
Abstract
It is now evident that elevated circulating levels of triglycerides in the non-fasting state, a marker for triglyceride (TG)-rich remnant particles, are associated with increased risk of premature cardiovascular disease (CVD). Recent findings from basic and clinical studies have begun to elucidate the mechanisms that contribute to the atherogenicity of these apoB-containing particles. Here, we review current knowledge of the formation, intravascular remodelling and catabolism of TG-rich lipoproteins and highlight (i) the pivotal players involved in this process, including lipoprotein lipase, glycosylphosphatidylinositol HDL binding protein 1 (GPIHBP1), apolipoprotein (apo) C-II, apoC-III, angiopoietin-like protein (ANGPTL) 3, 4 and 8, apoA-V and cholesteryl ester transfer protein; (ii) key determinants of triglyceride (TG) levels and notably rates of production of very-low-density lipoprotein 1 (VLDL1) particles; and (iii) the mechanisms which underlie the atherogenicity of remnant particles. Finally, we emphasise the polygenic nature of moderate hypertriglyceridemia and briefly discuss modalities for its clinical management. Several new therapeutic strategies to attenuate hypertriglyceridemia have appeared recently, among which those targeted to apoC-III appear to hold considerable promise.
Similar content being viewed by others

Introduction
Whether plasma triglycerides, or more specifically, the lipoprotein particles in which they are transported between sites of absorption, lipolysis and remodelling and catabolism constitute an independent risk factor for CVD has been the subject of debate for decades [1]. Significant progress has been made of late in resolving this question as a result of three elements: firstly, the availability of prospective data focusing on the relationship between circulating TG levels and cardiovascular risk in large cohorts, secondly, observations made in the postprandial, non-fasting period, allowing analysis of this relationship over a substantially greater range of TG concentrations as compared to the fasting state and thirdly, analytical approaches which allow an estimation of the cholesterol burden carried in potentially atherogenic remnant particles [2–5, 6•]. Thus, accumulating evidence demonstrates a strong correlation between the risk of CVD and both non-fasting (postprandial) and fasting plasma TG levels. Furthermore, large prospective epidemiologic studies focused on non-fasting TG in response to normal food intake have demonstrated significant associations between increased CVD events with elevated concentrations of non-fasting TG [2, 3, 7]. Indeed, a meta-analysis of 17 prospective studies with 2900 CHD endpoints revealed that an increment of 1 mmol/L in fasting TG levels was associated with a 14 % increase in CVD risk [5]. However, this strong correlation is often lost or attenuated in multivariate analysis, principally as a consequence of the strong link between hypertriglyceridemia and other cardiovascular risk factors such as low high-density lipoprotein (HDL) cholesterol, obesity and insulin resistance [8, 9•]. Further support for a causative role of triglyceride-rich lipoproteins (TRLs) in CVD arises from genetic studies [4, 10–12]; such studies equally indicate that remnant particles, which represent the partially degraded products of TRLs (i.e. chylomicrons and very-low-density lipoprotein (VLDL)), play a key role in the pathophysiology of atherosclerotic vascular disease.
Hypertriglyceridemia is generally defined and diagnosed as fasting plasma TG > 1.7 mmol/L or >150 mg/dL and is the consequence of environmental, behavioural and genetic factors, among which lifestyle is prominent (alcohol use, smoking, a high carbohydrate diet and obesity) [13••]. Severe hypertriglyceridemia with plasma TG levels > 10 mol/L (885 mg/dL) is typically of genetic origin, notably in the pediatric age group, and is associated with elevated risk of pancreatitis [14]. Further understanding of the pathobiology which underlies the atherogenicity of TRLs and their remnants will undoubtedly enable us to identify novel therapeutic targets; the translation of such targets into innovative therapeutic agents may significantly decrease cardiovascular risk in large numbers of hypertriglyceridemic individuals who currently remain at high risk despite optimal treatment according to current guideline recommendations.
Production and Intravascular Metabolism of Triglyceride-Rich Lipoproteins and Remnants
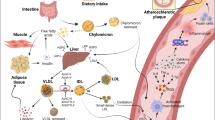
Triglycerides represent the transport module for fatty acids which provide an essential source of energy upon oxidation in mitochondria. A major source of TG is derived from dietary fat consumption. Dietary TGs are transported in intestinally derived apolipoprotein (apo) B48-containing chylomicrons, which enter the systemic circulation through the lymphatic system and target the heart as the first organ for delivery of fatty acids to fulfil energy requirements. The liver plays a cental role in TG homeostasis and maintains a steady state between TG synthesis, secretion and oxidation. In contrast to adipose tissue, the liver does not serve as an organ for TG storage under normal physiologic conditions. The liver can take up fatty acids derived from lipolysis in adipose tissue or from circulating lipoproteins but may equally synthesise fatty acids from carbohydrates in the process of de novo lipogenesis. In the liver, fatty acids can be partly stored as TG in lipid droplets or oxidised to generate energy in mitochondria in the process of beta-oxidation or packaged in apo B100-containing VLDL particles and secreted into the systemic circulation where they serve as a source for energy for peripheral tissues. The molecular pathway involved in the packaging of TG into both chylomicron and VLDL particles is remarkably similar, involving microsomal triglyceride transfer protein (MTTP) as described in detail elsewhere [15, 16].
