
Abstract
This work reports the development and testing of a magnetic polymer (Polyamide 6, PA6) nanocomposite capable of melting when exposed to an external magnetic field. Addition of high concentrations of iron oxide nanoparticles (NPs) can induce quick melting but is detrimental to the mechanical properties of the polymer. To reduce the amount of NPs required for achieving efficient melting, they should be well dispersed in the polymer. In this study, the oleic acid loading on the surfaces of the NPs was varied to study the effect of variations in coatings on the dispersion in the polymer and on the polymer melting time. The NPs functionalized with oleic acid were added to melted monomer ε-caprolactam and polymerized using ring-opening polymerization. The resulting PA6 nanocomposite was characterized by Fourier-transform infrared spectroscopy, differential scanning calorimetry, x-ray diffraction and transmission electron microscopy. The results confirmed that the PA6 nanocomposite showed a decrease of 8–10% in its glass-transition temperature compared to commercial PA6. The crystallinity of the synthesized samples were found to vary between 42% and 57%. The 55 wt.% oleic acid-loaded NPs were found to disperse most efficiently in the PA6 matrix; however, some large agglomerates were formed due to excessive oleic acid. Therefore, the 22 wt.% oleic acid coating showed overall superior dispersion. Additionally, the magnetic induction response was tested by observing a melt-characteristic of the magnetic polymer composite using a model set-up. Oleic acid concentration is found to affect the dispersion, melting time and crystallinity of the nanocomposite.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Polymer nanocomposites (PNCs) that can respond to external stimuli, namely electric, optical or magnetic, are proving promising for various applications, such as drug delivery, information storage, electromagnetic interference shielding, magnetic resonance imaging, environmental remediation, microscale manufacturing and self-healing.1,2,3,4,–5 Nanoparticles (NPs) naturally exhibit a strong tendency to agglomerate, as this minimizes the energy interactions linked with their high surface area to volume ratio. Moreover, increasing NP loading reduces the inter-particle distances, allowing the formation of the agglomerates due to short range van der Waals or hydrogen bonds, as observed in particle aggregation at the air–liquid interface in bubbles.3,4 The presence of NP agglomerates in the polymer matrix causes an uneven material heating in response to the applied stimulus, as the region with agglomerated particles will quickly absorb much of the stimulus energy and will melt the surrounding polymer excessively or undergo pyrolysis.5 Therefore, mechanical and chemical dispersion techniques can be employed to achieve uniform heating of the polymer in nanocomposites.
To improve the NP dispersion efficacy, it is critical to adjust the level of interaction and bonding strength between the NPs and the surrounding polymer matrix.6 Various mechanical techniques such as melt compounding,7,8 high shear mixing,9 three roll mills,10 and solution blending 11 are used to obtain uniform dispersion and to limit the formation of agglomerates. However, these methods do not promote a strong enough interaction between phases such that the agglomerates break down to a desired nanoscale. For example, the solution blending method still faces a major shortcoming due to the need to find an appropriate polymer–solvent pair for each filler–polymer system.12 Functionalization of NPs involving ionic charge, polymer coating (brushes), small molecules, and core–shell arrangement are efficient ways to promote their dispersion in the hydrophobic polymer matrix, when employed with electrostatic or steric mechanisms.
