
Abstract
Purpose of the review
Macrophage accumulation and activation function as hallmarks of atherosclerosis and have complex and intricate dynamics throughout all components and stages of atherosclerotic plaques. In this review, we focus on the regulatory roles and underlying mechanisms of macrophage phenotypes and metabolism in atherosclerosis. We highlight the diverse range of macrophage phenotypes present in atherosclerosis and their potential roles in progression and regression of atherosclerotic plaque. Furthermore, we discuss the challenges and opportunities in developing therapeutic strategies for preventing and treating atherosclerotic cardiovascular disease.
Recent findings
Dysregulation of macrophage polarization between the proinflammatory M1 and anti-inflammatory M2 phenotypealters the immuno-inflammatory response during atherosclerosis progression, leading to plaque initiation, growth, and ultimately rupture. Altered metabolism of macrophage is a key feature for their function and the subsequent progression of atherosclerotic cardiovascular disease. The immunometabolism of macrophage has been implicated to macrophage activation and metabolic rewiring of macrophages within atherosclerotic lesions, thereby shifting altered macrophage immune-effector and tissue-reparative function.
Summary, Insights and Perspective
Targeting macrophage phenotypes and metabolism are potential therapeutic strategies in the prevention and treatment of atherosclerosis and atherosclerotic cardiovascular diseases. Understanding the precise function and metabolism of specific macrophage subsets and their contributions to the composition and growth of atherosclerotic plaques could reveal novel strategies to delay or halt development of atherosclerotic cardiovascular diseases and their associated pathophysiological consequences. Identifying biological stimuli capable of modulating macrophage phenotypes and metabolism may lead to the development of innovative therapeutic approaches for treating patients with atherosclerosis and coronary artery diseases.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Atherosclerosis is a metabolic and inflammatory disease characterized by the accumulation of cholesterol-rich lipoproteins and inflammatory cells in the arterial wall, which primarily occurs in regions with disturbed laminar flow [1]. The build-up of lipoproteins containing apolipoprotein B is the initiating step. Atherosclerosis is the leading cause of cardiovascular events such as myocardial infarctions and strokes, resulting in 17.9 million deaths annually around the world [1]. Over the last two decades, macrophages have been identified as significant pathological features in the progression of atherosclerosis and thrombotic complications. Monocytes differentiate into macrophages at these sites, engulfing the accumulated lipoproteins. While initially beneficial, this process ultimately leads to the formation of foam cells laden with cholesterol contributing to plaque formation. The subsequent progression of plaques depends on a continuous influx of monocytes from the bloodstream, which fuels the accumulation of macrophages within the plaque. Moreover, Under the stimulation of pathological conditions such as hypertension, macrophages secret various cytokines to promote the formation and progression of atherosclerosis [2]. Precise regulation of macrophage activation is crucial for controlling atherosclerosis and maintenance the stability of plaques. Macrophages play a dual role as sources of inflammation and regulators of homeostasis, adapting to the complex and dynamic metabolism changes at different stages of atherosclerotic plaque progression. Importantly, metabolic pathways not only provide energy but also influence the macrophage polarization and function. In this article, we focus on the regulatory roles and underlying mechanisms of macrophage phenotypes, metabolism and activation in atherosclerosis and discuss the potential of targeting macrophages for therapeutic intervention in atherosclerosis and atherosclerotic cardiovascular diseases (ASCVD).
Origins of Lesion Macrophages
Macrophages widely distributes throughout nearly every tissue of our bodies and play a crucial role in the innate immune system. It is now known that macrophages have diverse and context-specific functions in various physiological and pathophysiological contexts [3]. During the progression of atherosclerosis, under the influence of proinflammatory stimuli, hematopoietic stem and progenitor cells originating from the bone marrow migrate to the spleen, where they differentiate into monocytes. These splenic monocytes are subsequently released into the blood circulation and then recruited to organs with inflammation, such as the atherosclerotic artery wall [4]. The process of monocyte extravasation commences with rolling along the endothelium mediated by P- and E-selectin, followed by firm adhesion mediated by intercellular cell adhesion molecules (ICAM), vascular cell adhesion molecules (VCAM), and integrins [5]. Subsequent transendothelial migration requires interaction between C–C motif chemokine receptor (CCR) 2 and monocyte chemoattractant protein 1/ chemokine (C–C motif) ligand 2 (MCP1/CCL2) as well as the involvement of CCR5 and CX3C chemokine receptor (CX3CR) 1. Deficiency or reduction of these receptors has been linked to a significant decrease of atherosclerosis. Monocytes penetrate the arterial vessel wall through the extensive network of highly permeable neovascularization developing during progression of plaque [6]. Within the intima, bone marrow derived-monocytes differentiate into macrophages under the simulation of M-CSF or GM-CSF. These macrophages proliferate and sustain the sterile inflammation. The macrophages in the mouse aorta wall during atherogenesis are derived from circulating monocytes and local proliferation, leading to 20-fold increase in macrophage numbers [7]. Before reaching the artery wall, monocytes are exposed to various pathophysiological stimuli in the systemic context. Evidence from studies of monocytes isolated from patients as well as mouse models of atherosclerosis indicates that this exposure modulates both cellular metabolism and function, promotes monocytes polarized to an inflammatory phenotype well before their differentiation within the atherosclerotic microenvironment. Among the various monocyte subpopulations in circulation, the CD14+ subpopulation in humans and the Ly6Chi subpopulation in mice, are significantly associated with macrophage differentiation in the progression of atherosclerosis [8].
Macrophages Polarization is Depending on Stimuli and Activated Signaling Pathways
Macrophages play a crucial role in innate immunity by expressing multiple pattern recognition receptors, including Toll-like receptors (TLRs), Ig superfamily (FcR), GPI-anchored (CD14), scavenger (CD36) and lectin like receptors (Dectin-1) (Table 1), which are strategically located in the macrophage cell membrane, cytoplasm, and endosomal compartment [9]. Traditionally, macrophage activation and metabolism have been implicated in two main subtypes: M1 macrophages, induced by various stimuli such as tumor necrosis factor α (TNFα), IFNγ, lipopolysaccharides (LPS), TLRs, pathogen-associated molecular complexes, lipoproteins as well as M2 macrophages, alternatively activated by IL-4 and IL13 stimulation. However, it is now understood that these macrophage phenotypes represent extremes and serve as starting point for understanding macrophage activation and metabolism in atherosclerosis (Fig. 1). Macrophages have diverse functions including maintaining of tissue homeostasis, inducing and resolving immune responses during pathogen infection and participating in tissue repair [10, 11]. Upon sensing various stimuli, macrophages become activated and polarized to different cellular states. For example, microbial stimuli activate M1 macrophages, which secrete pro-inflammatory factors such as IL-1β, IL-6, TNF-α, IL-12, IL-23, nitric oxide (NO), and reactive oxygen species (ROS), contributing to tissue destruction [12,13,14]. M1-like macrophages express inducible nitric oxide synthase (iNOS), TLR2, TLR4, CD68, CD80, and CD86, along with pro-inflammatory transcription factors like nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and signal transducer and activator of transcription (STAT)-1. They play a predominant role in pathogen defense, phagocytosis, destruction of foreign bodies, as well as antigen presentation to T cells, triggering an adaptive immune response [15, 16]. In terms of metabolism, M1 macrophages exhibit increased aerobic glycolysis and decreased oxidative phosphorylation (OXPHOS). They kill intracellular pathogens and prime their polarization to this phenotype in atherosclerosis. M1 macrophages generated reactive oxygen and nitrogen species to eliminate bacterial, fungal, and viral infections, which may worsen oxidative stress in the plaque [17]. Additionally, M1 macrophages express different chemokine (C-X-C motif) receptor ligands such as CXCL9, CXCL10, and CXCL5, which recruit Th1 and natural killer cells to eradicate intracellular pathogens [18]. While these functions are beneficial in acute infection, they can cause tissue damage and impair wound healing in sterile inflammation generated within atherosclerosis [19].