Triglycerides cannot pass through cell membranes freely. Consequently, intravascular lipolysis is an essential process for release of free fatty acids, which can then be taken up via specific fatty acid transporters or other yet unknown mechanisms. The underlying mechanism is still only partly understood, but identification of some of the key proteins involved has allowed progress in our understanding. Lipoprotein lipase (LPL) is the key enzyme which drives TG hydrolysis along the luminal surface of capillaries, whereas the recently identified protein glycosylphosphatidylinositol HDL binding protein 1 (GPIHBP1) provides the platform to allow lipolysis to occur at the endothelial cell surface [17, 18]. Lipoprotein lipase is synthesised in macrophages, adipocytes and myocytes and must be transferred to the luminal site of the endothelial cell to become active, a process which is facilitated by GPIHBP1 [18, 19]. Strict regulation of LPL production and activity is critical in different tissues and organs. LPL can be regulated at the level of transcription by both peroxisome proliferator-activated receptor (PPAR)α and PPARγ through binding to a PPRE element in the 5′ regulatory region of the LPL gene [20, 21]. PPARα is intimately involved in lipid metabolism in the liver, whereas PPARγ is more closely involved in adipose tissue lipid homeostasis thereby regulating LPL action in a tissue-specific manner. More recently, different microRNAs (miR29-a, 497b, 1277, 410) have been found to be involved in posttranslational regulation of LPL; the exact mechanism(s) has not as yet been elucidated [22, 23]. Both nutritional (fasting vs fed state) and hormonal status play central roles in the regulation of LPL expression in adipose tissue. In fed conditions, LPL activity is high due to the effect of insulin, thereby resulting in increased uptake of fatty acids. Interestingly, LPL is more strictly regulated in heart and skeletal muscle since these tissues need a continuous supply of fatty acids for energy production [24].
Regulation of the Lipolytic Process
In vivo LPL action is regulated by several proteins including apolipoprotein (apo) C-II, apoA-V, apoC-III and angiopoietin-like protein (ANGPTL) 3, 4 and 8 [25] (Fig. 1). ApoC-II is a 79 amino acid peptide of hepatic origin containing a C-terminal domain involved in LPL activation and an N-terminal domain involved in lipid binding. Very little is known of the regulation of apoC-II synthesis. ApoC-II circulates in plasma on TG-rich lipoprotein particles as well as on HDL and is the rate-limiting protein required for normal LPL activity to occur. Patients with complete loss-of-function mutations in APOC2 have severe hypertriglyceridemia similar to LPL deficiency [26]. On the other hand, increased plasma apoC-II levels are associated with increased plasma TG levels suggesting that a surplus may have an inhibitory effect on LPL function. ApoC-II may be important for guiding TG-rich lipoproteins to the active site of LPL at the endothelial cell surface [27, 28]. The proposed working model for apoC-II involves a mechanical process occurring during hydrolysis of lipoprotein TG and results in increased surface pressure with concomitant conformational change in apoC-II structure, followed by the release of an apoC-II-phosphatidyl choline complex to HDL [29]. The amino acid residues tyr63, ile66, asp69 and gln70 in the C-terminal helix of apoC-II are essential for LPL activation and have now been used to create an apoC-II mimetic peptide that promotes lipolysis on TG-rich lipoproteins by LPL and may represent a new therapeutic target [30, 31].

LPL is synthesised in parenchymal cells in muscle and adipose tissue and then transported to the endothelial cell surface. LPL-mediated TG lipolysis at this surface is the first essential step in TG homeostasis. TGs are hydrolysed by LPL bound to GPIHBP1 in a process that is dependent on apoC-II. ApoC-III and apoA-V are potential inhibitors of LPL-mediated lipolysis. Upon TG hydrolysis, free fatty acids are taken up by surrounding tissues
ApoC-III is a potent inhibitor of LPL function and is a 99 amino acid protein containing three sialic acid residues; it is synthesised mainly in the liver and to a small extent in the intestine. Different transcription factors may regulate APOC3 gene expression such as PPARα and Fox01 [20, 32]. ApoC-III circulates on TG-rich lipoprotein particles as well as on HDL [33]. The apoC-III protein undergoes O-linked glycosylation by GALNT2 resulting in the presence of three isoforms: apoC-III 0, 1 and 2, which is impacting on apoC-III function [34]. Genetic studies have provided insight into the function of apoC-III. With respect to the potential relationship of circulating apoC-III levels to cardiovascular risk, a null mutation, p.R19X (rs56353203), was found to be associated with low plasma TG levels and attenuated subclinical atherosclerosis in the Amish population [35, 36]. Additional evidence was provided in a number of epidemiological studies showing the causal relationship between genetic variants in APOC3, plasma TG and CVD risk [37–39]. ApoC-III is now recognised as a multifaceted protein involved in different metabolic processes related to TG homeostasis. Firstly, apoC-III may inhibit hepatic clearance of TG-rich remnant particles by interfering with receptor binding sites [33]. Secondly, apoC-III has been recognised to inhibit LPL-mediated lipolysis in vitro; kinetic studies in human subjects do not however favour this concept [40]. Apparently, the ratio of apoC-III to apoC-II molecules on the surface of VLDL particles is the main determinant for LPL inhibition to occur. In vitro studies have shown that apoC-III/apoC-II ratios >5.0 are effective in inhibiting LPL action [41], a molar ratio which does not occur in human physiology. Finally, recent clinical trials in hypertriglyceridemic LPL-deficient patients using an allele-specific oligonucleotide (ASO) against apoC-III are in line with the concept that apoC-III has a major role in the hepatic uptake of remnant particles [42]. Thus, apoC-III emerges as an important drug target for reducing residual cardiovascular risk in hypertriglyceridemic subjects [43].