These techniques enhance the polymer–nanocomposite interactions at the nanoscale to confine agglomerate formation and hence many studies have analyzed the effect of NP functionalization on its adhesion with the base polymer matrix.13,14,15,–16 The in situ polymerization method of dispersion has been successful for polyamide- and epoxy-based composites17,18 as it enhances covalent bonding between the base polymer matrix and the NP surfaces.19 However, a high viscosity and non-Newtonian rheology of the melted polymer in the melt-blending method still poses a challenge in dispersing NPs homogenously. In in situ polymerization,20 the inorganic precursors are added in a low-viscosity melt-monomer to achieve optimum dispersion. Additionally, an in situ polymerization method has further opportunities for attaining rapid and controlled cross-linking density, and therefore it provides new properties to the PNC. The methods such as an interfacial polymerization, free radical polymerization, anionic ring-opening polymerization, frontal polymerization, micellar copolymerization, and network polymerization have already been studied for in situ polymerization.21
Considerable variations in the size to surface area ratios of NPs and their agglomerates affect the magnetic response nonlinearly, hence the functionalization of such NPs can help tune the anomalous behavior of the derived nanocomposites.22 The favoured NP morphology and crystalline phases obtained after functionalization can significantly affect the magnetic properties and crystallinity of the synthesized nanocomposites.23
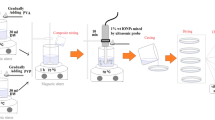
The use of oleic acid as a capping agent results in hydrophobic coating, as polar end groups attach to the NP surface and form a strongly bonded protective monolayer, which enhances the uniform dispersion of the NPs.24 In order to study the effect of oleic acid (OA) loading in the coating of iron oxide NPs on nanocomposite properties, a magnetic nanocomposite was prepared by keeping the 1 wt.% iron oxide NP concentration constant but varying the OA loading on the surface of the NPs. To prepare the nanocomposite, the 1 wt.% NPs were blended in a low-viscosity melted monomer, ε-caprolactam (CL). Rapid polymerization was then achieved by a bulk anionic ring-opening polymerization.25 The use of a strong Grignard reagent ensured the rapid confinement of the NPs and the promotion of a uniform dispersion state.26 A total of three different bulk samples of the nanocomposite were prepared with 1 wt.% iron oxide NPs, but the OA loading was kept at 22 wt.%, 55 wt.%, and 0 wt.% (Table I). The prepared materials have promising applications in diverse scientific fields.27
Materials and Methods
Materials
The following materials were used as received from Sigma-Aldrich, Dorset, UK: iron (III) oxide NPs (< 50 nm size BET), oleic acid (OA) (99% purity), N-acetylcaprolactam (NACL) (99% purity), 3.0 M ethyl magnesium bromide (EtMgBr) solution in diethyl ether, and ε-caprolactam (CL) (99% purity).
Experimental Methods
Functionalization of Iron Oxide NPs
The nanoparticle suspensions were prepared by dispersing 0.225 g of iron oxide NPs in 10 mL of methanol. Subsequently, OA was added into two suspension batches of 10 mL to make up 22 wt.% and 55 wt.% solutions. The mixture was rigorously stirred for 10 min at full power using a probe-type 150-W sonicator (Soniprep 150; MSE., UK). The NPs were thoroughly washed three times using methanol and decanted following centrifugation at 4000 rpm for 5 min. The NPs were then calcined at 80°C overnight in a furnace.
In Situ Synthesis of Iron Oxide Polymer
The first step in the polymerization process was to melt 30 g of CL at 60°C. Iron oxide NPs were introduced into the melted monomer to prepare a 1 wt.% iron oxide nanocomposite. The mixture of NPs and monomer was sonicated at 20 kHz for 30 min to ensure effective dispersion of the NPs. The temperature of the mixture was then raised to 150°C and 0.86 mL of EtMgBr (at a concentration of 2.5 mol.% mol−1 CL) was introduced under an inert gas atmosphere. To complete the polymerization, 0.94 mL (2.5 mol.%) of NACL was added at 160°C. The mixture instantly polymerized and captured the dispersed positions of the NPs. Before characterization, the nanocomposite was thoroughly washed with deionized water (18 MΩ cm) at 100°C to remove any unreacted monomer, activator and initiator. The samples prepared for characterization are shown in Table I.
Characterization Methodology
The nanocomposite samples were characterized using a Perkin-Elmer ATR-FTIR (Attenuated Total Reflection–Fourier-Transform Infrared Spectroscope) using a DGS-KBr sensor to identify functional groups and structural changes after the addition of the iron oxide NPs. Nanocomposite films averaging 0.1 mm thickness were prepared and a total of 30 scans in the range of 525–4000 cm−1 wavenumber were carried out at a resolution of 4 cm−1.
DSC was performed on a TA Instruments DSC Q100 at a heating rate of 10°C/min under a nitrogen purge within the temperature range of 20–250°C. The glass transition temperature (Tg) and melting temperature (Tm) were determined.
A PANalytical X’Pert Pro multipurpose diffractometer fitted with an X’Celerator detector and using monochromated Cu-Kα radiation (λ = 1.54 Å) was used to detect the crystalline phases present in the samples. X-ray diffraction (XRD) datasets were collected over a range of 10°–65° 2θ with a step size of 0.117° 2θ and nominal time per step of 1099.82 s. Phase identification was carried out using the HighScore Plus software (v.4.7) in conjunction with the ICDD Powder Diffraction File 4 Minerals (2017) and the ICDD Powder Diffraction File 2 (2004) databases.