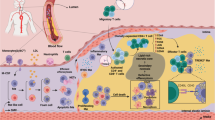
Phenotypes and metabolic mechanism of macrophages in the progression of atherosclerotic plaque and related cardiovascular diseases. Macrophages with different functional phenotypes are likely to perform different roles in the development of atherosclerosis. Macrophages in early atherosclerotic lesions are predominantly derived from monocytes recruited from the blood cycle. Monocyte adhesion to endothelial cells and migration into the arterial vessel wall, whereas macrophage proliferation and local inflammation are the primary features. With the progression of atherosclerosis, macrophage engulf lipoprotein-derived cholesterol, leading to foam cell formation, macrophage apoptosis, the plaque lesions towards to a vulnerable type known as unstable atherosclerotic plaques with large necrotic cores and thin fibrous caps. Unstable plaques are prone to rupture, which may cause thrombus formation and arterial occlusion contributing to myocardial infarction and sudden cardiac death
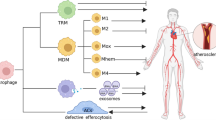
In the context of worm and parasite infection, macrophages are activated into an alternative state known as M2 macrophages, which play an active role in tissue repair and remodeling [20]. M2 macrophages have been categorized into four distinct subtypes: M2a, M2b, M2c and M2d macrophages. M2a macrophages are stimulated by IL-4 and IL-13 and express high levels of the mannose receptors [21, 22]. These macrophages secrete pro-fibrotic factors such as fibronectin, insulin-like growth factor (IGF), and transforming growth factor β (TGF-β), contributing to tissue repair [23, 24]. On the other hand, M2b macrophages are activated by immune complexes, such as TLR ligands, IL-1 receptor agonists and they produce both anti-inflammatory (IL-10) and pro-inflammatory (IL-1, IL-6, TNFα) cytokines along with TGF-β1 [25]. Furthermore, M2c macrophages are induced by IL-10 and glucocorticoids, while M2d macrophages are activated by IL-6 and express inflammatory cytokines (TGF- β, VEGF and IL-10) [26]. Traditionally, M2a macrophages are known as “wound healing macrophages”, whereas M2b and M2c macrophages are referred as “regulatory macrophages” [27]. They are characterized by the expression of arginase 1 (Arg1), mannose receptor C type 1 (CD206), chitinase-like protein 3 (Chil3), Chil4, and found in inflammatory zone-1 (Fizz1). The expression of Arg1 and Fizz1 depends on signal transducer and STAT6 signaling [28]. M2 macrophages also secrete multiple chemokines such as CCL17, CCL18, CCL22, and CCL24. They exhibit anti-inflammatory cytokine profile with low production of IL-12 and high production of IL-10or TGF-β [29]. Furthermore, M2 macrophages prevent cholesterol from accumulation due to increased expression of cholesterol efflux receptors, especially adenosine triphosphate (ATP)-binding cassette transporters (ABC), ABCA1 and ABCG1. Certain subsets of heme-dependent macrophages are associated with efficient phagocytosis of red cells [30]. However, due to the overlapping expression of certain markers among different subtypes of M2 macrophages, their phenotypic differentiation can be challenging. Therefore, it is crucial to categorize macrophage phenotypes based on surface marker expression and their specific functions. Nevertheless, the phenotypic spectrum of macrophages in vivo may be more complex than this classification suggests. However, how this subclassification of M2 macrophages mentioned above corresponds to their specific roles in atherosclerosis remains largely unclear. Apart from the classical subsets, there are other phenotypes. Mox macrophages are induced by oxidized LDL (oxLDL) and promote atherogenesis. M4 macrophages are induced by platelet chemokine CXCL4 and can be identified by the absence of the hemoglobin–haptoglobin scavenger receptor CD163, while they express a combination of CD68, S100A8 and MMP7. Similar to M1 macrophages, M4 macrophages exhibit proinflammatory characteristics, but lack phagocytotic capacity. Moreover, vulnerable plaques can develop intraplaque hemorrhage (IPH), leading to the release of free hemoglobin (Hb) and subsequent uptake haptoglobin binding to the CD163 receptor, which triggers differentiation into M (Hb) or Mhem macrophage. M (Hb) macrophages. Compared to foam cells, M (Hb) macrophage lacks lipid accumulation and exhibit high surface expression of CD163 and CD206 (Table 2), located in areas of angiogenesis and hemorrhage, produce both anti-inflammatory (IL-10, IL-1ra) and pro-inflammatory (IL-6, IL-1β) factors.
M1-like macrophages are predominantly associated with inflammatory processes and often observed in unstable plaques, while M2-like macrophages are predominantly present in stable plaques and the adventitia of arteries [31]. We hypothesize that the phenotype of lesional macrophages cannot be neatly classified into predefined subsets but rather emerges as a result of the lesional microenvironment and the activation of specific signaling pathways. For example, the activation of TLR4 triggers the activation of NF-κB, ERK, p38 mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase (JNK), and interferon response genes, thereby leading to different downstream effects. Conversely, stimulation of IL-4 receptor triggers the activation of signal transducer and STAT6, which can suppress TLR4 signaling [32]. Notably, STAT6 shifts macrophages from an inflammatory M1-like phenotype to a more stable M2-like phenotype [33]. Consequently, the phenotype of lesional macrophages rapidly changes in response to alterations of the microenvironment and intracellular signaling pathways. Metabolic pathways play a crucial role in immune cell function. In vitro studies have revealed significant metabolic difference between M1 and M2 macrophages.M2 macrophages more rely on fatty acid oxidation (FAO), whereas M1 macrophages mainly rely on an increase in glycolysis [34,35,36]. Additionally, IL − 4 and IL − 10 enhance lipid uptake of macrophages and promote foam cell formation, while inflammatory M1 and Mox macrophages display lower foam cell activity. Functionally, there are key differences between the M1 and Mox macrophages phenotypes that account for the balance of foam cell formation. M1 exhibit reduced lipid uptake, whereas Mox macrophages compensate for this by increasing lipid efflux activity [37]. Increased glycolysis just through overexpression of the glucose transporter 1 (GLUT1) in myeloid cells is insufficient to promote inflammatory activation in atherosclerosis [38]. However, inhibiting glycolysis in activated macrophages can lead to increased apoptosis, potentially contributing to the formation of necrotic cores. The microenvironment within lesions is likely to vary across different regions and stages of the lesion development and is affected by systemic factors dyslipidemia, inflammation associated with diabetes mellitus, autoimmune disease and infection. Therefore, we propose that immunometabolism would change with macrophage metabolic pathways and play important role in cell phenotypes and function. The interconnected pathways of glycolysis, tricarboxylic acid (TCA) cycle, the pentose-phosphate pathway (PPP), FAO, and amino acid metabolism contribute to generation of energy required for cellular maintenance, proliferation and modulation of cellular signaling [35]. Thus, we provide a brief overview of these highly interdependent metabolic contributors to macrophage phenotype: glucose, lipid and amino acid metabolism and how they are interconnected and intersect in macrophage activation and effector function (Fig. 2, Table 3).