ApoA-V is a 366 amino acid protein primarily of hepatic origin. Circulating plasma apoA-V concentrations are very low which means that at most only 4 % of VLDL particles carry one apoA-V molecule [44]. Despite such low abundance, evidence supporting an essential role for apoA-V in TG metabolism is accumulating. Rare variants in or close to the APOA5 gene locus are consistently associated with plasma TG levels and risk for CVD [45, 46]. However, plasma apoA-V levels are positively associated with plasma TG in humans, an observation which to date has not been fully understood [44, 47, 48]. Most studies on apoA-V function have been performed in mice overexpressing human apoA-V or in in vitro models using apoA-V liposomes [49]. In all of these models, apoA-V concentration is elevated in comparison to the physiological concentrations typically seen in humans. Moreover, the physiological context in which apoA-V is functional, i.e. in the presence of apoC-II, apoC-III or apoE on the same lipoprotein particle, and which all compete for similar functions, is missing. Interestingly, injection of apoA-V rHDL into Apoa5 −/− mice induces a rapid decline in plasma TG levels whereas a similar injection in Gpihbp1 −/− mice had no effect [50], suggesting that apoA-V might be involved in binding of TG-rich lipoproteins to GPIHBP1, thereby allowing LPL-mediated TG hydrolysis to proceed [51]. Whether apoA-V is involved in LPL-mediated TG lipolysis in humans has not yet been established.
ANGPTL3, 4 and 8 have been implicated in TG homeostasis. ANGPTL proteins contain a signal peptide, an N-terminal coiled-coil domain and a C-terminal fibrinogen-like domain [52]. ANGPTL4 is produced in adipose tissue, whereas ANGPTL3 is produced in the liver and ANGPTRL8 in both adipose tissue and the liver [53]. ANGPTL4 expression is increased under fasting conditions, whereas ANGPTL8 levels are increased in the fed state. ANGPTL3, 4 and 8 have been implicated in LPL action. In fasting conditions, in which ANGPTL4 expression in adipose tissue is upregulated, LPL action may be suppressed by ANGPTL4, resulting in rerouting of fatty acids towards other organs for supply of energy. ANGPTL4 effectively inhibits LPL action by converting the active dimer into an inactive monomer [54]. However, evidence has accumulated to show that ANGPTL4 could also act as a reversible, non-competitive inhibitor of LPL [55]. Such inhibition occurs solely when LPL is in a complex with ANGPTL4 and leads to restoration of LPL activity upon dissociation of the complex. Alternatively, ANGPTL4 may directly bind LPL that is bound to GPIHBP1 and in this manner inactivate the protein, whereafter dissociation from GPIHBP1 occurs [56]. Although the role of ANGPTL3 in regulation of lipolysis is less well understood, mutations in the ANGPTL3 gene in humans are associated with reduced TG and cholesterol levels and elevated LPL activity [57]. ANGPTL3 is activated by proteolytic cleavage, leading to the release of the N-terminal domain, which has been shown to inhibit LPL and therefore result in reduced TG clearance. Recent data have emerged that ANGPTL8, also known as betatrophin or lipasin and a paralog of ANGPTL3, is able to interact with ANGPTL3, facilitating the cleavage of its N-terminal domain and thereby regulating its activity [58, 59]. In conclusion, all three ANGPTL isoforms are able to impact LPL activity and thereby lead to altered plasma TG levels.
Determinants of Plasma Triglyceride Levels and Heterogeneity of Triglyceride-Rich Lipoprotein Particles
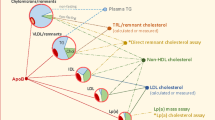
Very-low-density lipoprotein particles of hepatic origin can be subdivided on the basis of their size and role in TG metabolism; the larger, less dense particles are defined as VLDL1 (Sf 60–400) and the smaller as VLDL2 (Sf 20–60). Elevation of levels of large TG-rich VLDL1 is the major determinant of plasma TG concentrations in both normal and insulin-resistant individuals [60]. Increased plasma concentrations of VLDL1 can result either from hepatic oversecretion and/or impaired clearance of TRL remnants from the circulation [61, 62]. Hepatic oversecretion of VLDL1 particles is linked to increased liver fat and hyperglycemia [61, 63, 64]. Increased liver fat is equally associated with impaired suppression of VLDL1 secretion and results in oversecretion of VLDL1 particles [64, 65]. Recent findings in in vivo kinetic studies have shown that kinetic indices for VLDL1-TG catabolism are stronger determinants of circulating plasma TG concentration than kinetic parameters for the increased secretion of VLDL1 [66, 67•]. In particular, these studies revealed strong correlations between the catabolism of VLDL1-TG levels and the plasma concentration of apoC-III [66, 68]. The principal mechanism underlying the impairment of the catabolism of TRLs by apoC-III remains unclear, since apoC-III impairs both intravascular lipolysis by LPL and LPL-independent clearance of TRLs [69]. The significance of apoC-III in the hepatic clearance of TRL was recently illustrated by the markedly accelerated catabolism of TG-rich remnant particles in human subjects with deficiency of apoC-III [70].
