negative binding energy than ZINC08234189 in both complexes.
Among the different energy terms that contributed to the protein-ligand binding energy, van der Waals (E vdw), electrostatic, (E ele), and SASA energy played a crucial role in binding energy and complex stability. Nevertheless, polar solvation energy (G ps) has an opposite effect, causing binding energy to depend on its unfavorable positive value [38]. In this regard, van der Waals energy contributed more negative free energy than electrostatic energy in all protein–ligand complexes. Besides, in ZINC03871891-bound complexes, van der Waals interaction was predominant in total binding-free energy, whereas in ZINC08234189-bound complexes, the unfavorable contribution of polar solvation energy was significant.
Conclusion
We used the consensus docking approach to virtually screen 1574 compounds retrieved from NPACT database against both active (2RFS) and inactive (1R0P) state of the c-Met kinase domain, yielding a selection of two hit molecules. Using a 20-ns MD simulation, the stability of each complex was evaluated. Our results showed that both protein and ligand backbone of ZINC08234189 achieved stability after 5 ns. Nevertheless, ZINC03871891 experienced stable conformation in each case during the entire simulation process. Given that hydrogen bond with residues of the hinge region (P1158, M1160) is a hallmark of kinase domain inhibitors, our analysis showed that both hit molecules formed hydrogen bonds with key residues in the active form. In summary, based on hydrogen bond analysis and MM-PBSA studies, we predicted that ZINC08234189 is a plausible inhibitor for the active state of c-Met. In line with our docking results, ZINC03871891 showed more effectiveness toward active c-Met kinase domain than the inactive form due to higher binding energy. The results disclosed here can be useful to overcome Met-addicted cancers.
References
Maroun, C. R., & Rowlands, T. (2014). The Met receptor tyrosine kinase: A key player in oncogenesis and drug resistance. Pharmacology & Therapeutics, 142, 316–338.
Tai, W., Lu, T., Yuan, H., Wang, F., Liu, H., Lu, S., Leng, Y., Zhang, W., Jiang, Y., & Chen, Y. (2012). Pharmacophore modeling and virtual screening studies to identify new c-Met inhibitors. Journal of Molecular Modeling, 18, 3087–3100.
Elnagar, A. Y., Sylvester, P. W., & El Sayed, K. A. (2011). (−)-Oleocanthal as a c-Met inhibitor for the control of metastatic breast and prostate cancers. Planta Medica, 77, 1013–1019.
Ye, L., Ou, X., Tian, Y., Yu, B., Luo, Y., Feng, B., Lin, H., Zhang, J., & Wu, S. (2013). Indazoles as potential c-met inhibitors: Design, synthesis and molecular docking studies. European Journal of Medicinal Chemistry, 65, 112–118.
Li, C., Wu, J. J., Hynes, M., Dosch, J., Sarkar, B., Welling, T. H., Pasca di Magliano, M., & Simeone, D. M. (2011). c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology, 141, 2218–2227.
Akl, M. R., Ayoub, N. M., Mohyeldin, M. M., Busnena, B. A., Foudah, A. I., Liu, Y.-Y., & El Sayed, K. A. (2014). Olive phenolics as c-Met inhibitors: (−)-Oleocanthal attenuates cell proliferation, invasiveness, and tumor growth in breast cancer models. PLoS One, 9, e97622.
Gherardi, E., Birchmeier, W., Birchmeier, C., & Woude, G. V. (2012). Targeting MET in cancer: rationale and progress. Nature Reviews Cancer, 12, 89–103.
Yan, X.-J., Gong, L.-H., Zheng, F.-Y., Cheng, K.-J., Chen, Z.-S., & Shi, Z. (2014). Triterpenoids as reversal agents for anticancer drug resistance treatment. Drug Discovery Today, 19, 482–488.
Carocho, M., & Ferreira, I. C. (2013). The role of phenolic compounds in the fight against cancer-a review. Anti-cancer Agents in Medicinal Chemistry, 13, 1236–1258.
Vahedi, F., Najafi, M. F., & Bozari, K. (2008). Evaluation of inhibitory effect and apoptosis induction of Zyzyphus Jujube on tumor cell lines, an in vitro preliminary study. Cytotechnology, 56, 105–111.
Shi, M.-D., Liao, Y.-C., Shih, Y.-W., & Tsai, L.-Y. (2013). Nobiletin attenuates metastasis via both ERK and PI3K/Akt pathways in HGF-treated liver cancer HepG2 cells. Phytomedicine : International Journal of Phytotherapy and Phytopharmacology, 20, 743–752.
Lee, W.-J., Chen, W.-K., Wang, C.-J., Lin, W.-L., & Tseng, T.-H. (2008). Apigenin inhibits HGF-promoted invasive growth and metastasis involving blocking PI3K/Akt pathway and β4 integrin function in MDA-MB-231 breast cancer cells. Toxicology and Applied Pharmacology, 226, 178–191.
Ashtawy, H. M., & Mahapatra, N. R. (2015). Machine-learning scoring functions for identifying native poses of ligands docked to known and novel proteins. BMC Bioinformatics, 16, S3.
Kumar, V., Krishna, S., & Siddiqi, M. I. (2015). Virtual screening strategies: Recent advances in the identification and design of anti-cancer agents. Methods (San Diego, Calif.), 71, 64–70.
Ferreira, L. G., dos Santos, R. N., Oliva, G., & Andricopulo, A. D. (2015). Molecular docking and structure-based drug design strategies. Molecules (Basel, Switzerland), 20, 13384–13421.