To prepare samples for TEM study, an ultrathin section of the nanocomposite obtained using a microtome was placed on a 400-mesh-size gilder grid. A Philips CM100 TEM with an accelerating voltage of 100 kV and a spot size of 10 nm was used to capture the images. The exposure time was varied from 0 to 50 s, whereas the direct magnifications used ranged from × 7900 to × 245,000.
Small-angle x-ray scattering (SAXS) patterns were obtained using the polymer or nanocomposite films of 250–500 µm thickness, on a Bruker AXS Nanostar equipped with a 2D Hi-STAR multi-wire gas detector and modified with a Xenocs GeniX 3D x-ray source (CuKα radiation with x-ray wavelength λ = 1.54 Å) and motorized collimating scatterless slits. The patterns were obtained over a scattering vector length within the range of 0.008 Å−1 < q < 0.18 Å−1, where \( q = \frac{4\pi }{\lambda }\sin \theta \) and θ is a half of the scattering angle. One-dimensional scattering curves were obtained by an azimuthal binning and averaging of corresponding two-dimensional scattering patterns using software packages supplied with the SAXS instrument.
Results and Discussion
In order to determine the effect of OA loading in the coating of the NPs on the PA6 properties during in situ polymerization, a PNC with 1 wt.% iron oxide was prepared.
Transmission Electron Microscopy
The organic OA layer on the NPs was initially characterized by comparing TEM images of bare and coated NPs with different weight percentages of OA. A droplet of low-concentration aqueous NP suspension was placed on the TEM grid and dried before imaging. Care was taken to disperse the NPs well in the water using a sonicator. In addition, the degree of dispersion and/or agglomeration in the synthesized nanocomposite samples were studied using TEM images.
In order to measure the average diameter of NPs using TEM images (Fig. 1a, b and c, image analysis was performed (Supplementary material). The results show that the bare iron oxide NPs are highly agglomerated with sizes in the range of 35–50 nm, while OA-coated NP diameters were found to be in range of 25–40 nm for 22 wt.% and 30–60 nm for 55 wt.% OA loading. The hydrophobic nature of the organic OA layer on the NP surfaces restricted the interaction between the NPs and, hence, the mean diameter of the agglomerates in the polymer.28 The adsorption of OA on the NP surface is less transparent to the electron beam and, therefore, the coating on the NPs was observed as a thick opaque layer, as seen in Fig. 1a, b and c.

(a) TEM image of uncoated iron oxide NPs (scale bar 100 nm). TEM images of OA-coated NPs: (b) 22% OA and (c) 55% OA (scale bar 100 nm)
The TEM images of 50-μm-thick microtome samples of the PNC were analyzed to investigate the extent of dispersion. As seen in Fig. 2a, b and c, the dispersion was found to improve with increasing OA loading as the density of adsorbed hydrophobic molecules on the surface of the NPs increases. With 1-μm and 60-nm agglomerates were found in the PNC containing uncoated NPs and 55 wt.% OA-coated NPs, respectively, albeit overloading of the OA coating led to the formation of many oversized agglomerates with overall smaller mean diameters. Hence, as observed from the TEM images, the NP agglomerates in the 55 wt.% OA samples are larger in comparison to those observed in the 22 wt.% OA samples. This is comparable with the size calculated from the XRD peaks and the SAXS results.

TEM images show dispersion of NPs in (a) PNC with uncoated NPs, (b) PNC with NPs coated with 22 wt.% OA loading, and (c) PNC with NPs coated with 55 wt.% OA loading (scale bar 100 nm)
Fourier-Transform Infrared Spectroscopy
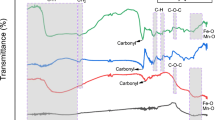
The FTIR peaks observed have been found to match with all the standard template chemical group peaks of the commercial PA6, which confirmed its successful synthesis. As seen in Fig. 3, a peak attributed to hydrogen bonds and N–H stretching was observed at 3293 cm−1. Further, the CH2 asymmetric and symmetric stretch of PA6 can be assigned to the peaks observed at 2934 cm−1 and 2861 cm−1,29,30 respectively.