Remnants are generated when chylomicrons and VLDL particles are remodelled during TG hydrolysis by LPL and are concomitantly enriched in cholesteryl esters by the action of the cholesteryl ester transfer protein (CETP). Thus, as TGs are removed, remnant particles become enriched with cholesteryl esters [71].
Subendothelial Accumulation of Atherogenic Lipoproteins Induces Atherogenesis
Lipoproteins in the circulation normally flux into and out of the arterial wall by transcytosis, a transport system in which lipoproteins and other macromolecules are transported across the endothelial cell in specialised clathrin-coated vesicles (Fig. 2) [72]. The transcytosis pathway has not previously attracted attention, but recent studies indicate that the process is responsive to LDL levels in the blood [73–75]. The transport vesicles are about 100 nm in diameter, and therefore, the transcytotic transport system is restricted to lipoproteins smaller than approximately 70 nm in diameter. Thus, larger lipoproteins, such as chylomicrons and large VLDL particles, cannot transverse the endothelium [76, 77]. This size limitation explains why individuals with lipoprotein disorders involving accumulation of large lipoproteins, such as chylomicrons in LPL-deficient patients, do not develop atherosclerosis. The capacity of the transcytotic transport system is very high; indeed, it has been estimated that about 2500 transport vesicles leave the plasma membrane every minute. Therefore, it is not the influx of lipoproteins into the artery wall that is rate limiting and thus determines the concentration of atherogenic lipoproteins in the artery wall but rather the selective subendothelial retention of lipoproteins in the artery wall [78]. Such retention is mediated by ionic interactions between positively charged residues in apoB and apoE on the atherogenic lipoproteins [79–81] and negatively charged sugar and sulphate groups in the glycosaminoglycan chains of the arterial wall proteoglycans.

Transcytosis enable the influx of lipoproteins over the vessel wall. This process is mediated by clathrin. The average diameter of these transport vesicles are around 100 nm, which only allows transport of lipoproteins with the size of 70 nm or smaller, thereby excluding chylomicrons and large VLDL remnant particles. The average transport speed is around 2500 vesicles per minute. The retention of lipoproteins in the subendothelial space is mediated by the interaction between positively charged residues on apoB and apoE and the negatively charged sulphate groups in the glycosaminoglycans chains of HSPG expressed on the vessel wall
Postprandial Hypertriglyceridemia and Atherogenicity of Remnant Particles
For many years, accumulation of chylomicron and chylomicron remnants in plasma was believed to be the essential cause of postprandial hypertriglyceridemia [82, 83]. However, it is now established that although approximately 80 % of the increase in postprandial TG is due to chylomicrons [84], approximately 80 % of the increase in particle number is accounted for by VLDL particles [85, 86]. The underlying reason is that chylomicrons and VLDL particles are cleared from the circulation by common pathways and therefore compete for clearance [76], even if chylomicrons seem to be preferentially cleared [87]. Increased secretion of liver-derived VLDL is therefore causatively linked to postprandial accumulation of chylomicron remnants [87]. As discussed above, remnant particles contain significant amount of cholesteryl esters and can enter the arterial wall, even if their size results in attenuated transport across the endothelium as compared to smaller LDL particles. However, since each remnant particle contains approximately 40 times more cholesterol compared with LDL, elevated levels of remnants may lead to accelerated atherosclerosis and CVD [77].
Interestingly, the importance of postprandial lipoproteins in the development of atherosclerotic vascular disease was initially proposed almost 70 years ago by Moreton who wrote “the lipid particles must be assumed to be retained and deposited from the plasma-derived nutrient lymph stream which normally passes from the lumen through the intramural structures towards the adventitial venules and lymphatics. It may be theorised that the increased particle size of the lipids in sustained or alimentary hyperlipemia is the stimulus to the phagocytosis in the intima by macrophages and the formation of the typical foam cells” [88, 89]. It is now clear that Moreton’s work has not, until now, received the attention it deserves.
Triglyceride-Rich Lipoproteins and Remnants as Therapeutic Targets in Hypertriglyceridemia
As discussed above, recent epidemiological studies have unequivocally demonstrated that elevated levels of postprandial TG and remnant particles are clinically significant risk factors for CVD [2, 3, 6•]. Furthermore, postprandial TG concentrations have been shown to be a superior risk predictor for CVD than fasting TG [2, 3, 90]. Epidemiological data providing insight into the frequency of mild to moderate hypertriglyceridemia (approx. 150 to 800 mg/dL) in the general population as a function of age, gender and ethnicity is lacking; nonetheless, findings in the NHANES survey suggest that at least one third of the US population can be classified as hypertriglyceridemic [91]. The degree to which such hypertriglyceridemia reflects the impact of elements of dietary habits and lifestyle relative to genetic factors is indeterminate. In this context, it is however especially relevant that recent studies from several laboratories suggest that mild to moderate hypertriglyceridemia is frequently of polygenic origin, arising as a result of a cumulative burden of common and rare variants in more than 30 genes coding for proteins of the complex lipolytic system, each of these polymorphisms generating proteins with mildly attenuated biological activity [13••]. A genetic score approach is therefore meaningful. Clearly then, an emerging body of evidence supports the contention that, from a therapeutic perspective, efforts to efficaciously reduce circulating concentrations of TRLs and their remnants have become critical.