Houston, D. R., & Walkinshaw, M. D. (2013). Consensus docking: Improving the reliability of docking in a virtual screening context. Journal of Chemical Information and Modeling, 53, 384–390.
Ren, W., Truong, T. M., & Ai, H.-w (2015). Study of the binding energies between unnatural amino acids and engineered orthogonal tyrosyl-tRNA synthetases. Science Reports, 5, 12632.
Berry, M., Fielding, B. C., & Gamieldien, J. (2015). Potential broad spectrum inhibitors of the coronavirus 3CLpro: A virtual screening and structure-based drug design study. Viruses, 7, 6642–6660.
Tuccinardi, T., Poli, G., Romboli, V., Giordano, A., & Martinelli, A. (2014). Extensive consensus docking evaluation for ligand pose prediction and virtual screening studies. Journal of Chemical Information and Modeling, 54, 2980–2986.
Irwin, J. J., Sterling, T., Mysinger, M. M., Bolstad, E. S., & Coleman, R. G. (2012). ZINC: A free tool to discover chemistry for biology. J Chem Inf Model 52, 1757–1768.
Lipinski, C. A., Lombardo, F., Dominy, B. W., & Feeney, P. J. (1997). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews, 23, 3–25.
Veber, D. F., Johnson, S. R., Cheng, H.-Y., Smith, B. R., Ward, K. W., & Kopple, K. D. (2002). Molecular properties that influence the oral bioavailability of drug candidates. Journal of Medicinal Chemistry, 45, 2615–2623.
Liang, Z., Zhang, D., Ai, J., Chen, L., Wang, H., Kong, X., Zheng, M., Liu, H., Luo, C., Geng, M., Jiang, H., & Chen, K. (2011). Identification and synthesis of N′-(2-oxoindolin-3-ylidene)hydrazide derivatives against c-Met kinase. Bioorganic & Medicinal Chemistry Letters, 21, 3749–3754.
Schiering, N., Knapp, S., Marconi, M., Flocco, M. M., Cui, J., Perego, R., Rusconi, L., & Cristiani, C. (2003). Crystal structure of the tyrosine kinase domain of the hepatocyte growth factor receptor c-Met and its complex with the microbial alkaloid K-252a. Proceedings of the National Academy of Sciences of the United States of America, 100, 12654–12659.
Peach, M. L., Tan, N., Choyke, S. J., Giubellino, A., Athauda, G., Burke, Jr, T. R., Nicklaus, M. C., & Bottaro, D. P. (2009). Directed discovery of agents targeting the Met tyrosine kinase domain by virtual screening. Journal of Medicinal Chemistry, 52, 943–951.
Šali, A., & Blundell, T. L. (1993). Comparative protein modelling by satisfaction of spatial restraints. Journal of Molecular Biology, 234, 779–815.
Trott, O., & Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31, 455–461.
Goodsell, D. S Morris, G. M,. & Olson, A. J. (1996). Automated docking of flexible ligands: applications of AutoDock. Journal of Molecular Recognition, 9, 1–5
Koes, D. R., & Camacho, C. J. (2012). ZINCPharmer: Pharmacophore search of the ZINC database. Nucleic Acids Research, 40, W409–W414.
Pronk, S., Páll, S., Schulz, R., Larsson, P., Bjelkmar, P., Apostolov, R., Shirts, M. R., Smith, J. C., Kasson, P. M., & van der Spoel, D. (2013). GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics (Oxford, England), 29, 845–854.
Humphrey, W., Dalke, A., & Schulten, K. (1996). VMD: visual molecular dynamics. Journal of Molecular Graphics, 14, 33–38.
Kumari, R., Kumar, R., & Lynn, A. (2014). g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. Journal of Chemical Information and Modeling, 54, 1951–1962.
Gu, M., Yu, Y., Gunaherath, G. M., Gunatilaka, A. A., Li, D., & Sun, D. (2014). Structure-activity relationship (SAR) of withanolides to inhibit Hsp90 for its activity in pancreatic cancer cells. Investigational New Drugs, 32, 68–74.
Johnson, J. J. (2011). Carnosol: A promising anti-cancer and anti-inflammatory agent. Cancer letters, 305, 1–7.
Mohyeldin, M. M., Busnena, B. A., Akl, M. R., Dragoi, A. M., Cardelli, J. A., & El Sayed, K. A. (2016). Novel c-Met inhibitory olive secoiridoid semisynthetic analogs for the control of invasive breast cancer. European Journal of Medicinal Chemistry, 118, 299–315.
Anwar, M. A., Panneerselvam, S., Shah, M., & Choi, S. (2015). Insights into the species-specific TLR4 signaling mechanism in response to Rhodobacter sphaeroides lipid A detection. Science Reports, 5, 7657.
Chang, M. W., Ayeni, C., Breuer, S., & Torbett, B. E. (2010). Virtual screening for HIV protease inhibitors: a comparison of AutoDock 4 and Vina. PLoS One, 5, e11955.
He, J.-Y Li, C., & Wu, G. (2014). Discovery of potential drugs for human-infecting H7N9 virus containing R294K mutation. Drug Design, Development and Therapy, 8, 2377
Funding
This research has been supported by Grant Number 93013896 from Iran National Science Foundation (INSF) and 94-01-33-28717 from Deputy of Research, Tehran University of Medical Science.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Aliebrahimi, S., Montasser Kouhsari, S., Ostad, S. et al. Identification of Phytochemicals Targeting c-Met Kinase Domain using Consensus Docking and Molecular Dynamics Simulation Studies. Cell Biochem Biophys 76, 135–145 (2018). https://doi.org/10.1007/s12013-017-0821-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12013-017-0821-6