Schematic representation of macrophage immunometabolism pathways. (A) LPS/IFN-γ induced macrophage. The enhanced glycolysis and pentose phosphate pathway, generating NADPH for reactive oxygen species, nitric oxide and a disturbed TCA cycle. (B) IL-4 induced macrophage. An intact TCA cycle resulting in sustained ATP production via oxidative phosphorylation (OXPHOS). ROS, reactive oxygen species; NO, nitric oxide; OXPHOS: oxidative phosphorylation; PPP: pentose phosphate pathway; IL4, interlukin-4; FAO: fatty acid oxidation; TCA, tricarboxylic acid
Mechanism of Glycolysis in Macrophage during Atherosclerosis
The conventional perspective widely acknowledges that glycolysis is a prominent characteristic of M1 macrophages [39], while FAO and OXPHOS are the main metabolic characteristics of M2 macrophages. Although substantial efforts have been made to unravel the mechanisms linking metabolic reprogramming and macrophage function, numerous questions remained. Glycolysis drives inflammatory macrophage responses in different ways. The utilization of glucose through glycolysis generates ATP as a form of energy and crucial intermediates, which functions as substrates for other metabolic pathways. For example, pyruvate can be further converted to lactate or Acetyl-CoA, two major metabolites important for macrophage metabolic adaptations. In addition, the generation of serine from glycolysis to sustain cellular one-carbon metabolism may regulate epigenetic reprogramming during macrophage activation [40]. This one-carbon metabolism is essential for nucleotide synthesis and methylation reactions, influencing gene expression patterns in activated macrophages. Although glycolysis produces only a small amount of ATP (2 per glucose), it can be activated swiftly to meet immediate energy demands. On the other hand, OXPHO, induced by signals such as IL-4 in macrophages, is a slower process but produces greater amounts of ATP (30 per glucose). This metabolic shift enables M1 macrophages to rapidly respond to inflammatory stimuli, while M2 macrophages, with their reliance on OXPHOS, are better suited for long-term, energy-efficient responses involved in tissue repair and anti-inflammatory functions.
Atherosclerotic plaques often harbor regions of hypoxia where oxygen availability is limited. In response to hypoxia and pro-inflammatory factors cell regulate hypoxia-inducible factor-1 (HIF-1), triggering a cascade of metabolic adaptations. One notable effect is the increased expression of GLUT1 and the glycolytic enzyme pyruvate kinase M2 (PKM2). This contributes to enhanced glucose uptake, glycolysis, glycolytic flux as well as upregulated expression of the key glycolysis proteins GLUT1, hexokinase II (HK-II), and 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase (PFKFB3) [41]. In patients with coronary artery disease (CAD), elevated glycolysis drives mitochondrial ROS production and promote the expression increased levels of pro-IL-1β and inflammasome activation. It then phosphorylates and activates signal transducer and activator of transcription (STAT3), to promote the production of inflammatory cytokines like IL-1 β and IL-6 [42]. A recent study unveiled that HIF-1α in orchestrating glycolytic reprogramming through induction of pyruvate dehydrogenase lipoamide kinase isozyme 1 (PDK1) and inhibition of HIF-1α-PDK1 axis promote systemic inflammation, suggesting that glycolysis contributes to macrophage migratory activity [43]. Notably, cellular response to hypoxia, including degradation of HIF proteins and HIF-dependent transcriptional programs, can occur as quickly as 24 h after exposure to hypoxia conditions [44, 45].
Contrary to conventional beliefs, recent studies have shed new light on the significance of glycolysis in M2 macrophage activation. IL-4 increased glucose metabolism through AKT and mechanistic target of rapamycin (mTOR) complex (mTORC) 1 [46]. This was evidenced by the blockage of glycolysis with 2-deoxyglucose (2-DG) strikingly which attenuated the IL-4 induced expression of Arg1, Fizz1, and CD301b, pivotal in sustaining glucose metabolism and overall energy balance [47]. Accordingly, inhibition of mitochondrial ATP synthase with oligomycin and glycolysis with 2-DG both suppress IL-4-regulated genes, surface markers and functions, implying that glucose fuels the TCA cycle for mitochondrial respiration in M2 macrophages [48, 49]. Meanwhile, recent study reassessed the role of glycolysis in M2 macrophages and found that glucose depletion, leaving OXPHOS intact, does not affect M2 activation. However, simultaneous impairment of glycolysis and OXPHOS by 2-DG resulted in reduced intracellular ATP levels and JAK-STAT6 signaling, ultimately impairing M2 differentiation. This study indicated that while glycolysis may not play a mandatory regulatory role in M2 differentiation, but it assumes an obligatory role once OXPHOS is compromised. Furthermore, suppressing both glycolysis and OXPHOS at higher doses of 2-DG led to reduced intracellular ATP content and M2 differentiation [49]. Notably, OXPHOS rather than glycolysis was the most discriminative process among macrophages in various tissues under homeostasis conditions [50]. This indicates that targeting OXPHOS may offer a more selective therapeutic approach of for macrophage-related diseases.
The Pentose Phosphate Pathway (PPP) in Macrophage
The PPP is another crucial pathway for glucose utilization in macrophage, pivotal for various cellular functions including ROS management and anti-oxidative defense by generating NADPH. While both M1 and M2 macrophages exhibit increased glycolysis, its noteworthy that only M1 macrophages activate the PPP. In M1 macrophages the PPP is bolstered by increased glucose intermediates flux and downregulation of carbohydrate kinase-like protein (CARKL) expression [39]. Conversely, M2 macrophages display decreased levels of glycolysis and PPP-associated metabolites such as lactate and sedoheptulose-7-phosphate. Macrophages rely on robust PPP flux and NADPH during respiratory bursts to combat extracellular bacteria and produce antioxidants like glutathione and thioredoxin, crucial for mitigating oxidative damage to cells. NADPH acts as a vital co-factor for lipid metabolism and other metabolic pathways, akin to ATP as a universal energy carrier utilized by diverse enzymes throughout the cellular metabolic networks. PPP activity play a vital role in redox-sensitive protein signaling during macrophage activation. Moreover, the PPP yields pentose molecules serving as precursors for nucleotide synthesis or reconvert to glycolytic intermediates [51]. Additionally, macrophages employ a noncanonical PPP loop involving repeated cycling of PPP intermediates back to glucose-6-phosphate (G6P) through a process resembling gluconeogenesis. this loop is essential for NADPH generation, ensuring appropriate phagolysosome maturation and facilitating apoptotic cell degradation [52].
The functional significance of GLUT1-mediated glucose uptake has been demonstrated through the selective ablation of GLUT1, leading to impaired glucose entry [53]. The causal relationship between hyperglycemia and atherosclerosis remains unclear. A recent study showed that sodium-glucose cotransporter 2 (SGLT2) inhibitors reduce blood glucose levels in diabetic mice and concomitantly prevent the stimulating effects of diabetes mellitus on myelopoiesis and the inflammatory activation of lesional macrophages [54]. These effects of are likely attribute to glucose reduction. Alternatively, it is possible that SGLT2 inhibitors exert their effects through indirect mechanisms, such as hemodynamic changes, rather than by solely preventing the direct effects of glucose on bone marrow cells or lesional cells [55]. It is crucial to determine whether monocytes, pro-inflammatory macrophages and foamy cells within the atherosclerotic plaque rely on specific and exclusive glucose dependent pathways. Furthermore, it is important to ascertain whether there is a metabolic reprogramming of macrophage glucose metabolism at a specific stage of disease progression, in a specific localization, and within a specific cell type [56].
Fatty Acid Oxidation (FAO) May Regulate Bioenergetics of Macrophage
Proinflammatory macrophages rely on glycolysis for their metabolic processes, whereas anti-inflammatory macrophages are thought to dependent on FAO. FAO occurs primarily within the mitochondrial matrix, serving as a significant ATP source, especially when glucose availability is limited. Besides bioenergetics, FAO may also generated metabolites that influence signal transduction and gene regulation [57]. IL-4 signaling activates the transcription factor STAT6, leading to increased expression of genes regulating FAO and mitochondrial biogenesis, such as CD36 for fatty acids (FAs) transport into the cell, carnitine palmitoyl transferase 1 (CPT1) for FAs transport into the mitochondria, and the nuclear receptor peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1β (PGC1β). Inhibition of FAO prevents M2 from activation, whereas overexpression of PGC1β is sufficient to attenuate M1 activation in response to IFN-γ or LPS [58]. However, it is now apparent that FAO is not just anti-inflammatory as it also supports inflammasome activation. Intriguingly, the oxidation of palmitate via CPT1A promoted mitochondrial ROS production by fueling mitochondrial respiration, leading to the activation of NOD-like receptor (NLR) family pyrin domain-containing 3 (NLRP3) [59, 60]. The pharmacologic FAO inhibitor trimetazidine reduced progression of atherosclerotic plaques in a mouse model by reducing IL-1β and NLRP3 activation. Although FAO is largely dispensable for M2 macrophage activation and regulates memory and regulatory T cell formation [61], dietary FA supplementation directly affects the FA composition of atherosclerotic plaques. For instance, omega 3 FA supplementation leads to a significant increase in docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) in carotid plaques as well as decreased inflammation [62, 63]. FAO is known to support cellular longevity in other immune cells, but its role in macrophages remains unclear [64, 65].