Management of Hypertriglyceridemia and New Therapeutic Options
Following exclusion of secondary causes, treatment of mild to moderate hypertriglyceridemia should follow guideline recommendations, the initial step involving counselling on dietary habits, smoking and exercise [92]. The objective in such individuals is clearly to diminish their cardiovascular risk. In the event that pharmacotherapy is required, statins, fibrates and omega-3 fatty acids are all effective agents for reduction of TG levels, but only the use of statins is supported by a solid evidence base derived from multiple randomised control intervention trials [92].
New therapeutic options are currently under development that have been based on genetic evidence for reduced cardiovascular risk in families with phenotypes involving markedly diminished TG levels and rare causative monogenic mutations. Of the candidate proteins involved, apoC-III stands out as an elegant example. Indeed, a null mutation in human APOC3 was discovered in 2008 in the Amish community in the USA and found to provide apparent cardioprotection [35]. Abundant data now support the working hypothesis that pharmacotherapeutic reduction in circulating apoC-III levels may represent a valid target for hypotriglyceridemic therapy and ultimately for reduction of cardiovascular risk and potentially pancreatitis. Thus, loss-of-function mutations in APOC3 are associated with low TG levels and reduced risk of ischemic heart disease in two general population studies involving more than 75,000 participants [37]. Similar findings were made in the exome sequencing project [38].
As discussed above, apoC-III is a key factor on the surface of TRL and is a critical modulator of the lipolytic activity of lipoprotein lipase. Circulating levels of apoC-III are elevated however and of the order of 10–20 mg/dL, suggesting that hepatic production of apoC-III may be more viable as a target compared to a monoclonal antibody approach to remove apoC-III protein, allowing maintenance of low plasma levels over extended periods of time [93]. It is in this context that the development of anti-sense oligonucleotides targeted to the hepatic mRNA of apoC3 hold considerable promise, as dose-dependent reductions in TG levels of up to 80 % are attainable [94].
Conclusion
The number of patients with hypertriglyceridemia will grow significantly over the coming years, partly due to the increase in patients with diabetes mellitus type 2 and metabolic syndrome. Indeed, hypertriglyceridemia poses a major emerging challenge for public health and requires adequate targeting. As current therapies are not optimal for normalisation of elevated TG levels, the development of novel therapeutic agents is therefore warranted.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Nordestgaard BG, Varbo A. Triglycerides and cardiovascular disease. Lancet. 2014;384:626–35.
Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007;298:299–308.
Bansal S, Buring JE, Rifai N, et al. Fasting compared with nonfasting triglycerides and risk of cardiovascular events in women. JAMA. 2007;298:309–16.
Jorgensen AB, Frikke-Schmidt R, West AS, et al. Genetically elevated non-fasting triglycerides and calculated remnant cholesterol as causal risk factors for myocardial infarction. Eur Heart J. 2013;34:1826–33.
Hokanson JE, Austin MA. Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J Cardiovasc Risk. 1996;3:213–9.
Varbo A, Benn M, Tybjaerg-Hansen A, et al. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. 2013;61:427–36. This excellent paper describes the association between plasma levels of remnant cholesterol and ischemic heart disease in a large population cohort in Denmark and illustrates the importance of remnant cholesterol in the pathokogy of CVD.
Lindman AS, Veierod MB, Tverdal A, et al. Nonfasting triglycerides and risk of cardiovascular death in men and women from the Norwegian Counties Study. Eur J Epidemiol. 2010;25:789–98.
Emerging Risk Factors C, Di Angelantonio E, Gao P, et al. Lipid-related markers and cardiovascular disease prediction. JAMA. 2012;307:2499–506.
Chapman MJ, Ginsberg HN, Amarenco P, et al. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. 2011;32:1345–61. This is an important paper describing new guidelines with regard to the treatment of patients with elevated levels of remnant cholesterol and triglycerides.
Do R, Willer CJ, Schmidt EM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45:1345–52.
Holmes MV, Asselbergs FW, Palmer TM, et al. Mendelian randomization of blood lipids for coronary heart disease. Eur Heart J. 2015;36:539–50.
Triglyceride Coronary Disease Genetics C, Emerging Risk Factors C, Sarwar N, et al. Triglyceride-mediated pathways and coronary disease: collaborative analysis of 101 studies. Lancet. 2010;375:1634–9.
Hegele RA, Ginsberg HN, Chapman MJ, et al. The polygenic nature of hypertriglyceridaemia: implications for definition, diagnosis, and management. Lancet Diabetes Endocrinol. 2014;2:655–66. This is an overview of all the current knowledge.
Johansen CT, Hegele RA. Genetic bases of hypertriglyceridemic phenotypes. Curr Opin Lipidol. 2011;22:247–53.
