
Abstract
Purpose of Review
Recombinant adeno-associated virus (rAAV) is the commonest viral vector used in gene therapies. With the increase in the number of such therapies being employed in human clinical trials and approved clinical use, the associated adverse events are increasingly observed. Hepatotoxicity is the most common adverse event.
Recent Findings
Although mild in most, hepatotoxicity may affect gene therapy efficacy, lead to acute liver failure, death and persisting hepatitis necessitating prolonged use of immunosuppressants. There has been an increase in the referrals of such cases to hepatologists.
Summary
There is a lack of good quality evidence on the use of reactive immunosuppressants. There is a need to devise empiric protocols to diagnose and treat hepatotoxicity based on the side-effect profile of individual gene therapy, the present understanding of immunological basis of hepatotoxicity, the experience with various immunosuppressants in other disorders and the unique challenges and requirements of gene therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gene therapies using viral vectors are increasingly being used to treat monogenic genetic defects. Recombinant adeno-associated virus (rAAV) is the commonest vector being used to deliver these therapies. Amongst the various adverse clinical effects which pose challenges in their clinical use, hepatotoxicity being the commonest [1, 2]. Although mild in many patients, hepatotoxicity may affect efficacy of gene therapy (GT), lead to acute liver injury and death [2,3,4, 5•, 6•]. This has resulted in termination of some clinical trials of rAAV vector-based GT [6•]. As clinical hepatologists, we and others note the increase in referrals to our services with the increased clinical use of approved medications [7••]. There is a need for clarity on available options for patients unresponsive to steroids or in those requiring longer term and high dose steroids. This review summarises the approved rAAV-vector based gene therapies available that are associated with hepatotoxicity, proposed mechanisms of liver injury, challenges in management with pragmatic recommendations into the use of various immunosuppressive agents.
rAAV Vector and GT
In vivo gene therapy is aimed at delivering genetic material into the target tissues as opposed to ex vivo therapy where the target tissue is taken from the patient, genetically modified and the tissue delivered back into the patient. Viral vectors are the main mode of gene delivery for in vivo therapy. rAAV has become well established as the main viral vector for in-vivo therapy in comparison to retroviral and lentiviral vectors [8]. The clinical preference for rAAV in GT stems from its (i) broad tropism, (ii) lack of established pathogenicity, (iii) the rarity of integration of viral genome into the host genome, (iv) presumed low immunogenicity relative to other vectors and (v) the ease of production. Although AAV is considered to cause asymptomatic infections, a few recent reports suggest its possible role in the causation of indeterminate acute liver failure [9, 10]. A number of GT products with rAAV have received approval or conditional approval for clinical use from the United States Food and Drugs Administration and European Medicines Agency (Table 1).
A preliminary understanding of adeno-associated virus (AAV) structure helps understand rAAV vectors and their immunogenicity. AAV is a non-enveloped single-stranded DNA parvovirus. It is a dependovirus that depends on helper virus such as adenovirus for replication. The viral capsid consists of 3 proteins viral proteins (VP1, VP2 and VP3). Each virion is composed of 60 VP subunits with 9 variable regions in each subunit. The variable regions in the capsid determine the tropism of the virus to organs and thereby its transduction efficiency and the specificity of neutralising antibodies (Nab). The virus has a wide host range and tropism towards various tissues including the skeletal muscle, cardiac muscle, eyes, lungs, liver and central nervous system. The tropism may vary between different species [17]. Thirteen well-studied natural serotypes are known to exist with numerous variants [17]. Depending on the tissues targeted, various serotypes including serotypes from rhesus (rh) monkeys and mutants have been used to create rAAV in clinical trials (AAV1, AAV2, AAV5, AAV6, AAV8, AAV9, AAVrh10, mutants—AAV2.5, AAV Spark100, AAV.7m8 and AAVtYF). The AAV genome is composed of two inverted terminal repeats (ITR) flanking the rep and cap genes. The rep gene aids in genome replication and virion assembly while the cap gene produces the capsids. While the ITRs are necessary for propagation and transduction, the bulk of the AAV genome (cap and rep) can be replaced to create a rAAV. The rep and cap genes are replaced by a promoter, transgene and a poly-A tail. This replacement results in rAAV which can infect and transduce but not lead to replication or capsid production. Innovations in engineering the AAV cassette and AAV capsid, production and purification of rAAV and use of small molecules and peptides have improved the specificity and efficiency of transduction enable delivery of large transgenes and decrease immunogenicity.
The rAAV when delivered as GT enters the cell by endocytosis after binding to receptors and co-receptors on the cell surface. It enters the nucleus and releases the genome. Single-stranded DNA is converted to a double stranded DNA followed by transcription and translation of the transgene. This initial step may not be required if a self-complementary DNA has been used. The virion released from endosomes may be ubiquitinated in the cell and targeted for degradation by the proteosome [18].
Immunogenicity of rAAV
Clinical trials and post-marketing data have shown that the immunogenicity of rAAV affects the transduction and adverse events of the GT [2, 19, 20]. The immune response to rAAV needs to be understood in the context of the various components of the immune system and their interplay (Fig. 1, supplementary table 1). The immunological mechanisms in rAAV GT are briefly summarised below [18].

Immune response to aAAV. 1 Pre-existing neutralising antibodies (Nab) to capsid antigen bind to rAAV and inhibit cellular entry, trafficking and uncoating of rAAV. 2 Binding of rAAV by Toll-like receptor-2 (TLR-2) activates innate immunity. 3 Binding of unmethylated cytosine–guanine dinucleotide (CpG) of rAAV genome by Toll-like receptor-9 (TLR-9) activates innate immunity. 4 Double stranded RNA derived from RNA generated from inverted terminal repeat sequences activate innate immune response by binding to Melanoma differentiation associated protein 5 (MDA-5). 5 Capsid peptide from proteosome presented to cytotoxic T-cells (CTL) with major histocompatibility complex-1 (MHC-1). 6 Transgene protein derived peptide from proteosome presented to CTL with MHC-1. 7 Transgene peptide presented on MHC-2 to elicit a humoral response to the transgene protein. 8 Capsid peptide presented on MHC-2 to elicit a humoral response to the capsid protein. Created with BioRender.com
Pre-existing Antibodies and B-Cell Response
Pre-existing antibodies to AAV in circulation can be neutralising or non-neutralising antibodies [21]. Nab against the capsid antigen bind to rAAV and prevent efficient transduction [8]. The Nabs appear due to prior exposure to wtAAV, often early in life [22]. The seroprevalence of Nab varies from 5 to 60% depending on the serotype, with seroprevalence of Nab against AAV2 being one of the highest [23]. Older children tend to have higher rates of seropositivity of Nab compared to infants. These antibodies have wide-cross reactivity across serotypes [24]. Thus, the frequency of such Nab in the population is a function of age, serotype and the geographical location [20]. In view of the negative effect on transduction, patients with Nab beyond certain titres are excluded from studies, even though lower titres of Nab can also significantly affect transduction [2, 25]. Any attempt to mop up these Nab with addition of empty capsids to the rAAV with the transgene expression cassettes as an antibody decoy, would lead to a risk of high capsid dose exposure resulting in higher innate and adaptive cell response to the capsid antigen [26, 27]. Attempts at engineering the capsid structure to minimise the effect of Nab tend to affect transduction [18]. An adaptive immune response to the capsid derived peptides released by the proteosome are presented with major histocompatibility complex class II receptors leading to an adaptive immune response. Nab thus formed often preclude or decrease the efficacy of subsequent administrations of the same rAAV GT. The humoral immunity to capsid is more relevant in the context of efficacy of transduction rather than a predominant direct role in hepatotoxicity, even though a temporal relationship between the humoral response and transaminase elevation has been alluded to in literature [2].