Cholesterol and Lipid Affect Macrophage Phenotype in Atherosclerosis
Cholesterol is a component of cell membranes and a precursor to viarious biological molecules. Cholesterol metabolism influences the ability of macrophages to mount inflammatory responses in atherosclerosis. Moreover, cholesterol overload directly triggers the release of mitochondrial DNA and activates the AIM2 inflammasome in stimulated macrophages, leading to IL-1β production [66]. Additionally, the accumulation of cholesterol crystals in the artery during the early stages of atherosclerosis has been proposed to activate the inflammasome pathway in local macrophages, resulting in the generation of IL-1β. Furthermore, 25-hydroxycholesterol (25-HC), a metabolic intermediate in cholesterol metabolism, exacerbated inflammatory responses in lipid-laden macrophages by accelerating progression of atherosclerosis and promoting plaque instability. Moreover, 25-HC produced by activated lipid-loaded macrophages amplifies the inflammatory response by increasing TLR4 dependent signaling, enhancing apoptosis sensitivity, reducing efferocytotic capacity, and promoting paracrine effects in neighboring cells [67]. Additionally, cholesterol crystals could promote nuclear translocation of PKM2 to facilitate metabolic reprogramming and M1 macrophage polarization in primary human macrophages [68]. This suggests that accumulation of cholesterol crystals during the initial phases of atherosclerotic lesions may contribute to the modulation of macrophage phenotype and function (Fig. 1). In macrophages, cholesterol can be synthesized de novo or taken up in the form of LDL particles which transport cholesterol synthesized in the liver [69]. Cholesterol synthesis occurs in the cytosol, using acetyl-CoA as a substrate and is mediated by a series of enzymes targets by drugs designed to reduce blood cholesterol levels [70]. Excess cholesterol can be esterified, stored in lipid droplets, or removed from the cell through reverse cholesterol transport, a process in which surplus cholesterol is efflux to high-density lipoprotein (HDL) for transport back to the liver by ABC transporters on the cell surface [71]. Dysregulated cholesterol metabolism in arterial macrophages has long been recognized as pathogenic in atherosclerosis. Elevated levels of modified cholesterol, particularly oxLDL are important risk factors for the development of atherosclerosis. Macrophages uptake oxLDL within atherosclerotic plaques, leading to the formation of cholesterol-laden foam cells. The role of the cholesterol synthesis pathway in oxLDL-induced training remains to be established. OxLDL generates signals recognized by pattern recognition receptors (PRR) on macrophages and other immune cells [72]. Scavenger receptors such as, scavenger receptor A1 (SR-A1), CD36, and lectin-like oxLDL receptor-1 (LOX-1) mediate the uptake of oxLDL by macrophages, resulting in high cholesterol levels and the foam-like of macrophages. In vitro, SR-A1 and CD36 mediate over 75% of the degradation of modified LDL with combined deficiency of both receptors reducing but not totally preventing foam cell formation in mice, suggesting other mechanisms of LDL uptake in vivo. A recent study has showed that deficiency of nuclear factor of activated T cells 3 (NFATc3) in macrophages promotes foam cell formation by enhancing SR-A1 and CD36-meditated lipid uptake [73]. Foam cells contribute to plaque pathogenesis by producting numerous pro-inflammatory cytokines, chemokines and matrix metalloproteinases (MMP) which degrade extracellular matrix in plaques [74]. The accumulation of lipids and inflammatory cytokines induces endoplasmic reticulum (ER) stress through the unfolded protein response (UPR) to increase the sensitivity of arterial macrophages to apoptosis, contributing to plaque inflammation and destabilization. Accumulation of oxidized lipoproteins can also induce a M1 proinflammatory phenotype in human macrophages by inhibiting the transcription factor Kruppellike factor 2. OxLDL promotes an inflammatory macrophage phenotype by activating a TLR4 mediated pathway in mice [75]. Cholesteryl esters induce M1 polarization by activating the TLR4 or NFκB signaling pathways [76]. Regulation of cholesterol influx and efflux is crucial for the atherosclerosis progression. Liver X receptor (LXR) enhances the expression of cholesterol efflux transporters, controlling the cholesterol outflow. LXR activation exerts anti-inflammatory effects, partly due to the inhibition of TLR2, TLR4, and TLR9 signaling to their downstream NF-κB and MAPK effectors achieved by changes in membrane lipid composition mediated by ABCA1 [77, 78]. On the other side, HDL attenuates macrophage necroptosis by activating Akt signaling in macrophages in an SR-B1 and PDZK1-dependent manner, suppressing the formation of necrotic cores within atherosclerotic plaques [79].
Amino Acid Metabolism and Atherosclerosis
The metabolism of amino acid plays a critical role in the inflammation during atherogenesis. A recent study aimed to assess the impact of various amino acids on the macrophage in order to screen anti- or pro-atherogenic amino acids by a macrophage model system. Among the 20 amino acids, six specific amino acids including cysteine, histidine, methionine, lysine, arginine and tryptophan exhibited significantly cytotoxic effects on macrophages. The observed cytotoxicity was associated with increased generation of ROS and accumulation of cholesterol or triglycerides in macrophages. Histidine and tryptophan exhibit the most prominent efffects [80]. Homoarginine, a endogenous amino acid, has been identified as a strong biomarker with low circulating levels associated with increased risk of cardiovascular diseases [81]. In atherosclerosis, the metabolism of arginine, and its byproduct NO, is vitally important for the early stages of the disease [82]. Arginine can be used for synthesis of polyamines through the action of Arg1, which support macrophage proliferation and collagen production for IL-4 induced tissue repair [83]. Decreased bioavailability of NO and endothelial dysfunction are associated with risk factors for CAD [84, 85]. The oxidation of arginine produces NO and citrulline, which play crucial roles in maintaining the homeostasis of vascular tissue preventing abnormal proliferation of vascular smooth muscle cells, regulating leukocyte interaction with the vascular wall, presenting antigens, and maintaining vascular tone and growth. NO downstream of iNOS has been shown to suppress oxidative phosphorylation in inflammatory macrophages and the interaction of NO with ROS leads to the formation of even more reactive nitrogen species (RNS) such as peroxynitrit [86,87,88,89]. Amino acids leucine could activate mTORC1 in macrophages, raising questions about whether dietary proteins or amino acids exacerbate macrophage apoptosis induced by atherogenic lipids, a process involving mTORC1-dependent inhibition of mitophagy, accumulation and apoptosis of dysfunctional mitochondria [90]. Amino acids metabolism plays significant roles in inflammation and is important for the induction of IL-1 by macrophages in response to LPS stimulation as well as contributing NO generation by feeding into the arginine synthesis pathway [91]. Furthermore, glutamine deprivation has been shown to have profound functional consequences in M2 macrophages immunity, as glutamine flux into both the TCA cycle and hexosamine pathway promotes M2 macrophage polarization [92, 93].