Hussain MM. Intestinal lipid absorption and lipoprotein formation. Curr Opin Lipidol. 2014;25:200–6.
Cohen DE, Fisher EA. Lipoprotein metabolism, dyslipidemia, and nonalcoholic fatty liver disease. Semin Liver Dis. 2013;33:380–8.
Beigneux AP, Davies BS, Gin P, et al. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 2007;5:279–91.
Goulbourne CN, Gin P, Tatar A, et al. The GPIHBP1-LPL complex is responsible for the margination of triglyceride-rich lipoproteins in capillaries. Cell Metab. 2014;19:849–60.
Davies BS, Beigneux AP, Barnes 2nd RH, et al. GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. 2010;12:42–52.
Fruchart JC, Staels B, Duriez P. PPARS, metabolic disease and atherosclerosis. Pharmacol Res. 2001;44:345–52.
Schoonjans K, Peinado-Onsurbe J, Lefebvre AM, et al. PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J. 1996;15:5336–48.
Caussy C, Charriere S, Meirhaeghe A, et al. Multiple microRNA regulation of lipoprotein lipase gene abolished by 3’UTR polymorphisms in a triglyceride-lowering haplotype harboring p.Ser474Ter. Atherosclerosis. 2016;246:280–6.
Ahn J, Lee H, Chung CH, Ha T. High fat diet induced downregulation of microRNA-467b increased lipoprotein lipase in hepatic steatosis. Biochem Biophys Res Commun. 2011;414:664–9.
Wang H, Eckel RH. Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab. 2009;297:E271–288.
Kersten S. Physiological regulation of lipoprotein lipase. Biochim Biophys Acta. 1841;2014:919–33.
Surendran RP, Visser ME, Heemelaar S, et al. Mutations in LPL, APOC2, APOA5, GPIHBP1 and LMF1 in patients with severe hypertriglyceridaemia. J Intern Med. 2012;272:185–96.
LaRosa JC, Levy RI, Herbert P, et al. A specific apoprotein activator for lipoprotein lipase. Biochem Biophys Res Commun. 1970;41:57–62.
Kei AA, Filippatos TD, Tsimihodimos V, Elisaf MS. A review of the role of apolipoprotein C-II in lipoprotein metabolism and cardiovascular disease. Metabolism. 2012;61:906–21.
Meyers NL, Larsson M, Olivecrona G, Small DM. A pressure-dependent model for the regulation of lipoprotein lipase by apolipoprotein C-II. J Biol Chem. 2015;290:18029–44.
Amar MJ, Sakurai T, Sakurai-Ikuta A, et al. A novel apolipoprotein C-II mimetic peptide that activates lipoprotein lipase and decreases serum triglycerides in apolipoprotein E-knockout mice. J Pharmacol Exp Ther. 2015;352:227–35.
Sakurai T, Sakurai A, Vaisman BL et al. Creation of ApoC-II mutant mice and correction of their hypertriglyceridemia with an ApoC-II mimetic peptide. J Pharmacol Exp Ther. 2016;356:341–353.
Sparks JD, Dong HH. FoxO1 and hepatic lipid metabolism. Curr Opin Lipidol. 2009;20:217–26.
Sacks FM. The crucial roles of apolipoproteins E and C-III in apoB lipoprotein metabolism in normolipidemia and hypertriglyceridemia. Curr Opin Lipidol. 2015;26:56–63.
Holleboom AG, Karlsson H, Lin RS, et al. Heterozygosity for a loss-of-function mutation in GALNT2 improves plasma triglyceride clearance in man. Cell Metab. 2011;14:811–8.
Pollin TI, Damcott CM, Shen H, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322:1702–5.
Crawford DC, Dumitrescu L, Goodloe R, et al. Rare variant APOC3 R19X is associated with cardio-protective profiles in a diverse population-based survey as part of the Epidemiologic Architecture for Genes Linked to Environment Study. Circ Cardiovasc Genet. 2014;7:848–53.
Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjaerg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371:32–41.
TG, HDL Working Group of the Exome Sequencing Project NHL, Blood I, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371:22–31.
Natarajan P, Kohli P, Baber U, et al. Association of APOC3 loss-of-function mutations with plasma lipids and subclinical atherosclerosis: The Multi-Ethnic BioImage Study. J Am Coll Cardiol. 2015;66:2053–5.
Mendivil CO, Zheng C, Furtado J, et al. Metabolism of very-low-density lipoprotein and low-density lipoprotein containing apolipoprotein C-III and not other small apolipoproteins. Arterioscler Thromb Vasc Biol. 2010;30:239–45.
Larsson M, Vorrsjo E, Talmud P, et al. Apolipoproteins C-I and C-III inhibit lipoprotein lipase activity by displacement of the enzyme from lipid droplets. J Biol Chem. 2013;288:33997–4008.
Gaudet D, Brisson D, Tremblay K, et al. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med. 2014;371:2200–6.
Ginsberg HN, Brown WV. Apolipoprotein CIII: 42 years old and even more interesting. Arterioscler Thromb Vasc Biol. 2011;31:471–3.
Dallinga-Thie GM, van Tol A, Hattori H, et al. Plasma apolipoprotein A5 and triglycerides in type 2 diabetes. Diabetologia. 2006;49:1505–11.