A similar process may result in production of antibodies to the transgene protein, especially in patients whose immune system has never encountered the protein due to the nature of their mutation [28]. This may result in failure of the GT and possible failure of any protein replacement therapy.
Immunomodulation strategies including rapamycin (mToR inhibitor), rituximab (anti-CD20 antibody), imlifidase (cleaves IgG antibodies), proteosome inhibitors and selective plasmapheresis to remove AAV-specific antibodies have been used in animal models and humans to decrease the impact of Nab on transduction [20, 29,30,31,32,33,34,35,36,37,38,39,40].
Innate Immunity
The innate immunity response in rAAV may be responsible for the early adverse events a week after GT administration [41,42,43]. It is invariably a modest response unlike other viruses such as adenovirus even though serious adverse events could be elicited in non-human primates due to an innate immune response [44]. Although this response has not resulted in serious adverse events in humans at doses used in GT along with prophylactic immunosuppressants, it leads to an early peak in transaminases after GT administration and has a role in driving the adaptive immune response [2].
The innate immune response to rAAV is triggered by.
-
(i)
Binding of rAAV by Toll-like receptor-2 (TLR-2) in Kupffer cells (not present in the human primary hepatocyte) [42]
-
(ii)
Binding of unmethylated cytosine–guanine dinucleotide (CpG) of rAAV genome by Toll-like receptor-9 (TLR-9) [45]
-
(iii)
Double stranded RNA (dsRNA) derived from mRNA generated from inverted terminal repeat sequences that activate the immune system by binding to cytoplasmic dsRNA sensor [43]
The activation of the innate immune response by the binding of rAAV to these pathogen recognition receptors leads to the release of pro-inflammatory cytokines through the NF-KB and type-1 interferons. This leads to maturation of antigen presenting cells and activation of neutrophils, macrophages and natural-killer cells [41, 46,47,48].
The activation of TLR-9 by the unmethylated CpG motifs in rAAV can be blunted in GT by removal of the CpG rich sequences from most of the vector genome but the CpG sequences in inverted terminal repeats persist and continue to activate the innate immune system [49].
T-Cell Response
The capsid antigen from the lysosomes is processed by the proteosomes and the resulting capsid derived peptides are presented by MHC-1, resulting in a T-cell response. The T-cell response in rAAV GT is mediated by CD8 + T-cells. The T-cell response in humans was initially attributed to an exclusive memory T-cell response in those individuals exposed earlier to AAV [50]. The onset of this response in humans is often after a month of rAAV GT thus indicating a primary T-cell response rather than an early memory T-cell response [2, 4]. The CD8 + cellular response results in destruction of the transduced cells which was not noted in animal studies [42, 51, 52]. The chemokines produced by the innate immune response have a role in priming the CD8 + cells towards a cellular response. The T-cell response in liver results in hepatocellular dysfunction. The hepatotoxicity may result in reduced transgene expression if the target of transduction was the liver [4]. Despite the inability of rAAV to produce new capsids than what has been already administered, the T-cell response to the administered capsid proteins that undergo degradation continues to persist for months [53]. The CD8 + T-cell response may resolve over time with the infiltration of CD4 + CD25 + FoxP3 + T-regulatory cellular infiltration in the transduced tissue and with the CD8 + cells acquiring an exhausted T-cell phenotype [54].
Host genetic factors also influence the variability in immune response between individuals. Polymorphism in the IL6R gene may be a factor in variable immune response between individuals and this is known to contribute to long term factor IX expression in rAAV GT despite CpG enrichment [55].
Although the rAAV vector in GT is not capable of proliferation, AAV capsid has been detected for longer periods in tissues depending on the route of administration. Studies on NHP have shown that 6 years after subretinal injections, rAAV2 and rAAV5 capsids were still detected in the retina [56]. rAAV1 capsids have been demonstrated in muscles at 12-month follow-up after intramuscular injection of the vector in phase II gene transfer study for AAT deficiency. Immunofluorescent staining of the capsid was demonstrated within myofibers of the muscle, mostly in distinct foci at the perinuclear region [54].
Complement System
The complement system is implicated in the causation of thrombotic microangiopathy when rAAV is used in very high doses and has been shown to be important in the development of a humoral response [57, 58]. It is not known to be involved in the causation of hepatotoxicity.
Effect of the Route of Administration on Immune Response
Although the liver is not immunologically privileged or isolated from the systemic immune system unlike the eye, intrahepatic rAAV GT may offer immunological advantages in terms of antigenic tolerance to the transgene and the capsid [59]. The liver and its draining lymph nodes provide a microenvironment conducive to tolerance induction. The antigen presentation by Kupffer cells, dendritic cells in the liver and the liver sinusoidal endothelial cells results in tolerance induction by inducing FoxP3 + Tregs, apoptosis of effector T-cells and other mechanisms including T-cell exhaustion [60,61,62,63].
Pattern of Hepatotoxicity in rAAV-GT
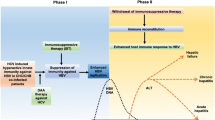
The general pattern of liver injury is of a hepatocellular liver injury. Children with an underlying cholestatic liver disease as in myotubular myopathy may have an exacerbation of the cholestasis. The liver injury may have a dual peak in its severity as is the case in onasemnogene abeparvovec (AVXS-101) for spinal muscular atrophy (SMA). The early peak in transaminases in the first week because of the innate immune response is modest and dampened by the high doses of prophylactic immunosuppression used. As the month-long steroid dosing is tapered, the liver injury flares up to a second peak. This flare can result in acute liver failure if the steroid tapering is too rapid. The flare tends to respond to increase in immunosuppression. The transaminases normalise eventually requiring a variable duration of immunosuppression.
Hepatotoxicity—a Double Jeopardy
The hepatic dysfunction seen in patients receiving rAAV GT is always not due to an immune response exclusively. Some of the disorders treated with rAAV have underlying liver abnormalities that are exacerbated by the immune response. Patients receiving GT are screened for significant liver injury to preclude them from trials and clinical use. The parameters that determine the exclusion of patients from GT is likely to be different for different disorders and these parameters are ill-defined and empirical at present [2, 64]. The inherent liver abnormalities described in some of the disorders receiving GT are detailed below. Irrespective of such associations, all such patients will need screening for other liver co-morbidities (Table 2).
Spinal Muscular Atrophy
Liver dysfunction in patients with SMA is indicated often by mild elevation in the liver transaminases. Although SMA is not myopathy, release of enzymes from muscles could also lead to elevation of AST and ALT. Chand et al. had suggested that the associated elevation of gamma glutamyl transpeptidase (GGT) may indicate the source of enzyme elevation to be the liver [2]. This may not always be the case and it may be worthwhile investigating an elevation in transaminases assuming the source to be the liver.
In an analysis of patients enrolled in clinical trials, 61% had elevations in ALT and/or AST and/or bilirubin concentrations prior to receiving the drug. These were below the threshold for exclusion from the study and normalised over time [2]. These abnormal liver function tests are possibly related to hepatic steatosis. Children with SMA are known to have dyslipidaemia with a propensity for hepatic steatosis. This is attributed to abnormal fatty acid metabolism. An abnormal beta-oxidation (e.g. dicarboxylic aciduria) is unique to SMA compared to non-SMA denervating disorders [65,66,67]. The underlying mechanism of this defect has been hypothesised to be either the deficiency of SMN protein or the dysfunction of genes adjacent to SMN gene. This may result in a higher sensitivity to superimposed liver injury or metabolic stress. Reye-syndrome like acute liver failure requiring liver transplantation has been reported in SMA resulting from a combination of risk factors including possible underlying abnormality of beta-oxidation, prolonged intraoperative propofol and sevoflurane exposure, post-operative acetaminophen, stress induced lipolysis and reduced perioperative glucose intake [68]. Children with SMA are also reported to be susceptible to paracetamol toxicity with therapeutic doses, which could be related to reduced muscle mass, glutathione stores and volume of distribution of the drug [69].