Macrophage Roles in Progression and Regression of Plaques
Macrophages of distinct functional phenotypes play varying roles in atherosclerosis development. In mice, macrophages in early atherosclerotic lesions are mainly derived from recruited monocytes, while advanced plaques exhibit prominent macrophage proliferation influenced by signals from microenvironment. Consequently, differences in resident macrophage phenotypes can impact their local proliferation, altering the abundance of specific phenotype macrophages [93, 94]. In atherosclerosis, the pivotal feature is the ingestion and accumulation of lipoproteins by arterial macrophages, leading to the formation of foam cells [95]. The phenotype of macrophages in the carotid plaques of symptomatic patients during acute ischemic stroke has been compared with those from asymptomatic patients [93, 96]. The majority of the macrophages in the plaques of symptomatic patients exhibit M1 phenotype, whereas those from asymptomatic patients predominantly express markers of M2 polarization. Additionally, macrophages located in the lesion shoulder, considered one of the most unstable areas express M1 markers, whereas macrophages in the fibrous cap surrounding the necrotic core express both M1 and M2 markers [97,98,99]. In the adventitia, M2 macrophages are more abundant than M1 macrophages. Recent evidence suggests that macrophages exist on a continuum of activation and the M1/M2 classification system oversimplifies macrophage heterogeneity and functions [100, 101]. Furthermore, macrophage significantly impacts atherosclerotic lesion development and progression, contributing to cell death and plaque necrosis promoting lesion disruption and acute thrombosis lesioned arteries [102]. Under extensive oxidative stress, macrophage death occurs both in vivo and in vitro, accompanied by enhanced lipid accumulation. Leucine-enriched macrophage lipid accumulation could drive macrophage activation and atherogenesis. As the lesion progresses, the number of macrophages increases with symptomatic plaque containing more macrophages than asymptomatic plaques [95, 103]. Macrophages in shoulder regions of plaque fibrous cap mainly express M1 markers. Ruptured plaques express transcriptional markers of both M1 and M2 macrophages without preferential polarization and upregulate markers of both M1[e.g., CD68, HLA-DP/Q/R and macrophage receptor with collagenous structure (MARCO)] and M2 [(e.g. dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN) and PPARγ] [99]. Additionally, markers of IPH such as CD163 and heme oxygenase-1 (HO-1) are highly upregulated in ruptured plaques, accumulating as plaques severity increases. Lesional foam cells exhibit markers are of both M1 (HLA-DP/Q/R and iNOS as well as dectin-1) and M2 phenotypes. Experimental data indicate that macrophage phenotype can switch to differential subtypes in response to the microenvironment. Suppressing M1 polarization and promoting enhanced M2 activation can accelerate plaque regression [104]. Overall, recent evidence supports the central role of macrophages in all stages of atherosclerosis and targeted lesional macrophage could be beneficial in protecting against atherosclerotic inflammation and lesion formation.
Therapeutic Targeting of Plaque Macrophages
Considering the pivotal role of macrophages in both the progression and regression of atherosclerotic lesions (Fig. 1), these cells present a crucial target for therapeutic intervention in ASCBD. Over the past few decades, the widespread implementation of lipid-lowering strategies, particularly statin therapy, has significantly revolutionized the management of cardiovascular disease, reducing the risk of acute cardiovascular events by 25–35%. As plaque macrophages plays a crucial role in atherogenesis, the primary mechanisms associated with macrophage activity, such as inflammation, oxidative stress, lipid metabolism and proliferation, are being explored as potential therapeutic targets [5]. Promoting cholesterol efflux is a key strategy to encourage atherosclerosis regression. LXR served as a transcription factor that triggers the expression of genes is involved in cholesterol transport and efflux and plays an essential role in impeding atherosclerosis progression and facilitating its regression [105, 106]. The nonspecific efflux molecule, cyclodextrin, promotes atherosclerosis regression through LXR-mediated reprogramming, leading to enhanced cholesterol efflux and exert anti-inflammatory effects. Yu and colleagues utilized biodegradable collagen type IV targeted polymeric nanoparticles to deliver the LXR agonist GW3965 to atherosclerotic lesions in LDLR –/– mice [107].
Furthermore, preclinical and clinical observations indicate that the reprogramming of plaque macrophages to an anti-inflammatory M2 phenotype can facilitate the regression and stabilization of atherosclerosis. Treatment of LDLR −/− mice with helminth-derived antigens derived form a eukaryotic parasitic worm has been shown to robustly stimulate anti-inflammatory immune responses, leading to the suppression of myeloid cell activation, i inflammation and reduced macrophages recruitment to lesions [108]. Combining this approach with lipid lowering therapies may represent a viable strategy for mitigating residual inflammatory cardiovascular disease risk in humans and expediting plaque regression. Nanomedicine provides an attractive strategy for locally targeting macrophage activity within atherosclerotic plaques [109]. A recently study showed that pterostilbene, T09 and simvastatin exhibited potent overall anti-atherogenic effects in macrophages, exerting beneficial impacts through multiple pathways simultaneously. Notably, monocyte recruitment to the arterial wall represents one of the earliest processes accelerating atherosclerosis progression [110]. In addition, siRNA-encapsulated polymeric nanoparticles are employed to inhibit monocyte recruitment to atherosclerotic lesions in ApoE–/– mice after myocardial infarction, concurrently inhibiting matrix-degrading protease activity in plaques by silencing endothelial cell adhesion molecules [111]. Furthermore, DPP-4 inhibitors can largely inhibit plaque formation in coronary arteries, resulting in a pronounced reduction in macrophages. Previous study utilized reconstituted HDL nanoparticles to deliver a small-molecule inhibitor targeting the tumor necrosis factor receptor-associated factor 6 interaction to circulating Ly6Chi monocytes, suppressing their pro-inflammatory response [112].
Apart from lipid metabolism and inflammation, the immune response plays a pivotal role in the pathogenesis of atherosclerosis. Modulation the immune response through the use of a vaccine containing antigens pertinent to atherogenesis holds promise for inducing a targeted immune response against these antigens while preserving overall host immunity. Intriguingly, administrating vaccines containing apoB100 peptides derived from oxLDL has shown therapeutic efficacy in mice atherosclerotic models by promoting the activation of regulatory T lymphocytes in the plaques. Furthermore, the emerging application of mRNA as a therapeutic tool has been utilized to modulate both innate and adaptive immunity responses through activating specific signaling pathways. Notably, the effective delivery of mRNA via nanoparticle-mediated strategies has revolutionized vaccine development, with mRNA vaccines being successfully employed to combat coronavirus disease 2019 (COVID-19) [113, 114]. Overall, we anticipate that technologies focusing on early targeting of macrophages to inhibit or diminish the inflammatory response hold potential for preventing the initiation and progression of atherosclerosis.
Conclusion and Perspectives
Macrophages are integral to the initiation and progression of atherosclerotic lesions, where phenotypes exhibit plasticity in response to changes in the microenvironment and specific stimuli. Macrophages undergo metabolic reprogramming in response to environmental cues such as hypoxia, apoptotic cells, and cytokines, leading to metabolic heterogeneity among plaque-resident macrophages. Metabolic changes of macrophages are likely to be dynamic due to the evolving microenvironment in plaque progression and disease management. Within the realm of fundamental macrophage biology, it is necessary to thoroughly comprehend how metabolic signatures of macrophages evolve in early atherogenesis and to elucidate the effects of specific metabolites in modulating distinct macrophage functions. More importantly, the exact relationship between immunometabolism in macrophages and atherosclerosis remains largely unclear. Further mechanistic in vivo investigations employing monocyte-macrophage targeted methodologies are warranted to furnish evidence of the causal roles of macrophage-derived metabolites in various stages of atherosclerosis lesion formation, encompassing early foam cell lesions, advanced plaques, and vulnerable plaques predisposed to rupture and subsequent acute myocardial infarction. There is increasing interest in exploiting the adaptability and polarization of macrophage, as well as their metabolic activities, to balance inflammatory responses and instate protective immune functions in atherosclerosis. A comprehensive comprehension of the specific metabolic reprogramming undergone by activated macrophages throughout the progression and regression of atherosclerosis is imperative for the discovery of innovative therapeutic approaches. Furthermore, the continued exploration of novel phenotypic and functional markers, coupled with the application of advanced expression profiling approaches as well as imaging technologies, is anticipated to refine and deepen the characterization and understanding of distinct roles played by various macrophage subtypes in atherosclerosis. The identification of biological triggers capable of affecting macrophage phenotypes holds promise for the development of novel therapeutic interventions for atherosclerosis and atherosclerotic cardiovascular diseases.