Do R, Stitziel NO, Won HH, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518:102–6.
Johansen CT, Wang J, Lanktree MB, et al. Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat Genet. 2010;42:684–7.
Schaap FG, Nierman MC, Berbee JF, et al. Evidence for a complex relationship between apoA-V and apoC-III in patients with severe hypertriglyceridemia. J Lipid Res. 2006;47:2333–9.
Vaessen SF, Schaap FG, Kuivenhoven JA, et al. Apolipoprotein A-V, triglycerides and risk of coronary artery disease: the prospective Epic-Norfolk Population Study. J Lipid Res. 2006;47:2064–70.
Sharma V, Forte TM, Ryan RO. Influence of apolipoprotein A-V on the metabolic fate of triacylglycerol. Curr Opin Lipidol. 2013;24:153–9.
Shu X, Nelbach L, Weinstein MM, et al. Intravenous injection of apolipoprotein A-V reconstituted high-density lipoprotein decreases hypertriglyceridemia in apoav−/− mice and requires glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1. Arterioscler Thromb Vasc Biol. 2010;30:2504–9.
Gin P, Beigneux AP, Voss C, et al. Binding preferences for GPIHBP1, a glycosylphosphatidylinositol-anchored protein of capillary endothelial cells. Arterioscler Thromb Vasc Biol. 2011;31:176–82.
Hato T, Tabata M, Oike Y. The role of angiopoietin-like proteins in angiogenesis and metabolism. Trends Cardiovasc Med. 2008;18:6–14.
Nidhina Haridas PA, Soronen J, Sadevirta S, et al. Regulation of angiopoietin-like proteins (ANGPTLs) 3 and 8 by insulin. J Clin Endocrinol Metab. 2015;100:E1299–1307.
Sukonina V, Lookene A, Olivecrona T, Olivecrona G. Angiopoietin-like protein 4 converts lipoprotein lipase to inactive monomers and modulates lipase activity in adipose tissue. Proc Natl Acad Sci U S A. 2006;103:17450–5.
Lafferty MJ, Bradford KC, Erie DA, Neher SB. Angiopoietin-like protein 4 inhibition of lipoprotein lipase: evidence for reversible complex formation. J Biol Chem. 2013;288:28524–34.
Chi X, Shetty SK, Shows HW, et al. Angiopoietin-like 4 modifies the interactions between lipoprotein lipase and its endothelial cell transporter GPIHBP1. J Biol Chem. 2015;290:11865–77.
Musunuru K, Pirruccello JP, Do R, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med. 2010;363:2220–7.
Fu Z, Yao F, Abou-Samra AB, Zhang R. Lipasin, thermoregulated in brown fat, is a novel but atypical member of the angiopoietin-like protein family. Biochem Biophys Res Commun. 2013;430:1126–31.
Quagliarini F, Wang Y, Kozlitina J, et al. Atypical angiopoietin-like protein that regulates ANGPTL3. Proc Natl Acad Sci USA. 2012;109:19751–6.
Hiukka A, Fruchart-Najib J, Leinonen E, et al. Alterations of lipids and apolipoprotein CIII in very low density lipoprotein subspecies in type 2 diabetes. Diabetes. 2005;48:1207–15.
Taskinen MR, Adiels M, Westerbacka J, et al. Dual metabolic defects are required to produce hypertriglyceridemia in obese subjects. Arterioscler Thromb Vasc Biol. 2011;31:2144–50.
Lewis GF, Xiao C, Hegele RA. Hypertriglyceridemia in the genomic era: a new paradigm. Endocr Rev. 2015;36:131–47.
Adiels M, Olofsson SO, Taskinen MR, Boren J. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:1225–36.
Adiels M, Taskinen MR, Packard C, et al. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia. 2006;49:755–65.
Adiels M, Westerbacka J, Soro-Paavonen A, et al. Acute suppression of VLDL1 secretion rate by insulin is associated with hepatic fat content and insulin resistance. Diabetologia. 2007;50:2356–65.
Boren J, Watts GF, Adiels M, et al. Kinetic and related determinants of plasma triglyceride concentration in abdominal obesity: Multicenter Tracer Kinetic Study. Arterioscler Thromb Vasc Biol. 2015;35:2218–24.
Taskinen MR, Boren J. New insights into the pathophysiology of dyslipidemia in type 2 diabetes. Atherosclerosis. 2015;239:483–95. Excellent review on the underlying pathophysiology of the dyslipidemia in type 2 diabetes mellitus.
Taskinen MR, Adiels M, Westerbacka J, et al. Dual metabolic defects are required to produce hypertriglyceridemia in obese subjects. Arterioscler Thromb Vasc Biol. 2011;31:2144–50.
Norata GD, Tsimikas S, Pirillo A, Catapano AL. Apolipoprotein C-III: from pathophysiology to pharmacology. Trends Pharmacol Sci. 2015;36:675–87.
Ginsberg HN, Le NA, Goldberg IJ, et al. Apolipoprotein B metabolism in subjects with deficiency of apolipoproteins CIII and AI. Evidence that apolipoprotein CIII inhibits catabolism of triglyceride-rich lipoproteins by lipoprotein lipase in vivo. J Clin Invest. 1986;78:1287–95.