AVXS-101 as GT is given as a single infusion in patients with a bi-allelic mutation in the SMN1 gene and up to 3 copies of the SMN2 gene. It delivers a nonintegrating, fully functional copy of SMN1 into motor neuron cells using self-complementary adeno-associated viral vector (AAV) serotype 9 (AAV9) [2].
A total of 90% of the children treated with AVXS-101 had some degree of ALT and/or AST elevation during therapy irrespective of the baseline elevation in serum transaminases. 90% of these children had transaminase elevation that was < 3 × ULN while 9% were categorised as mild elevation (3 to 5 times ULN), 6% as moderate (5 to 20 × ULN) and 5% as severe (> 20 × ULN). The elevation in liver enzymes was noted a week after drug infusion with a second peak a month after infusion when steroid tapering commenced and later when steroids was stopped [2]. In the managed access programme, two children developed acute liver failure 3 to 8 weeks after infusion, especially when the steroid taper was rapid. Both these children responded to high dose steroids. The liver biopsy in these patients show expanded portal triad, interface hepatitis with neutrophils and CD8 + T-cells, with occasional CD4 + T-cells, B-cells and eosinophils, lobular lymphocytic infiltration with CD8 + T-cells, diffuse hepatocellular swelling/ballooning and mild portal fibrosis, fibrous septae and lobular pericellular fibrosis [5•]. The transaminase elevation was shown to follow the course of elevation of antibodies to AAV-9 vector but this does not necessarily attribute the adverse event to a humoral response rather than a CD8 + T-cell mediated response.
X‑Linked Myotubular Myopathy (XLMTM)
Patients with XLMTM are known to have raised transaminases, alkaline phosphate and GGT, hepatomegaly, cholestatic jaundice, pruritis, cholelithiasis, acute cholecystitis, hepatic steatosis and peliosis hepatis. Although earlier retrospective series had underestimated the frequency of liver abnormalities in XLMTM, a series from Italy showed that elevated serum transaminases with gallstones and abnormal liver structure (including abnormal echogenicity or haemorrhagic spaces) on imaging were detected in 42% and 58% of patients, respectively. The abnormalities were not progressive without correlation to age, disease duration, clinical severity, or type of MTM1 mutation [70]. In the INCEPTUS cohort, 91% of patients had a history of hepatobiliary disease at enrolment or had evidence of hepatic disease in follow-up. This was present in participants across all three mutation types. Patients with nonsense mutations had the most prevalent and pronounced abnormalities [71]. Amburgey et al. studied 33 patients with XLMTM for one year who demonstrated hepatic manifestations in the form of abnormal serum liver enzymes in 22.5%, hepatomegaly in 11.8%, jaundice in 14.7% and liver haemorrhage in 5.9% [72]. Hepatic peliosis can lead to death due to bleeding in the liver [71]. Hypothesis on the mechanism of liver manifestations include defects in autophagy and the impaired lipid layer trafficking [73].
Although not approved for clinical use, the outcomes in the ASPIRO clinical trial on AT132 (resamirigene bilparvovec) to treat MTM provided valuable lessons on the role of vector doses and the influence of an underlying liver pathology intrinsic to the disease being treated and the limited value of animal studies [6•]. AT132 was a rAAV-8 vector expressing MTM1 gene. The ASPIRO study recruited 24 XLMTM patients in total who received AT132. More than half of those recruited had baseline evidence of liver disease including intermittent direct hyperbilirubinemia, transaminase elevation, or cholestasis in the past. Seven received a lower dose of 1 × 1014 vg/kg dose and 17 dose of 3 × 1014 vg/kg dose. Three children in the high-dose cohort died within 20–40 weeks of receiving AT-132. They were older children who were over 5 years of age at the time of death and thus had received high vector doses (range 4.8–7.7 × 1015 vg) compared to younger and lighter children in the high dose cohort. All three children had pre-existing hyperbilirubinemia and while on the drug developed direct hyperbilirubinemia leading to a decompensated liver disease. Though the immediate cause of death was sepsis in two patients and gastrointestinal bleed in the other, the underlying mechanism of their hepatotoxicity is not understood. The autopsy showed intrahepatic and canalicular cholestasis, bile ductular reaction, fibrosis and absence of any prominent liver parenchymal inflammatory infiltrates. One child showed lesions that may be consistent with hepatic peliosis. The study was held by FDA and then recommenced with only with the lower dose cohort as the deaths were attributed to the higher dose. The first patient administered the lower dose (1.3 × 1014 vg/kg) on recommencing the trial died, leading to termination of the study. It is important to note that the dose labelled as a ‘low dose’ was still a significantly high vector dose by the standards of trials in other diseases [6•, 19, 74, 75].
Haemophilia
Liver disease in haemophilia may be a result of parenterally transmitted chronic hepatitis C virus infection in adult patients who had received contaminated plasma-derived coagulation factors. The association of human immunodeficiency virus infection in some of these patients can add to the liver injury [76].
Endogenous production of factor VIII and factor IX happens in the liver sinusoidal endothelial cells and hepatocytes, respectively. Regardless of the endogenous source of the proteins, the cellular target for transgene expression in AAV GT of haemophilia A and B is the hepatocyte for both haemophilia A (factor VIII deficiency) and haemophilia B (factor IX deficiency) [14,15,16].
Clinical trials in haemophilia have provided valuable information on the role of the innate and adaptive immune system in hepatotoxicity and durability of transgene expression in the liver. They highlighted the limitations in the animal models while drawing attention to the fact that hepatotoxicity in rAAV GT may not necessarily parallel the measurable biomarkers of adaptive immune response in the serum. Apart from the importance of screening patients for neutralising antibodies to allow for transduction, the data underscored the persistence of high-titre, multi-serotype cross-reactive AAV NAbs following GT [50, 77,78,79]. Nathwani et al. demonstrated dose dependent expression of factor IX levels, with higher levels in those who received higher doses of the vector [80]. Liver transaminases elevation was noted with higher vector doses. The rise in transaminases was associated with a concomitant drop in factor IX levels due to the destruction of the transduced hepatocytes. This phenomenon was demonstrated to be associated with a cytotoxic T-cell response in early trials [4]. Transient courses of rescue corticosteroids lead to normalisation of liver enzymes and continued expression of factor IX with demonstration of long-term benefit from the GT after 8 years [81]. This experience could not be replicated in all studies as the factor IX levels were sustained only for 5–11 weeks in most patients [55, 78, 82]. The fall in factor IX levels was not correlated to an immune response to the transgene protein. The fall in factor IX levels with rising transaminases could not be adequately rescued with corticosteroids. The role of an early innate immune response against CpG motifs was demonstrated, thus explaining the inability to calm the T-cell response by corticosteroids and the eventual lack of clinically significant response [55]. Subsequent studies by George et al. utilizing vector codon optimization with reduced CpG content and high specific activity factor IX Padua variant that enabled use of lower vector doses resulted in significant reduction in bleeding rates. Immune mediated transaminase elevation was amenable to treatment by steroids [83, 84]. Studies using a factor IX Padua variant of the transgene, which have a higher activity than the wild type of factor IX, have been able to demonstrate sustained factor IX activity in those with low or intermediate levels of pre-existing Nab [85].