Key References
-
Wculek SK, Heras-Murillo I, Mastrangelo A, Mañanes D, Galán M, Miguel V, et al. Oxidative phosphorylation selectively orchestrates tissue macrophage homeostasis. Immunity. 2023;56(3):516-530.e9. (Identifies oxidative phosphorylation is critical for the maintenance of macrophages in tissues).
-
Kluck GEG, Qian AS, Sakarya EH, Quach H, Deng YD, Trigatti BL. Apolipoprotein A1 Protects Against Necrotic Core Development in Atherosclerotic Plaques: PDZK1-Dependent High-Density Lipoprotein Suppression of Necroptosis in Macrophages. Arterioscler Thromb Vasc Biol. 2023;43(1):45-63. (Identifies the new pathway to protect necroptosis of macrophages in atherosclerosis).
-
Edsfeldt A, Swart M, Singh P, Dib L. Interferon regulatory factor-5-dependent CD11c+ macrophages contribute to the formation of rupture-prone atherosclerotic plaques. Eur Heart J. 2022;43(19):1864-1877. (Demonstrates macrophage phenotype may play important role in the rupture of atherosclerosis plaque).
Data Availability
No datasets were generated or analysed during the current study.
References
Williams JW, Zaitsev K, Kim KW, Ivanov S, Saunders BT, Schrank PR, et al. Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat Immunol. 2020;21(10):1194–204.
Oppi S, Nusser-Stein S, Blyszczuk P, Wang X, Jomard A, Marzolla V, et al. Macrophage NCOR1 protects from atherosclerosis by repressing a pro-atherogenic PPARγ signature. Eur Heart J. 2020;41(9):995–1005.
Leuschner F, Nahrendorf M. Novel functions of macrophages in the heart: insights into electrical conduction, stress, and diastolic dysfunction. Eur Heart J. 2020;41(9):989–94.
Evren E, Ringqvist E, Tripathi KP, Sleiers N, Rives IC, Alisjahbana A, et al. Distinct developmental pathways from blood monocytes generate human lung macrophage diversity. Immunity. 2021;54(2):259-275.e7.
Galkina E, Ley K. Vascular adhesion molecules in atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27(11):2292–301.
Pourcet B, Staels B. Alternative macrophages in atherosclerosis: not always protective! J Clin Invest. 2018;128(3):910–2.
Moore KJ, Koplev S, Fisher EA, Tabas I, Björkegren JLM, Doran AC, et al. Macrophage trafficking, inflammatory resolution, and genomics in atherosclerosis: JACC macrophage in CVD series (Part 2). J Am Coll Cardiol. 2018;72(18):2181–97.
Narasimhan PB, Marcovecchio P, Hamers AAJ, Hedrick CC. Nonclassical monocytes in health and disease. Annu Rev Immunol. 2019;37:439–56.
Tang SCW, Yiu WH. Innate immunity in diabetic kidney disease. Nat Rev Nephrol. 2020;16(4):206–22.
Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44(3):450–62.
Vannella KM, Wynn TA. Mechanisms of organ injury and repair by macrophages. Annu Rev Physiol. 2017;79:593–617.
Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–69.
Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40(2):274–88.
Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177(10):7303–11.
Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. 2014;14(6):392–404.
Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233(9):6425–40.
Barrett TJ. Macrophages in atherosclerosis regression. Arterioscler Thromb Vasc Biol. 2020;40(1):20–33.
Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15(5):551–61.
Willenborg S, Injarabian L, Eming SA. Role of macrophages in wound healing. role of macrophages in wound healing. Cold Spring Harbor Perspect Biol. 2022;14(12):041216.
Locati M, Curtale G, Mantovani A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev Pathol. 2020;15:123–47.
Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunol Rev. 2014;262(1):153–66.
Abdelaziz MH, Abdelwahab SF. Alternatively activated macrophages; a double-edged sword in allergic asthma. J Transl Med. 2020;18(1):58.
Tang L, Zhang H, Wang C, Li H, Zhang Q, Bai J. M2A and M2C macrophage subsets ameliorate inflammation and fibroproliferation in acute lung injury through interleukin 10 pathway. Shock. 2017;48(1):119–29.
Sapudom J, Karaman S, Mohamed WKE, Garcia-Sabaté A. 3D in vitro M2 macrophage model to mimic modulation of tissue repair. npj Regen Med. 2021;6(1):83.
Chen MM, Xiao X, Lao XM, Wei Y, Liu RX, Zeng QH, et al. Polarization of Tissue-Resident TFH-Like Cells in Human Hepatoma Bridges Innate Monocyte Inflammation and M2b Macrophage Polarization. Cancer Discov. 2016;6(10):1182–95.
Rőszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm. 2015;2015:816460.
Liberale L, Dallegri F, Montecucco F, Carbone F. Pathophysiological relevance of macrophage subsets in atherogenesis. Thromb Haemost. 2017;117(1):7–18.
Tariq M, Zhang JQ, Liang GK, He QJ, Ding L, Yang B. Gefitinib inhibits M2-like polarization of tumor-associated macrophages in Lewis lung cancer by targeting the STAT6 signaling pathway. Acta Pharmacol Sin. 2017;38(11):1501–11.
Zhang J, Muri J, Fitzgerald G, Gorski T, Gianni-Barrera R, Masschelein E, et al. Endothelial Lactate Controls Muscle Regeneration from Ischemia by Inducing M2-like Macrophage Polarization. Cell Metabo. 2020;31(6):1136-1153.e7.
Habib A, Finn AV. The role of iron metabolism as a mediator of macrophage inflammation and lipid handling in atherosclerosis. Front Pharmacol. 2014;27(5):195.
Cochain C, Zernecke A. Macrophages and immune cells in atherosclerosis: recent advances and novel concepts. Basic Res Cardiol. 2015;110(4):34.
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889–96.
Chistiakov DA, Bobryshev YV, Nikiforov NG, Elizova NV, Sobenin IA, Orekhov AN. Macrophage phenotypic plasticity in atherosclerosis: the associated features and the peculiarities of the expression of inflammatory genes. Int J Cardiol. 2015;184:436–45.
Mouton AJ, Li X, Hall ME, Hall JE. Obesity, hypertension, and cardiac dysfunction: novel roles of immunometabolism in macrophage activation and inflammation. Circ Res. 2020;126(6):789–806.
Van den Bossche J, O’Neill LA, Menon D. macrophage immunometabolism: where are we (going)? Trends Immunol. 2017;38(6):395–406.
Liu PS, Chen YT, Li X, Hsueh PC, Tzeng SF, Chen H, et al. CD40 signal rewires fatty acid and glutamine metabolism for stimulating macrophage anti-tumorigenic functions. Nat Immunol. 2023;24(3):452–62.
Baidžajevas K, Hadadi É, Lee B, Lum J, Shihui F, Sudbery I, et al. Macrophage polarisation associated with atherosclerosis differentially affects their capacity to handle lipids. Atherosclerosis. 2020;305:10–8.
Renaudin F, Orliaguet L. Gout and pseudo-gout-related crystals promote GLUT1-mediated glycolysis that governs NLRP3 and interleukin-1β activation on macrophages. Ann Rheum Dis. 2020;79(11):1506–14.
Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 2012;15(6):813–26.
Yu W, Wang Z, Zhang K, Chi Z, Xu T, Jiang D, et al. One-carbon metabolism supports S-Adenosylmethionine and histone methylation to drive inflammatory macrophages. Mol Cell. 2019;75:1147-1160.e1145.
Wu SB, Wei YH. AMPK-mediated increase of glycolysis as an adaptive response to oxidative stress in human cells: implication of the cell survival in mitochondrial diseases. Biochim Biophys Acta. 2012;1822(2):233–47.
Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med. 2016;213(3):337–54.
Semba H, Takeda N, Isagawa T, Sugiura Y, Honda K, Wake M, et al. HIF-1α-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat Commun. 2016;7:11635.
Ginouvès A, Ilc K, Macías N, Pouysségur J, Berra E. PHDs overactivation during chronic hypoxia “desensitizes” HIFalpha and protects cells from necrosis. Proc Natl Acad Sci U S A. 2008;105(12):4745–50.
Jain IH, Calvo SE, Markhard AL, Skinner OS, To TL, Ast T, et al. Genetic screen for cell fitness in high or low oxygen highlights mitochondrial and lipid metabolism. Cell. 2020;181(3):716-727.e11.
Covarrubias AJ, Aksoylar HI, Horng T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin Immunol. 2015;27(4):286–96.
Kumamoto Y, Camporez JPG, Jurczak MJ, Shanabrough M, Horvath T, Shulman GI, et al. CD301b(+) Mononuclear Phagocytes Maintain Positive Energy Balance through Secretion of Resistin-like Molecule Alpha. Immunity. 2016;45(3):583–96.
Puleston DJ, Buck MD, Klein Geltink RI, Kyle RL, Caputa G, O’Sullivan D, et al. Polyamines and eIF5A hypusination modulate mitochondrial respiration and macrophage activation. Cell Metab. 2019;30(2):352-363.e8.
Wang F, Zhang S, Vuckovic I, Jeon R, Lerman A, Folmes CD, et al. Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell Metab. 2018;28(3):463-475.e4.
Wculek SK, Heras-Murillo I, Mastrangelo A, Mañanes D, Galán M, Miguel V, et al. Oxidative phosphorylation selectively orchestrates tissue macrophage homeostasis. Immunity. 2023;56(3):516-530.e9 Identifies oxidative phosphorylation is critical for the maintenance of macrophages in tissues.
Blagih J, Jones RG. Polarizing macrophages through reprogramming of glucose metabolism. Cell Metab. 2012;15(6):793–5.
Wang YT, Trzeciak AJ, Rojas WS, Saavedra P, Chen YT, Chirayil R, et al. Metabolic adaptation supports enhanced macrophage efferocytosis in limited-oxygen environments. Cell Metab. 2023;35(2):316-331.e6.
Cho H, Kwon HY, Sharma A. Visualizing inflammation with an M1 macrophage selective probe via GLUT1 as the gating target. Nat Commun. 2022;13(1):5974.
Kim SR, Lee SG. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat Commun. 2020;11(1):2127.
Heerspink HJ, Perkins BA, Fitchett DH, Husain M, Cherney DZ. Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes mellitus: cardiovascular and kidney effects, potential mechanisms, and clinical applications. Circulation. 2016;134(10):752–72.
Groh L, Keating ST, Joosten LAB, Netea MG, Riksen NP. Monocyte and macrophage immunometabolism in atherosclerosis. Seminars Immunopathol Semin Immunopathol. 2018;40(2):203–14.
Cader MZ, Boroviak K, Zhang Q, Assadi G, Kempster SL, Sewell GW, et al. C13orf31 (FAMIN) is a central regulator of immunometabolic function. Nat Immunol. 2016;17(9):1046–56.
Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15(9):846–55.
Moon JS, Nakahira K, Chung KP, DeNicola GM, Koo MJ, Pabón MA, et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat Med. 2016;22(9):1002–12.
Hall CJ, Boyle RH, Astin JW, Flores MV, Oehlers SH, Sanderson LE, et al. Immunoresponsive gene 1 augments bactericidal activity of macrophage-lineage cells by regulating β-oxidation-dependent mitochondrial ROS production. Cell Metab. 2013;18(2):265–78.
Van den Bossche J, van der Windt GJW. Fatty Acid Oxidation in macrophages and T Cells: time for reassessment? Cell Metab. 2018;28(4):538–40.
Thies F, Garry JM, Yaqoob P, Rerkasem K, Williams J, Shearman CP, et al. Association of n-3 polyunsaturated fatty acids with stability of atherosclerotic plaques: a randomised controlled trial. Lancet. 2003;361(9356):477–85.
Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med. 2019;380(1):11–22.
Pearce EL. Metabolism in T cell activation and differentiation. Curr Opin Immunol. 2010;22(3):314–20.
van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36(1):68–78.
Dang EV, McDonald JG, Russell DW, Cyster JG. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell. 2017;171(5):1057-1071.e11.
Canfrán-Duque A, Rotllan N, Zhang X, Andrés-Blasco I. Macrophage-derived 25-hydroxycholesterol promotes vascular inflammation, atherogenesis, and lesion remodeling. Circulation. 2023;147(5):388–408.
O’Rourke SA, Neto NGB, Devilly E, Shanley LC, Fitzgerald HK, Monaghan MG, et al. Cholesterol crystals drive metabolic reprogramming and M1 macrophage polarisation in primary human macrophages. Atherosclerosis. 2022;352:35–45.
Nurmohamed NS, Ditmarsch M. Cholesteryl ester transfer protein inhibitors: from high-density lipoprotein cholesterol to low-density lipoprotein cholesterol lowering agents? Cardiovasc Res. 2022;118(14):2919–31.
Goedeke L, Fernández-Hernando C. Regulation of cholesterol homeostasis. Cell Mol Life Sci. 2012;69(6):915–30.
Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang N. HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab. 2008;7(5):365–75.
Libby P, Aikawa M, Schönbeck U. Cholesterol and atherosclerosis. Biochim Biophys Acta. 2000;1529(1–3):299–309.
Liu X, Guo JW, Lin XC. Macrophage NFATc3 prevents foam cell formation and atherosclerosis: evidence and mechanisms. Eur Heart J. 2021;42:4847–61.
Di Gregoli K, Jenkins N, Salter R, White S, Newby AC, Johnson JL. MicroRNA-24 regulates macrophage behavior and retards atherosclerosis. Arterioscler Thromb Vasc Biol. 2014;34(9):1990–2000.
Xu Y, Kong X, Zhou H, Zhang X, Liu J, Yan J, et al. oxLDL/β2GPI/anti-β2GPI complex induced macrophage differentiation to foam cell involving TLR4/NF-kappa B signal transduction pathway. Thromb Res. 2014;134(2):384–92.
Ackerman D, Tumanov S, Qiu B, Michalopoulou E, Spata M, Azzam A, et al. Triglycerides promote lipid homeostasis during hypoxic stress by balancing fatty acid saturation. Cell Rep. 2018;24(10):2596-2605.e5.
Kong XN, Yan HX, Chen L, Dong LW, Yang W, Liu Q, et al. LPS-induced down-regulation of signal regulatory protein alpha contributes to innate immune activation in macrophages. J Exp Med. 2007;204(11):2719–31.
Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 2015;15(2):104–16.
Kluck GEG, Qian AS, Sakarya EH, Quach H, Deng YD, Trigatti BL. Apolipoprotein A1 protects against necrotic core development in atherosclerotic plaques: PDZK1-dependent high-density lipoprotein suppression of necroptosis in macrophages. Arterioscler Thromb Vasc Bio. 2023;43(1):45–63 Identifies the new pathway to protect necroptosis of macrophages in atherosclerosis.
Rom O, Grajeda-Iglesias C, Najjar M, Abu-Saleh N, Volkova N, Dar DE, et al. Atherogenicity of amino acids in the lipid-laden macrophage model system in vitro and in atherosclerotic mice: a key role for triglyceride metabolism. J Nutr Biochem. 2017;45:24–38.
Nitz K, Lacy M, Bianchini M, Wichapong K. The amino acid homoarginine inhibits atherogenesis by modulating T-Cell function. Circ Res. 2022;131(8):701–12.