ATR-FTIR spectra of pristine PA6, PNC containing uncoated NPs and PNC containing NPs with varying proportions of OA coatings
Also, the shift in the spectra observed for the OA-coated samples, i.e. the new shifted peaks observed at 2929 cm−1 (22 wt.% OA) and 2921 cm−1 (55 wt.% OA) and the peaks of 2860 cm−1 (22 wt.% OA) and 2862 cm−1 (55 wt.% OA) are attributed to the asymmetric and symmetric CH2 stretching, respectively. These correspond to the characteristic OA absorption peaks of 2924 cm−1 and 2854 cm−1, respectively.31,32 Even the slight peaks developed at 1716 cm−1 (22 wt.% OA) and 1715 cm−1 (55 wt.% OA) are due to the overlapping of carboxyl groups and the OA double-bond absorption bands. This confirms the presence of chemisorbed OA (compared to the standard 1710 cm−1 peak) that acts as a surfactant,32 improving the dispersion (which has been studied from the TEM, XRD and SAXS results). Also, the inset image shows that a slight OA peak developed, matching with the standard peak at 1710.32
The shift in the peaks from 1536 cm−1 to 1540 cm−1 can be attributed to a decrease in the degree of crystallinity.33 Additionally, this band at 1536–1635 cm−1 also corresponds to the primary amide band of amide II-type. The peaks around 1536 cm−1 and 1540 cm−1 correspond to the mentioned amide II of the primary type, resulting in vibration due to the bending of CO-NH (939–948 cm−1) and the N-H bond.34 The intensity and position of these crystalline bands determines the degree of crystallinity, whereas the broader peaks of these bands suggest the formation of an amorphous phase.35 Therefore, it can be inferred that the polymer with 22 wt.% OA has a moderately higher degree of crystallinity, as the amide II band was sensitive to the crystalline structure.
Hence, the increasing concentration of OA employed lowers the crystallinity. Furthermore, the polymer chain mobility is limited and the crystallization process slackened36 by the carboxylic end group at the end of the fatty acid chain in the OA coating interacting with NH2 of the polyamide chain. The wavelength fingerprint for OA at around 1629 cm−1 corresponding to the asymmetric and symmetric bands (COO-) of the oleic acid grafted at the surface of the iron oxide NPs were suppressed by PA6 and observed as flat peaks, as seen in Fig. 4.37,38 The peak near 686 cm−1 corresponds to both the Fe-O bond stretching of iron oxide and the amide V (α and β) of the pristine PA6.39 However, interaction between the peaks results in the suppression of the peak near 686 cm−1 for the 22 wt.% and 55 wt.% PNCs. The increased interaction between the NPs and the polymer matrix due to the increase in OA loading is further confirmed with DSC results.

ATR-FTIR spectra of polymer samples with pristine PA6, no coating PNC and PNC with varying proportions of OA coatings
All the nanocomposites contained 1 wt.% iron oxide NPs, and the Fe-O peaks are not clearly discernible in the FTIR plots. To clearly establish the effect of NP inclusions on PA6, the FTIR spectrum of PNC containing a high weight percentage, i.e., 20 wt.% of NPs, was obtained and compared with the pristine PA6 and the PNC containing uncoated iron oxide NPs (“No coating” legend in Figs. 3 and 4, and Figure S1) spectra (which had only 1 wt.% of NP inclusion). The spectra of 20 wt.% PNC, as seen in Figure S1, shows a distinctive broadening of the peaks around the absorption bands of 295–541 cm−1, a characteristic of Fe-O stretching,40 and also the peak at 587 cm−1 confirms the inclusion of Fe3O4,41 especially from the 20 wt.% sample. The distinguishing peak at 534 cm−1, a characterizing peak confirming the presence of iron oxide, is only observed in the 20 wt.% sample, whereas the 570 cm−1 is attributed to amide-V stretching.41 This implies that the Fe3O4 peak is not discernible due to the low wt.% of iron oxide NPs samples, as seen in Figs. 3 and 4. To confirm, energy dispersive X-ray spectroscopy (EDX) results were obtained (Supplementary Material Figure S2 a–d).
Differential Scanning Calorimetry and Degree of Crystallinity
Noticeable melting with endothermic peaks clearly depicts the glass transition (Tg) and the melting temperatures (Tm) of the polymer samples, as shown in Fig. 5. The Tg occurrence of the pristine polymer is observed at around 46°C, matching the previously reported values.42,43

DSC curves of the PA6 samples with varying percentages of OA coating
However, the observed Tm values are slightly lower than previously reported values, at around 220°C.42,43 Additionally, as seen in Table II, the Tm of the nanocomposite was found to increase with increased OA loading. However, the uncoated NP sample showed subsequent reductions in the Tm and Tg of the sample due to the uncoated inclusions leading to amorphous growth and hence reduced crystallinity, as summarized in Table II.