Clinical trials of rAAV GT in haemophilia A showed sustained medium term factor VIII levels. In the long term, there is gradual loss of factor VIII expression which is not related to an immune response. Any intermittent immune response measured by IFN-γ and TNF-α fluorospot with either AAV5 or factor VIII peptide stimulation may not necessarily be associated with liver transaminase elevation or decrease in transgene expression [77, 86]. Delayed initiation of an immune response and transaminase elevation after 3 to 4 months is a unique feature of factor VIII trials. The reactive and prophylactic courses of steroids used in factor VIII GT have been much longer courses compared to factor IX GT trials. The long-term decline in synthesis of factor VIII level could be related to its synthesis by liver sinusoidal endothelial cells rather than in hepatocytes, natural senescence and turnover of hepatocytes [77].
Risk Mitigation by Patient Selection
Risk mitigation by patient selection for clinical trials and approved clinical use of drugs usually focus on [1] pre-existing Nab to AAV capsid or antibodies towards the protein product of the transgene (pre-existing antibodies to coagulation factor in haemophilia) and [2] pre-existing liver abnormalities. The level of pre-existing antibodies that are a contraindication for GT vary between trials and more recent haemophilia trials have pushed the boundaries on the acceptable levels of Nab [85]. The focus on pre-existing antibodies is for the purpose of improving transduction rather than for alleviating adverse events. The absence of or lower levels of Nab do not preclude hepatotoxicity. The utility of measuring any pre-existing cellular immunity to AAV is of doubtful value in predicting hepatotoxicity. The timeline of onset of hepatoxicity is often indicative of an early innate immune response followed by a new onset T-cell response rather than a memory response [2, 81]. The latter patient selection criteria on the degree of hepatic dysfunction that would effectively mitigate hepatotoxicity is less well defined and variable between studies. The parameter used could be the serum transaminase levels, the serum bilirubin level or the degree of fibrosis. The parameter that would be best judge of an exclusion criteria could vary between diseases and the vector dose used [2, 64, 74]. The fine balance between safety and benefiting more patients with GT could be difficult to achieve.
Avoiding hepatotoxic medications is an essential part of risk mitigation. In addition, avoidance of rapid prophylactic steroid tapering could prevent severe flares of liver injury that could lead to liver dysfunction.
Clinical Use of Immunosuppression Protocols Used in Hepatotoxicity
Pre-clinical and clinical use of immunosuppressant protocols in rAAV GT have been for the following purposes:
-
(i)
Depleting circulating pre-existing Nabs to allow for improved transduction and expanding the recipient cohort that can receive the GT
-
(ii)
Prophylactic immunosuppression (PIS) to dampen innate immune response to decrease adverse event during the early post-administration phase
-
(iii)
Reactive immunosuppression (RIS) to suppress adaptive immune responses that lead to adverse events and inhibition of transgene expression
Considering the predominant role of innate and cellular adaptive immunity in hepatotoxicity, Nab depletion may have no effect on hepatotoxicity. In view of the rarity of hepatotoxicity in animal studies there is lack of data to draw from animal trials regarding the use of immunosuppressants, prophylactic and reactive immunosuppression for hepatotoxicity. While awaiting advances in rAAV GT that would avoid hepatotoxicity, the goal of PIS should be to preclude any hepatotoxicity with the least drug induced adverse events. Present PIS regimens that are mainly based on corticosteroids have not achieved this goal, including that of preventing severe adverse events. RIS has again used high dose steroids. Although short courses alleviate adverse events in many cases, the long courses required in some patients prompt a need to investigate second line immunosuppressants [2, 7••, 77]. There is also a need to explore options beyond steroids to treat serious adverse events that carry a risk of mortality and high morbidity [5•].
Corticosteroids
Corticosteroids are widely used drugs in rAAV GT in view of their wide impact on the immune system to include innate and adaptive immune system encompassing both the cellular and humoral systems. Corticosteroids are used in haemophilia GT as prophylactic drugs and also in reactive protocols whenever there was transaminase elevation with or without a decline in loss of transgene expression [3, 4, 84]. These reactive treatments were often short courses of steroids in factor IX GT [4]. The contrasting long courses of steroids used in factor VIII GT are associated with steroid-associated adverse events [77].
In XLMTM, AT132 was administered along with prednisolone as a prophylactic medication used at 1 mg/kg/day for 8 weeks after administering the drug and tapered over the subsequent 8 weeks. Corticosteroids were used to treat hepatoxicity along with ursodeoxycholic acid in patients who worsened but subsequently died. The benefit of corticosteroids in modifying outcomes in patients with hepatotoxicity in XLMTM is not known, considering that the inflammatory infiltrates were minimal in the liver at autopsy [6•, 19, 74, 75].
The initial protocol used for steroid use as a prophylactic and reactive therapy for hepatoxicity in AVXS-101 treatment of SMA is described in Table 3. Presently, the suggested regimen on the tapering of steroids was discontinued and the tapering regimen was left to the discretion of the treating clinician (Table 3). The reported mean duration of prednisolone treatment in the studies in these patients (n = 100) was 83 days (range 33–229 days; SD 60.6 days) [2]. The data does not differentiate those requiring a low-dose of prednisolone from those on higher dose steroids [2]. Acute liver failure was reported in two patients with INR > 1.5 with encephalopathy. The onset was between 3 and 8 weeks post infusion of AVXS-101, in spite of steroids. One of the patients had a rapid tapering of steroids. Both the patients received methylprednisolone starting at 20 mg/kg/day which was gradually tapered with improvement [5•].
The reported prolonged duration of transaminase elevation is concerning [2, 7••, 77]. A sizeable group of patients had steroid usage for more than 90 days. Individual variations in the practice of tapering steroids could contribute to variable duration of steroid usage [2]. The effect of long courses of steroids on muscle mass and function needs to be evaluated. A formal study of steroid adverse events in these patients is lacking.
Although self-limiting, the effect of prolonged liver inflammation is not known. Performing liver biopsies in patients with prolonged transaminase elevation and neuromuscular weakness is challenging and should be reserved for a select group of patients with acceptable anaesthetic risk. The role of non-invasive biomarkers of fibrosis is not well established in children in routine clinical practice. It is essential that these patients have their liver stiffness assessed by ultrasound elastography once the inflammation subsides.
There are no biomarkers that could identify patients who will need prolonged use of immunosuppressive agents. The time of initiation of second-line immunosuppressants in those who persist to have prolonged transaminase elevation is empirical at the most and, in the authors’ view, should be around 60 days. It would have to be balanced with the understanding that some patients might normalise their transaminases while drugs such as MMF take time to act from the time of their initiation. Second line immunosuppressants may not be justified when steroid weaning could be achieved in few months and without steroid toxicity.
Sirolimus
Sirolimus inhibits mTOR and thereby cytotoxic T-cells, T-helper cells, upregulate T-regulatory cells and also inhibit the B-cell response. The earlier of sirolimus in rAAV GT has been in combination with rituximab based on its use in animal studies. The primary aim of using sirolimus in these studies was to attenuate the antibody formation in combination with rituximab leading to a decrease in pre-existing Nabs and improving transduction [35]. The facilitation of autophagy by the drug is also known to enhance transduction [87]. Down-regulation of T-cell and B-cell response hampers the development of antibodies to rAAV capsid and transgene, thus improving efficacy of GT and allowing for re-administration of GT [30, 33]. Rituximab and sirolimus (serum trough level of 3–7 ng/ml) were used in a cross-reacting immunologic material-negative (CRIM-ve) child with Pompe disease who initially received enzyme replacement therapy followed by rAAV1-CMV-hGAA and was shown to not develop antibodies to rAAV capsid and along with the absence of a T-cell response [31]. Yet, the role of sirolimus on prevention of hepatotoxicity is not known. It could be argued that sirolimus upregulates T-regulatory cells and thereby encourage tolerance to rAAV capsid and the transgene [88]. More recent trials have used sirolimus for both PIS and RIS for rAAV-induced hepatotoxicity is sparse. The recently published clinical trial of rAAV GT in Crigler-Najjar syndrome used methylprednisolone, 8 weeks of prednisolone and 12 weeks of sirolimus as PIS followed by steroids and sirolimus also for RIS in the initial few patients. Eventually, this resulted in a change in their practice to the use to the use of sirolimus for a prolonged course of 48 weeks in patients who received GT later, rather than the 12-week course. Although this was effective in suppressing the hepatotoxicity and maintaining efficacy of the liver-targeted GT, the development of antibodies to AAV could not be prevented [89].