Nitz K, Lacy M, Atzler D. Amino acids and their metabolism in atherosclerosis. Arterioscler Thromb Vasc Biol. 2019;39(3):319–30.
Boutens L, Hooiveld GJ, Dhingra S, Cramer RA, Netea MG, Stienstra R. Unique metabolic activation of adipose tissue macrophages in obesity promotes inflammatory responses. Diabetologia. 2018;61(4):942–53.
Gutiérrez E, Flammer AJ, Lerman LO, Elízaga J, Lerman A, Fernández-Avilés F. Endothelial dysfunction over the course of coronary artery disease. Eur Heart J. 2013;34(41):3175–81.
Kratzer A, Giral H, Landmesser U. High-density lipoproteins as modulators of endothelial cell functions: alterations in patients with coronary artery disease. Cardiovasc Res. 2014;103(3):350–61.
Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. 2016;17(3):684–96.
Everts B, Amiel E, van der Windt GJ, Freitas TC, Chott R, Yarasheski KE, et al. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood. 2012;120(7):1422–31.
Tabas I, Bornfeldt KE. Intracellular and intercellular aspects of macrophage immunometabolism in atherosclerosis. Circ Res. 2020;126(9):1209–27.
Alef MJ, Vallabhaneni R, Carchman E, Morris SM Jr, Shiva S, Wang Y, et al. Nitrite-generated NO circumvents dysregulated arginine/NOS signaling to protect against intimal hyperplasia in Sprague-Dawley rats. J Clin Invest. 2011;121(4):1646–56.
Zhang X, Sergin I, Evans TD, Jeong SJ, Rodriguez-Velez A, Kapoor D, et al. High-protein diets increase cardiovascular risk by activating macrophage mTOR to suppress mitophagy. Nat Metab. 2020;2(1):110–25.
Wallace C, Keast D. Glutamine and macrophage function. Metabolism: Clin Exp. 1992;41(9):1016–20.
Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42(3):419–30.
Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332:1284–8.
Jenkins SJ, Ruckerl D, Thomas GD, Hewitson JP, Duncan S, Brombacher F, et al. IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. J Exp Med. 2013;210(11):2477–91.
Wang J, Liu H, Sun J, Xue H, Xie L, Yu S, et al. Varying correlation between 18F-fluorodeoxyglucose positron emission tomography and dynamic contrast-enhanced MRI in carotid atherosclerosis: implications for plaque inflammation. Stroke. 2014;45(6):1842–5.
van Dam-Nolen DHK, Truijman MTB, van der Kolk AG, Liem MI, Schreuder FHBM, Boersma E, et al. Carotid plaque characteristics predict recurrent ischemic stroke and TIA: the parisk (Plaque At RISK) Study. JACC Cardiovasc Imaging. 2022;15(10):1715–26.
Barlis P, Serruys PW, Devries A, Regar E. Optical coherence tomography assessment of vulnerable plaque rupture: predilection for the plaque “shoulder.” Eur Heart J. 2008;29(16):2023.
Edsfeldt A, Swart M, Singh P, Dib L. Interferon regulatory factor-5-dependent CD11c+ macrophages contribute to the formation of rupture-prone atherosclerotic plaques. Eur Heart J. 2022;43(19):1864–77 Demonstrates macrophage phenotype may play important role in the rupture of atherosclerosis plaque.
Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta. J Exp Med. 2001;194(6):809–21.
Stöger JL, Gijbels MJ, van der Velden S, Manca M, van der Loos CM, Biessen EA, et al. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis. 2012;225(2):461–8.
Williams JW, Giannarelli C, Rahman A, Randolph GJ, Kovacic JC. Macrophage biology, classification, and phenotype in cardiovascular disease: JACC macrophage in CVD Series (Part 1). J Am Coll Cardiol. 2018;72(18):2166–80.
Robinson N, Ganesan R, Hegedűs C, Kovács K, Kufer TA, Virág L. Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 2019;26:101239.
Golledge J, Greenhalgh RM, Davies AH. The symptomatic carotid plaque. Stroke. 2000;31(3):774–81.
Liu Y, Wang X, Pang J, Zhang H, Luo J, Qian X, et al. attenuation of atherosclerosis by protocatechuic acid via inhibition of M1 and promotion of M2 macrophage polarization. J Agric Food Chem. 2019;67(3):807–18.
Shrestha E, Hussein MA, Savas JN, Ouimet M, Barrett TJ, Leone S, et al. Poly (ADP-ribose) polymerase 1 represses liver X receptor-mediated ABCA1 expression and cholesterol efflux in macrophages. J Biol Chem. 2016;291(21):11172–84.
Lee SD, Tontonoz P. Liver X receptors at the intersection of lipid metabolism and atherogenesis. Atherosclerosis. 2015;242(1):29–36.
Yu M, Amengual J, Menon A, Kamaly N, Zhou F, Xu X, et al. Targeted nanotherapeutics encapsulating liver X receptor agonist GW3965 enhance antiatherogenic effects without adverse effects on hepatic lipid metabolism in Ldlr-/- Mice. Adv Healthc Mater. 2017;6(20). https://doi.org/10.1002/adhm.201700313
Wolfs IM, Stöger JL, Goossens P, Pöttgens C, Gijbels MJ, Wijnands E, et al. Reprogramming macrophages to an anti-inflammatory phenotype by helminth antigens reduces murine atherosclerosis. FASEB J. 2014;28(1):288–99.
Chen W, Schilperoort M. Macrophage-targeted nanomedicine for the diagnosis and treatment of atherosclerosis. Nat Rev Cardiol. 2022;19(4):228–49.
Alaarg A, Zheng KH, van der Valk FM, da Silva AE, Versloot M, van Ufford LC, Schulte DM, Storm G, Metselaar JM, Stroes ES, Hamers AA. Multiple pathway assessment to predict anti-atherogenic efficacy of drugs targeting macrophages in atherosclerotic plaques. Vascul Pharmacol. 2016;82:51–9.
Sager HB, Dutta P, Dahlman JE, Hulsmans M, Courties G, Sun Y, Heidt T, Vinegoni C, Borodovsky A, Fitzgerald K, Wojtkiewicz GR, Iwamoto Y, Tricot B, Khan OF, Kauffman KJ, Xing Y, Shaw TE, Libby P, Langer R, Weissleder R, Swirski FK, Anderson DG, Nahrendorf M. RNAi targeting multiple cell adhesion molecules reduces immune cell recruitment and vascular inflammation after myocardial infarction. Sci Transl Med. 2016;8(342):342ra80.
Braza MS, van Leent MMT, Lameijer M, Sanchez-Gaytan BL, Arts RJW, Pérez-Medina C, et al. Inhibiting inflammation with myeloid cell-specific nanobiologics promotes organ transplant acceptance. Immunity. 2018;49(5):819-828.e6.
Powles T, Rosenberg JE, Sonpavde GP, Loriot Y, Durán I, Lee JL, et al. Enfortumab vedotin in previously treated advanced urothelial carcinoma. N Engl J Med. 2021;384(12):1125–35.
Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N Engl J Med. 2020;383(27):2603–15.
Funding
This work was supported by in part, by the National Natural Science Fund (NSFC, China, 81900382, 92168117, 82370432). Beijing Municipal Natural Science Foundation (7222072, 7222068).
Author information
Authors and Affiliations
Contributions
J.W., WM.L., JC.Z., Q.W. and XY.W. wrote the main manuscript text and J.W., HB.L., ML.C, L.X. and KB.L. prepared Figs. 1–2, Z.Z. prepared Fig. 1. J.W., WM.L. and Z.Z. revised the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Juan Wang, Qiang Wu and Xinyu Wang were co-first authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, J., Wu, Q., Wang, X. et al. Targeting Macrophage Phenotypes and Metabolism as Novel Therapeutic Approaches in Atherosclerosis and Related Cardiovascular Diseases. Curr Atheroscler Rep 26, 573–588 (2024). https://doi.org/10.1007/s11883-024-01229-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11883-024-01229-z