Increased wetting of the surface-modified NPs by polymers is known to either increase or decrease the crystallinity and, hence, the Tg and Tm of the PNC. To confirm whether or not this is the case for the samples studied here, the percentage degree of crystallinity was calculated by dividing the total heat of fusion for each sample by the heat of fusion for 100% crystalline PA6, which is 190 J/g.44 The values for the degree of crystallinity, calculated from the areas under the curve for all the samples, and the glass transition (Tg) and melting temperature (Tm), are as listed in Table II.
The increase in Tg due to reductions in cooperative segmental mobility in the polymer is because the OA’s denser fatty acid chains on the surface of the NPs induced greater bonding with the polymer matrix, as observed with the DSC trend.45,46,–47 The strong attraction at the interface of the NPs and the polymer tends to decrease the crystallinity and Tm, as observed in the DSC results (Table II).
X-ray Diffraction
The XRD results confirmed the polymer as the semi-crystalline phase.48 Diffraction peaks observed at 30.2° 2θ (220), 35.6° 2θ (311), 43.3° 2θ (400), 57.25° 2θ (511) and 62.9° 2θ (440) (Fig. 6) were found to compare well with the JCPDS card number 01-089-5892 for the magnetic mineral γ-iron oxide (cubic maghemite), therefore confirming the spinel structure of the NP iron oxides. The crystal planes within the maghemite represented by the most intense diffraction peaks, described by the Miller indices above, were used in Eq. 1 to calculate the crystallite sizes of the NPs; Eq. 1 relates the diffraction peak width (B) to the crystallite size (L).
As seen in Fig. 6, with 22 wt.% OA loading, the prominent diffraction peaks showed a low intensity and maximum broadening, which suggests a decrease in NP agglomeration size. Similar to the TEM results, the standard deviation of the crystallite size for 55 wt.% OA loading is high, confirming the formation of few large agglomerates. However, the mean size of the particles was found to be lower than 32.41 nm, again with the large standard deviation (Table III).

XRD patterns of the PA6 samples with varying percentages of OA loading of coatings on the NPs. The triangle (∆) and circle (○) represents the peaks related to iron oxide and polymer, respectively
Ideally, PA6 contains two monoclinic crystalline phases: the α-phase and the γ-phase. With the entirety of the α-phase attaining a trans-chain conformity, a hydrogen bond appears between adjacent antiparallel chains in this phase; the thermodynamically most stable phase.48 Conversely, the chains appear twisted in the γ-phase, enabling the formation of hydrogen bonds between parallel chains.48
In the PA6 samples prepared during this study, two broad-based diffraction peaks were observed around 21° 2θ (4.2 Å) and 24° 2θ (3.7 Å) which almost certainly correspond to the α-phase of PA6. Figure 8 shows that the α-phase dominates the crystalline structure of the PA6. The peak at 21° 2θ (4.2 Å) is referred to as α1 and originated from the (200) plane, whereas the peak at 24° 2θ (3.7 Å) originated from the (002) plane.49 The minor peaks observed at 17° 2θ, 22° 2θ and 28° 2θ can be identified as the γ-phase of of the PA6 with the corresponding Miller indices of (020), (001) and (200)/(201).47
There are various factors contributing to the observed diffraction patterns (Fig. 6). The two prominent peaks related to the α crystalline phase of PA6 were observed to decrease with increased OA loading up to 22 wt.%, suggesting an increase in the crystalline to amorphous ratio, meaning the degree of crystallinity. The XRD plots presented here suggest that the maghemite iron oxide NPs tend to be more crystalline and ordered than the polymer PA6, which is reported as being semi-crystalline48 and therefore less well ordered.
Both the TEM and XRD results suggest that the 22 wt.% OA proportion of NP coating, the crystallite size, and therefore the NP agglomerate size, are the lowest. In summary, small crystal formation results due to improved dispersion of NPs.50,51
Small Angle X-ray Scattering
The filler dispersivity in a PNC has been studied using scattering techniques. Statistical mechanical theories relate to the dispersion state of the nano-inclusions as well as the stress carried by the polymer chains.52 The model-free approach of average particle size determination using the SAXS data is based on the implementation of the Guinier and Porod law.53,54
Figure S3 represents the scattering intensity I(q), plotted as a function of the scattering vector q. The SAXS profile shows two regions: the flat response from PA6 and the high gradient response from the nanoparticles in the samples. The cumulatively slope-dropping region is a characteristic of a Porod scattering response from the NPs.55 The Guinier region precedes the Porod region, which reflects the radius of gyration of the NPs according to the Guinier’s law:
where G is the Guinier prefactor and Rg is the radius of gyration. The slope of the regions in the Guinier plot represented in Fig. 7 are used to determine Rg.