Mycophenolate Mofetil (MMF)
MMF is a purine analogue that suppresses T-cell and B-cell proliferation by inhibiting type II Inosine monophosphate dehydrogenase that is essential for de novo purine synthesis. Based on the data in mice that MMF reduces vector genome second strand synthesis by depleting intracellular guanosine triphosphate, MMF was felt to be unsuitable as an immunosuppressant for the early phase of transduction using single-stranded rAAV. However, MMF did not impair transduction by a self-complementary rAAV vector, similar to that used in AVXS-101 [90]. Studies in non-human primates with MMF added 4 weeks after single-stranded rAAV infusion showed that in spite of the delayed introduction of MMF to allow for second strand synthesis, transgene expression was inhibited by MMF through an immunological mechanism [91]. In contrast, Jiang et al. did not demonstrate impaired transduction or FIX synthesis in rhesus macaques with coadministration of MMF and tacrolimus along with rAAV [92] MMF was used as prophylactic drug alongside cyclosporine with or without methylprednisolone in Glybera GT in humans but failed to prevent development of a T-cell and B-cell response to the capsid or the transgene product [11]. MMF may be used as a steroid sparing agent in reactive treatment of prolonged hepatotoxicity.
Calcineurin Inhibitors
Non-depleting anti-CD4 antibody and cyclosporine have been used in mice to decrease Nabs to allow for repeat administration [93]. Tacrolimus was effective in one patient to treat hepatotoxicity in rAAV GT for haemophilia B in the B-AMAZE study in addition to its use as a prophylactic agent in combination with prednisolone [3]. The use of calcineurin inhibitors (cyclosporin and tacrolimus) raises the concerns regarding downregulation of T-regulator cells and a potential loss of tolerance to the transgene product [88]. This has not been verified in the clinical context in humans.
Rituximab
Rituximab reduces Nabs to AAV in rheumatoid arthritis patients [32]. The present use of rituximab alone or in combination with sirolimus in rAAV GT decreases pre-existing Nabs and development of Nab resulting from therapy [30, 31, 34, 35]. Although a Chand et al. demonstrate a temporal correlation between the course of development of Nabs and transaminase elevation, Nabs are unlikely to be a major contributor to hepatotoxicity [2]. Rituximab may not be useful as a reactive therapy for hepatotoxicity.
Timing of Immunosuppression and T-Regulatory Cells
The relevance of timing of administration of immunosuppression was demonstrated in nonhuman primates by comparing early anti-thymocyte globulin (ATG) administration along with rAAV with delayed ATG administration at 5 weeks. Development of anti-FIX immune response was associated more often with the former regimen. The Th17/regulatory T-cell ratio determines transgene-product immunogenicity or tolerance [94]. Daclizumab, an anti-CD25 antibody, when used early along with sirolimus and MMF during AAV administration of rAAV vector leads to transient depletion of CD4+CD25+FoxP3+ T-regulatory cells in non-human primate models along with an increase in anti-AAV capsid antibodies and anti-factor IX antibodies when compared to the use of sirolimus and MMF without daclizumab [95]. The effects of the timing of immunosuppression on T-regulatory cells are important in understanding the implications of any prophylactic regimen that may affect T-regulatory cells. As a corollary, reactive immunosuppressants such as MMF administered for hepatotoxicity at a later phase should have minimal effect on development of tolerance to the transgene product.
Suggested Pharmacotherapy and Monitoring of rAAV GT Related Hepatotoxicity
Considering the widespread clinical use of rAAV GT, there is a need for empirical guidelines on RIS. Our clinical practice for the management of AVXS-101 related hepatotoxicity is as follows (Fig. 2) [96].

Algorithm for use of second-line reactive immunosuppression in children receiving AVXS-101
High dose intravenous methylprednisolone (10–20 mg/kg/day) may be indicated in.
-
(i)
Elevation of transaminases > 20 times upper limit of normal (ULN) or rise in bilirubin while on oral prednisolone at 2 mg/kg/day for more than 2 weeks or
-
(ii)
Alanine aminotransferase (ALT) or aspartate aminotransferase (AST) > 3 times ULN with liver dysfunction (INR > 1.5) or
-
(iii)
ALT or AST > 3 times ULN with associated fatigue, nausea, vomiting, right upper quadrant pain, or tenderness or fever [97]
Tacrolimus may be indicated in patients who have not responded adequately to high dose methylprednisolone. A target 12 h trough serum level could be 6–8 ng/dl.
Thresholds for second line steroid-sparing immunosuppressants could be.
-
(i)
AST or ALT > 3 times ULN despite > 2 weeks of prednisolone at 2 mg/kg/day or
-
(ii)
A need for prolonged (more than 60 days) requirement of prednisolone at more than acceptable doses (more than 0.25–0.5 mg/kg/day) during steroid tapering period to maintain AST or ALT < 3 times ULN
-
(iii)
Steroid toxicity
Mycophenolate mofetil (MMF) may be the first choice of second line steroid-sparing immunosuppressants with sirolimus being considered the second choice in those who do not tolerate MMF. In our experience in few children, where steroid weaning was not possible or in those who developed severe liver dysfunction like prolonged prothrombin time unresponsive to vitamin K, the use of MMF and tacrolimus was effective.
The management decisions for rAAV GT related hepatotoxicity should be the outcome of multidisciplinary team discussion which should include a hepatologist [7••, 96]. The threshold of serum transaminase elevation that warrants RIS is likely to much lower in children where the rAAV-GT is directed towards transgene expression in the liver to avoid loss of efficacy. There is a trend towards maintaining normal serum transaminase levels in these patients [89].
Conclusions
Hepatotoxicity with rAAV GT could be a challenging problem either due to profound or prolonged liver injury or compromised transgene expression in the liver. There is a need to better understand the mechanisms of immunological injury, the modalities for early and reliable identification of at-risk individuals, the effect and choice of prophylactic and reactive immunosuppressants and long-term effects of liver injury and immunosuppressants. Protocols for immunosuppressants will need to move beyond steroids. There is a need for wider partnership amongst all stake holders including patient groups on all aspects of GT-related hepatotoxicity and its management.
Abbreviations
- CK:
-
Creatinine kinase
- CpG:
-
Cytosine–guanine dinucleotide
- EMA:
-
European Medicines Agency
- GGT:
-
Gamma glutamyl transpeptidase
- GT:
-
Gene therapy
- MMF:
-
Mycophenolate mofetil
- Nab:
-
Neutralising antibodies
- PIS:
-
Prophylactic immunosuppression
- RIS:
-
Reactive immunosuppression
- rAAV:
-
Recombinant adeno-associated virus
- SMA:
-
Spinal muscular atrophy
- TLR-2:
-
Toll-like receptor-2
- TLR-9:
-
Toll-like receptor-9
- USFDA:
-
United States Food and Drugs Administration
- ULN:
-
Upper limit of normal
- XLMTM:
-
X-linked myotubular myopathy
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Shen W, Liu S, Ou L. rAAV immunogenicity, toxicity, and durability in 255 clinical trials: a meta-analysis. Front Immunol. 2022;13:1001263.
Chand D, Mohr F, McMillan H, Tukov FF, Montgomery K, Kleyn A, et al. Hepatotoxicity following administration of onasemnogene abeparvovec (AVXS-101) for the treatment of spinal muscular atrophy. J Hepatol. 2021;74(3):560–6.