Chapman MJ, Le Goff W, Guerin M, Kontush A. Cholesteryl ester transfer protein: at the heart of the action of lipid-modulating therapy with statins, fibrates, niacin, and cholesteryl ester transfer protein inhibitors. Eur Heart J. 2010;31:149–64.
Fogelstrand P, Boren J. Retention of atherogenic lipoproteins in the artery wall and its role in atherogenesis. Nutr Metab Cardiovasc Dis. 2012;22:1–7.
Bartels ED, Christoffersen C, Lindholm MW, Nielsen LB. Altered metabolism of LDL in the arterial wall precedes atherosclerosis regression. Circ Res. 2015;117:933–42.
Armstrong SM, Sugiyama MG, Fung KY, et al. A novel assay uncovers an unexpected role for SR-BI in LDL transcytosis. Cardiovasc Res. 2015;108:268–77.
Frank PG, Pavlides S, Cheung MW, et al. Role of caveolin-1 in the regulation of lipoprotein metabolism. Am J Physiol Cell Physiol. 2008;295:C242–248.
Boren J, Matikainen N, Adiels M, Taskinen MR. Postprandial hypertriglyceridemia as a coronary risk factor. Clin Chim Acta. 2014;431:131–42.
Boren J, Taskinen MR, Olofsson SO, Levin M. Ectopic lipid storage and insulin resistance: a harmful relationship. J Intern Med. 2013;274:25–40.
Skalen K, Gustafsson M, Rydberg EK, et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002;417:750–4.
Flood C, Gustafsson M, Pitas RE, et al. Molecular mechanism for changes in proteoglycan binding on compositional changes of the core and the surface of low-density lipoprotein-containing human apolipoprotein B100. Arterioscler Thromb Vasc Biol. 2004;24:564–70.
Flood C, Gustafsson M, Richardson PE, et al. Identification of the proteoglycan binding site in apolipoprotein B48. J Biol Chem. 2002;277:32228–33.
Boren J, Olin K, Lee I, et al. Identification of the principal proteoglycan-binding site in LDL. A single-point mutation in apo-B100 severely affects proteoglycan interaction without affecting LDL receptor binding. J Clin Invest. 1998;101:2658–64.
Zilversmit DB. A proposal linking atherogenesis to the interaction of endothelial lipoprotein lipase with triglyceride-rich lipoproteins. Circ Res. 1973;33:633–8.
Zilversmit DB. Atherogenesis: a postprandial phenomenon. Circulation. 1979;60:473–85.
Cohn JS, Johnson EJ, Millar JS, et al. Contribution of apoB-48 and apoB-100 triglyceride-rich lipoproteins (TRL) to postprandial increases in the plasma concentration of TRL triglycerides and retinyl esters. J Lipid Res. 1993;34:2033–40.
Karpe F, Bell M, Bjorkegren J, Hamsten A. Quantification of postprandial triglyceride-rich lipoproteins in healthy men by retinyl ester labeling and simultaneous measurement of apolipoproteins B-48 and B-100. Arterioscler Thromb Vasc Biol. 1995;15:199–207.
Schneeman BO, Kotite L, Todd KM, Havel RJ. Relationships between the responses of triglyceride-rich lipoproteins in blood plasma containing apolipoproteins B-48 and B-100 to a fat-containing meal in normolipidemic humans. Proc Natl Acad Sci U S A. 1993;90:2069–73.
Adiels M, Matikainen N, Westerbacka J, et al. Postprandial accumulation of chylomicrons and chylomicron remnants is determined by the clearance capacity. Atherosclerosis. 2012;222:222–8.
Moreton JR. Atherosclerosis and alimentary hyperlipidemia. Science. 1947;106:190–1.
Moreton JR. Physical state of lipids and foreign substances producing atherosclerosis. Science. 1948;107:371–3.
Freiberg JJ, Tybjaerg-Hansen A, Jensen JS, Nordestgaard BG. Nonfasting triglycerides and risk of ischemic stroke in the general population. JAMA. 2008;300:2142–52.
Beltran-Sanchez H, Harhay MO, Harhay MM, McElligott S. Prevalence and trends of metabolic syndrome in the adult U.S. population, 1999–2010. J Am Coll Cardiol. 2013;62:697–703.
Reiner Z, Catapano AL, De Backer G, et al. ESC/EAS Guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J. 2011;32:1769–818.
Cohen JC, Stender S, Hobbs HH. APOC3, coronary disease, and complexities of Mendelian randomization. Cell Metab. 2014;20:387–9.
Graham MJ, Lee RG, Bell 3rd TA, et al. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res. 2013;112:1479–90.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
M. Dallinga-Thie, J. Kroon and Jan Borén declare that they have no conflict of interest.
John Chapman reports grants and personal fees from Pfizer and Kowa, grants from CSL and personal fees from Amgen, Sanofi-Regeneron and Astrazeneca.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Lipid Abnormalities and Cardiovascular Prevention
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dallinga-Thie, G.M., Kroon, J., Borén, J. et al. Triglyceride-Rich Lipoproteins and Remnants: Targets for Therapy?. Curr Cardiol Rep 18, 67 (2016). https://doi.org/10.1007/s11886-016-0745-6
Published:
DOI: https://doi.org/10.1007/s11886-016-0745-6