Chowdary P, Shapiro S, Makris M, Evans G, Boyce S, Talks K, et al. Factor IX Expression within the normal range prevents spontaneous bleeds requiring treatment following FLT180a gene therapy in patients with severe hemophilia B: long-term follow-up study of the B-amaze program. Blood. 2021;138:3967.
Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365(25):2357–65.
• Feldman AG, Parsons JA, Dutmer CM, Veerapandiyan A, Hafberg E, Maloney N, et al. Subacute liver failure following gene replacement therapy for spinal muscular atrophy type 1. J Pediatr. 2020;225:252–8.e1. Describes serious adverse events following rAAV GT.
• Philippidis A. Fourth boy dies in trial of astellas gene therapy candidate 2021 [updated 15th September, 2021. Available from: https://www.genengnews.com/news/fourth-boy-dies-in-trial-of-astellas-gene-therapy-candidate/. Describes serious adverse events following rAAV GT.
•• Miesbach W, Foster GR, Peyvandi F. Liver-related aspects of gene therapy for haemophilia: call to action for collaboration between haematologists and hepatologists. J Hepatol. 2023;78(3):467–70. Describes the need for multidisciplinary approach and the need for involvement of hepatologists in the management of rAAV GT related hepatotoxicity.
Shirley JL, de Jong YP, Terhorst C, Herzog RW. Immune responses to viral gene therapy vectors. Mol Ther. 2020;28(3):709–22.
Ho A, Orton R, Tayler R, Asamaphan P, Tong L, Smollett K, et al. Adeno-associated virus 2 infection in children with non-A-E hepatitis. medRxiv. 2022:2022.07.19.22277425.
Morfopoulou S, Buddle S, Torres Montaguth OE, Atkinson L, Guerra-Assunção JA, Storey N, et al. Genomic investigations of acute hepatitis of unknown aetiology in children. medRxiv. 2022:2022.07.28.22277963.
Ferreira V, Petry H, Salmon F. Immune responses to AAV-vectors, the glybera example from bench to bedside. Front Immunol. 2014;5:82.
European Medicines Agency recommends first gene therapy for approval [updated 20th July 2012. Available from: https://www.ema.europa.eu/en/news/european-medicines-agency-recommends-first-gene-therapy-approval.
FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality [updated 24th May 2019. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease#:~:text=The%20U.S.%20Food%20and%20Drug,genetic%20cause%20of%20infant%20mortality.
FDA approves first gene therapy to treat adults with hemophilia B [updated 22nd November 2022. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapy-treat-adults-hemophilia-b.
First gene therapy to treat haemophilia B: European medicines agency; 2022 [updated 16/12/2022. Available from: https://www.ema.europa.eu/en/news/first-gene-therapy-treat-haemophilia-b.
First gene therapy to treat severe haemophilia A [updated 24th June 2022. Available from: https://www.ema.europa.eu/en/news/first-gene-therapy-treat-severe-haemophilia.
Pupo A, Fernández A, Low SH, François A, Suárez-Amarán L, Samulski RJ. AAV vectors: The Rubik’s cube of human gene therapy. Mol Ther. 2022;30(12):3515–41.
Li C, Samulski RJ. Engineering adeno-associated virus vectors for gene therapy. Nat Rev Genet. 2020;21(4):255–72.
Cellular, tissue, and gene therapies advisory committee September 2–3, 2021 Meeting Announcement [updated 24th November 2021. Available from: https://www.fda.gov/advisory-committees/advisory-committee-calendar/cellular-tissue-and-gene-therapies-advisory-committee-september-2-3-2021-meeting-announcement.
Yang TY, Braun M, Lembke W, McBlane F, Kamerud J, DeWall S, et al. Immunogenicity assessment of AAV-based gene therapies: an IQ consortium industry white paper. Mol Ther Methods Clin Dev. 2022;26:471–94.
Fitzpatrick Z, Leborgne C, Barbon E, Masat E, Ronzitti G, van Wittenberghe L, et al. Influence of pre-existing anti-capsid neutralizing and binding antibodies on AAV vector transduction. Mol Ther Methods Clin Dev. 2018;9:119–29.
Calcedo R, Vandenberghe LH, Gao G, Lin J, Wilson JM. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis. 2009;199(3):381–90.
Louis Jeune V, Joergensen JA, Hajjar RJ, Weber T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum Gene Ther Methods. 2013;24(2):59–67.
Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF, et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21(6):704–12.
Meadows AS, Pineda RJ, Goodchild L, Bobo TA, Fu H. Threshold for pre-existing antibody levels limiting transduction efficiency of systemic rAAV9 gene delivery: relevance for translation. Mol Ther Methods Clin Dev. 2019;13:453–62.
Mingozzi F, Anguela XM, Pavani G, Chen Y, Davidson RJ, Hui DJ, et al. Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci Transl Med. 2013;5(194):194ra92.
Mingozzi F, High KA. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122(1):23–36.
Herzog RW, Cooper M, Perrin GQ, Biswas M, Martino AT, Morel L, et al. Regulatory T cells and TLR9 activation shape antibody formation to a secreted transgene product in AAV muscle gene transfer. Cell Immunol. 2019;342:103682.
Chu WS, Ng J. Immunomodulation in administration of raav: preclinical and clinical adjuvant pharmacotherapies. Front Immunol. 2021;12:658038.
Corti M, Cleaver B, Clément N, Conlon TJ, Faris KJ, Wang G, et al. Evaluation of readministration of a recombinant adeno-associated virus vector expressing acid alpha-glucosidase in pompe disease: preclinical to clinical planning. Hum Gene Ther Clin Dev. 2015;26(3):185–93.
Corti M, Elder M, Falk D, Lawson L, Smith B, Nayak S, et al. B-cell depletion is protective against anti-AAV capsid immune response: a human subject case study. Mol Ther Methods Clin Dev. 2014;1:14033-.
Mingozzi F, Chen Y, Edmonson SC, Zhou S, Thurlings RM, Tak PP, et al. Prevalence and pharmacological modulation of humoral immunity to AAV vectors in gene transfer to synovial tissue. Gene Ther. 2013;20(4):417–24.
Velazquez VM, Meadows AS, Pineda RJ, Camboni M, McCarty DM, Fu H. Effective depletion of pre-existing anti-AAV antibodies requires broad immune targeting. Mol Ther Methods Clin Dev. 2017;4:159–68.
Corti M, Liberati C, Smith BK, Lawson LA, Tuna IS, Conlon TJ, et al. Safety of intradiaphragmatic delivery of adeno-associated virus-mediated alpha-glucosidase (rAAV1-CMV-hGAA) gene therapy in children affected by pompe disease. Hum Gene Ther Clin Dev. 2017;28(4):208–18.
Mueller C, Berry JD, McKenna-Yasek DM, Gernoux G, Owegi MA, Pothier LM, et al. SOD1 suppression with adeno-associated virus and MicroRNA in familial ALS. N Engl J Med. 2020;383(2):151–8.
Biswas M, Palaschak B, Kumar SRP, Rana J, Markusic DM. B Cell depletion eliminates FVIII memory B cells and enhances AAV8-coF8 immune tolerance induction when combined with rapamycin. Front Immunol. 2020;11:1293.
Leborgne C, Barbon E, Alexander JM, Hanby H, Delignat S, Cohen DM, et al. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat Med. 2020;26(7):1096–101.
Mitchell AM, Samulski RJ. Mechanistic insights into the enhancement of adeno-associated virus transduction by proteasome inhibitors. J Virol. 2013;87(23):13035–41.
Monahan PE, Lothrop CD, Sun J, Hirsch ML, Kafri T, Kantor B, et al. Proteasome inhibitors enhance gene delivery by AAV virus vectors expressing large genomes in hemophilia mouse and dog models: a strategy for broad clinical application. Mol Ther. 2010;18(11):1907–16.
Chicoine LG, Montgomery CL, Bremer WG, Shontz KM, Griffin DA, Heller KN, et al. Plasmapheresis eliminates the negative impact of AAV antibodies on microdystrophin gene expression following vascular delivery. Mol Ther. 2014;22(2):338–47.
Zhu J, Huang X, Yang Y. The TLR9-MyD88 pathway is critical for adaptive immune responses to adeno-associated virus gene therapy vectors in mice. J Clin Invest. 2009;119(8):2388–98.
Hösel M, Broxtermann M, Janicki H, Esser K, Arzberger S, Hartmann P, et al. Toll-like receptor 2-mediated innate immune response in human nonparenchymal liver cells toward adeno-associated viral vectors. Hepatology. 2012;55(1):287–97.
Shao W, Earley LF, Chai Z, Chen X, Sun J, He T, Deng M, Hirsch ML, Ting J, Samulski RJ, Li C. Double-stranded RNA innate immune response activation from long-term adeno-associated virus vector transduction. JCI Insight. 2018;3(12):e120474
Hinderer C, Katz N, Buza EL, Dyer C, Goode T, Bell P, et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum Gene Ther. 2018;29(3):285–98.
Martino AT, Suzuki M, Markusic DM, Zolotukhin I, Ryals RC, Moghimi B, et al. The genome of self-complementary adeno-associated viral vectors increases Toll-like receptor 9-dependent innate immune responses in the liver. Blood. 2011;117(24):6459–68.
Rogers GL, Suzuki M, Zolotukhin I, Markusic DM, Morel LM, Lee B, et al. Unique roles of TLR9- and MyD88-dependent and -independent pathways in adaptive immune responses to AAV-mediated gene transfer. J Innate Immun. 2015;7(3):302–14.
Shirley JL, Keeler GD, Sherman A, Zolotukhin I, Markusic DM, Hoffman BE, et al. Type I IFN Sensing by cDCs and CD4(+) T cell help are both requisite for cross-priming of AAV capsid-specific CD8(+) T Cells. Mol Ther. 2020;28(3):758–70.
Rogers GL, Shirley JL, Zolotukhin I, Kumar SRP, Sherman A, Perrin GQ, et al. Plasmacytoid and conventional dendritic cells cooperate in crosspriming AAV capsid-specific CD8(+) T cells. Blood. 2017;129(24):3184–95.
Faust SM, Bell P, Cutler BJ, Ashley SN, Zhu Y, Rabinowitz JE, et al. CpG-depleted adeno-associated virus vectors evade immune detection. J Clin Invest. 2013;123(7):2994–3001.
Mingozzi F, Maus MV, Hui DJ, Sabatino DE, Murphy SL, Rasko JE, et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med. 2007;13(4):419–22.
Wang L, Figueredo J, Calcedo R, Lin J, Wilson JM. Cross-presentation of adeno-associated virus serotype 2 capsids activates cytotoxic T cells but does not render hepatocytes effective cytolytic targets. Hum Gene Ther. 2007;18(3):185–94.
Pien GC, Basner-Tschakarjan E, Hui DJ, Mentlik AN, Finn JD, Hasbrouck NC, et al. Capsid antigen presentation flags human hepatocytes for destruction after transduction by adeno-associated viral vectors. J Clin Invest. 2009;119(6):1688–95.
Mingozzi F, Meulenberg JJ, Hui DJ, Basner-Tschakarjan E, Hasbrouck NC, Edmonson SA, et al. AAV-1-mediated gene transfer to skeletal muscle in humans results in dose-dependent activation of capsid-specific T cells. Blood. 2009;114(10):2077–86.
Mueller C, Chulay JD, Trapnell BC, Humphries M, Carey B, Sandhaus RA, et al. Human Treg responses allow sustained recombinant adeno-associated virus-mediated transgene expression. J Clin Invest. 2013;123(12):5310–8.
Konkle BA, Walsh CE, Escobar MA, Josephson NC, Young G, von Drygalski A, et al. BAX 335 hemophilia B gene therapy clinical trial results: potential impact of CpG sequences on gene expression. Blood. 2021;137(6):763–74.
Stieger K, Schroeder J, Provost N, Mendes-Madeira A, Belbellaa B, Le Meur G, et al. Detection of intact rAAV particles up to 6 years after successful gene transfer in the retina of dogs and primates. Mol Ther. 2009;17(3):516–23.
Zaiss AK, Cotter MJ, White LR, Clark SA, Wong NC, Holers VM, et al. Complement is an essential component of the immune response to adeno-associated virus vectors. J Virol. 2008;82(6):2727–40.
Guillou J, de Pellegars A, Porcheret F, Frémeaux-Bacchi V, Allain-Launay E, Debord C, et al. Fatal thrombotic microangiopathy case following adeno-associated viral SMN gene therapy. Blood Adv. 2022;6(14):4266–70.
Cao O, Dobrzynski E, Wang L, Nayak S, Mingle B, Terhorst C, et al. Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood. 2007;110(4):1132–40.
Perrin GQ, Zolotukhin I, Sherman A, Biswas M, de Jong YP, Terhorst C, et al. Dynamics of antigen presentation to transgene product-specific CD4(+) T cells and of Treg induction upon hepatic AAV gene transfer. Mol Ther Methods Clin Dev. 2016;3:16083.
Dobrzynski E, Mingozzi F, Liu YL, Bendo E, Cao O, Wang L, et al. Induction of antigen-specific CD4+ T-cell anergy and deletion by in vivo viral gene transfer. Blood. 2004;104(4):969–77.
Kumar SRP, Hoffman BE, Terhorst C, de Jong YP, Herzog RW. The balance between CD8(+) T cell-mediated clearance of AAV-encoded antigen in the liver and tolerance is dependent on the vector dose. Mol Ther. 2017;25(4):880–91.
Poupiot J, Costa Verdera H, Hardet R, Colella P, Collaud F, Bartolo L, et al. Role of regulatory T cell and effector T cell exhaustion in liver-mediated transgene tolerance in muscle. Mol Ther Methods Clin Dev. 2019;15:83–100.
Pipe SW, Leebeek FWG, Recht M, Key NS, Castaman G, Miesbach W, et al. Gene therapy with etranacogene dezaparvovec for hemophilia B. N Engl J Med. 2023;388(8):706–18.
Kelley RI, Sladky JT. Dicarboxylic aciduria in an infant with spinal muscular atrophy. Ann Neurol. 1986;20(6):734–6.
Tein I, Sloane AE, Donner EJ, Lehotay DC, Millington DS, Kelley RI. Fatty acid oxidation abnormalities in childhood-onset spinal muscular atrophy: primary or secondary defect(s)? Pediatr Neurol. 1995;12(1):21–30.
Crawford TO, Sladky JT, Hurko O, Besner-Johnston A, Kelley RI. Abnormal fatty acid metabolism in childhood spinal muscular atrophy. Ann Neurol. 1999;45(3):337–43.
Zolkipli Z, Sherlock M, Biggar WD, Taylor G, Hutchison JS, Peliowski A, et al. Abnormal fatty acid metabolism in spinal muscular atrophy may predispose to perioperative risks. Eur J Paediatr Neurol. 2012;16(5):549–53.
Brehm TT, Wehmeyer MH, Fuhrmann V, Schäfer H, Kluwe J. Severe acute liver injury following therapeutic doses of acetaminophen in a patient with spinal muscular atrophy. Am J Ther. 2019;26(4):e528–9.
D’Amico A, Longo A, Fattori F, Tosi M, Bosco L, Chiarini Testa MB, et al. Hepatobiliary disease in XLMTM: a common comorbidity with potential impact on treatment strategies. Orphanet J Rare Dis. 2021;16(1):425.
Dowling JJ, Müller-Felber W, Smith BK, Bönnemann CG, Kuntz NL, Muntoni F, et al. INCEPTUS natural history, run-in study for gene replacement clinical trial in X-linked myotubular myopathy. J Neuromuscul Dis. 2022;9(4):503–16.
Amburgey K, Tsuchiya E, de Chastonay S, Glueck M, Alverez R, Nguyen CT, et al. A natural history study of X-linked myotubular myopathy. Neurology. 2017;89(13):1355–64.
Fetalvero KM, Yu Y, Goetschkes M, Liang G, Valdez RA, Gould T, et al. Defective autophagy and mTORC1 signaling in myotubularin null mice. Mol Cell Biol. 2013;33(1):98–110.
Gene transfer clinical study in X-linked myotubular myopathy (ASPIRO) [updated 10th February, 2023. Available from: https://clinicaltrials.gov/ct2/show/NCT03199469.
Paulk N. Gene therapy: it’s time to talk about high-dose AAV [updated 7th July 2020. Available from: https://www.genengnews.com/commentary/gene-therapy-its-time-to-talk-about-high-dose-aav/.
Wilde JT. HIV and HCV coinfection in haemophilia. Haemophilia. 2004;10(1):1–8.
Monahan PE, Négrier C, Tarantino M, Valentino LA, Mingozzi F. Emerging Immunogenicity and Genotoxicity Considerations of Adeno-Associated Virus Vector Gene Therapy for Hemophilia. J Clin Med. 2021;10(11):2471.
Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-factor IX and limitations imposed by the host immune response. Nat Med. 2006;12(3):342–7.
George LA, Ragni MV, Rasko JEJ, Raffini LJ, Samelson-Jones BJ, Ozelo M, et al. Long-term follow-up of the first in human intravascular delivery of AAV for gene transfer: AAV2-hFIX16 for severe hemophilia B. Mol Ther. 2020;28(9):2073–82.
Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J, et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 2014;371(21):1994–2004.
Nathwani AC, Reiss U, Tuddenham E, Chowdary P, McIntosh J, Riddell A, et al. Adeno-associated mediated gene transfer for hemophilia b:8 year follow up and impact of removing “empty viral particles” on safety and efficacy of gene transfer. Blood. 2018;132(Supplement 1):491.
Pipe S, Stine K, Rajasekhar A, Everington T, Poma A, Crombez E, et al. 101HEMB01 Is a phase 1/2 open-label, single ascending dose-finding trial of DTX101 (AAVrh10FIX) in patients with moderate/severe hemophilia B that demonstrated meaningful but transient expression of human factor IX (hFIX). Blood. 2017;130:3331.
Wright JF. Quantification of CpG motifs in rAAV genomes: avoiding the toll. Mol Ther. 2020;28(8):1756–8.
George LA, Sullivan SK, Giermasz A, Rasko JEJ, Samelson-Jones BJ, Ducore J, et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N Engl J Med. 2017;377(23):2215–27.
Pipe SW, Recht M, Key NS, Leebeek FWG, Castaman G, Lattimore SU, et al. First data from the phase 3 HOPE-B gene therapy trial: efficacy and safety of etranacogene dezaparvovec (AAV5-Padua hFIX variant; AMT-061) in adults with severe or moderate-severe hemophilia B treated irrespective of pre-existing anti-capsid neutralizing antibodies. Blood. 2020;136(Supplement_2):LBA-6-LBA-.
Long BR, Veron P, Kuranda K, Hardet R, Mitchell N, Hayes GM, et al. Early phase clinical immunogenicity of valoctocogene roxaparvovec, an AAV5-mediated gene therapy for hemophilia A. Mol Ther. 2021;29(2):597–610.
Hösel M, Huber A, Bohlen S, Lucifora J, Ronzitti G, Puzzo F, et al. Autophagy determines efficiency of liver-directed gene therapy with adeno-associated viral vectors. Hepatology. 2017;66(1):252–65.
Levitsky J, Mathew JM, Abecassis M, Tambur A, Leventhal J, Chandrasekaran D, et al. Systemic immunoregulatory and proteogenomic effects of tacrolimus to sirolimus conversion in liver transplant recipients. Hepatology. 2013;57(1):239–48.
D’Antiga L, Beuers U, Ronzitti G, Brunetti-Pierri N, Baumann U, Di Giorgio A, et al. Gene therapy in patients with the crigler-najjar syndrome. N Engl J Med. 2023;389(7):620–31.
Montenegro-Miranda PS, ten Bloemendaal L, Kunne C, de Waart DR, Bosma PJ. Mycophenolate mofetil impairs transduction of single-stranded adeno-associated viral vectors. Hum Gene Ther. 2011;22(5):605–12.
Unzu C, Hervás-Stubbs S, Sampedro A, Mauleón I, Mancheño U, Alfaro C, et al. Transient and intensive pharmacological immunosuppression fails to improve AAV-based liver gene transfer in non-human primates. J Transl Med. 2012;10:122.
Jiang H, Couto LB, Patarroyo-White S, Liu T, Nagy D, Vargas JA, et al. Effects of transient immunosuppression on adenoassociated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood. 2006;108(10):3321–8.
McIntosh JH, Cochrane M, Cobbold S, Waldmann H, Nathwani SA, Davidoff AM, et al. Successful attenuation of humoral immunity to viral capsid and transgenic protein following AAV-mediated gene transfer with a non-depleting CD4 antibody and cyclosporine. Gene Ther. 2012;19(1):78–85.
Samelson-Jones BJ, Finn JD, Favaro P, Wright JF, Arruda VR. Timing of intensive immunosuppression impacts risk of transgene antibodies after AAV gene therapy in nonhuman primates. Mol Ther Methods Clin Dev. 2020;17:1129–38.
Mingozzi F, Hasbrouck NC, Basner-Tschakarjan E, Edmonson SA, Hui DJ, Sabatino DE, et al. Modulation of tolerance to the transgene product in a nonhuman primate model of AAV-mediated gene transfer to liver. Blood. 2007;110(7):2334–41.
Jagadisan B, Dhawan A. Multidisciplinary approach and reactive immunosuppression for gene therapy-related hepatotoxicity. J Hepatol. 2023;79(2):e67–e68
Guidance for Industry. Drug-Induced Liver Injury: Premarketing Clinical Evaluation 2009 [cited U.S. Department of Health and Human Services FDA, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Available from: https://www.fda.gov/media/116737/download. Accessed 2 Oct 2023
Author information
Authors and Affiliations
Contributions
Both authors contributed to the manuscript and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
AD – AD is on the scientific advisory boards of BitBio, Aspect Bio, MSD, Astellas, Kate Pharma, Univar, Alexion. No funding for the project was received.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Ethical Approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article has not been published elsewhere in any language and it is not currently under consideration for publication elsewhere.
Key Points
• Paediatric hepatologists are increasingly asked to manage hepatotoxicity related to recombinant adeno-associated virus gene therapy.
• Hepatoxicity in recombinant adeno-associated virus gene therapy is an immunological phenomenon while pre-existing liver disease appears to worsen the injury.
• There is a need for empiric protocols for prophylactic and reactive immunosuppression, early recognition of at-risk individuals and better understanding of the long-term effects of hepatotoxicity and immunosuppression.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jagadisan, B., Dhawan, A. Hepatotoxicity in Adeno-Associated Viral Vector Gene Therapy. Curr Hepatology Rep 22, 276–290 (2023). https://doi.org/10.1007/s11901-023-00624-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11901-023-00624-5