Guinier plot for all the samples, with the region fitting highlighted
Using Eq. 3, the mean diameter D is calculated:56,57
The results in Table IV are in proximity of those obtained with TEM and XRD above. This confirms that the 22 wt.% OA sample has the lowest NP size and also has the fewest agglomerates. The maximum standard deviation was found to be ± 8.9 nm for the uncoated particles.
Melt Characteristics
A magnetic field response test of the synthesized nanocomposite was carried out to determine its melt characteristics. The magnetic coil (30 V) set-up used (Figure S4) was designed58 and used for testing.
Preliminary melting times upon exposure to the magnetic field were recorded, as shown in Table V. A total of five sets of results for each nanocomposite film (200 μm thick), with 22% and 55 wt.% OA loading, were recorded. A representative surfaces of nanocomposite samples with defects are shown in Fig. 8a and c. The same defective nanocomposites were exposed to a magnetic field to observe the surface melting and therefore healing effect. Typical results showing surface crack healing in samples containing iron oxide NPs coated with 22% and 55 wt.% OA loading are shown in Fig. 8b and d.

Defective polymer nanocomposite films with (a) 22 wt.% and (c) 55 wt.% OA-loaded NPs. The same films after surface melting using magnetic induction with (b) 22 wt.% and (d) 55 wt.% OA-loaded NPs
Both samples confirmed the melting response as reported earlier.58 The cracks identified on the initial samples were observed to heal after the magnetic induction heating response. These are marked in Fig. 8b and d showing—the feasibility of material healing on damaged samples. The mean time periods for the melt response of—five sample surfaces were found to be similar in both cases. This is not surprising given the NP wt.% loading for both samples was the same at 1 wt.%. However, the variance in the time recorded was found to be higher in—samples with the 22 wt.% OA coating showing uncertainties.
The considerable variation in the melt response time for the 22 wt.% OA sample can be attributed to the dispersion of the NPs. The agglomerates in the 55 wt.% OA sample created localized heating effects. Hence, some samples melted quickly in certain regions.
Conclusion
An iron oxide nanocomposite in a PA6 polymer matrix was successfully synthesized using an in situ polymerization approach. Increases in the OA concentration of the NP coating, up to 22 wt.%, showed improvement in the dispersion combined with increased Tg, reduced Tm and decreased crystallinity, compared with a commercial PA6 polymer. The results confirmed changes in the properties of the polymer due to variation in OA loading in the coating of iron oxide NPs. In general, increases in OA concentration improved the dispersion of the iron oxide NPs and their chemical interaction with the host polymer chain. At 55 wt.%, OA loading led to the formation of some agglomerates, and, although the mean diameter was indicating improved dispersion, the standard deviation was large. Furthermore, the overall result showed that the 22 wt.% loading was optimum for improved dispersion of NPs. The magnetic response suggests that a higher weight percentage of NPs would improve the melting response time. Such nanocomposites can find use in biomedical applications. The effect of particle concentration and other polymerization catalysts will be researched in future work.
References
A.S. Perera, S. Zhang, S. Homer-Vanniasinkam, M. Coppens, and M. Edirisinghe, ACS Appl. Mater. Interfaces 10, 15524 (2018).
C. Cih, K. Kun-Cheng, and Y. Sen-Yeu, RSC Adv. 7, 6336 (2017).
J. Fiabane, P. Prentice, and K. Pancholi, BioMed Res. Int. (2016). https://doi.org/10.1155/2016/3572827.
K. Pancholi, P.K.J. Robertson, P. Okpozo, N.S. Beattie, and D. Huo, J. Phys. Energy 1, 015007 (2018).
K. Pancholi, R. Gupta, R. Prabhu, D. Huo, M. Pancholi, V. Jha, J. Latto, and N. Dodds, OCEANS’17 MTS/IEEE, (Aberdeen, 2017).
R. Gupta, R. Prabhu, D. Huo, M. Pancholi, J. Njuguna, and K. Pancholi, 3rd Defence and Security Doctoral Symposium (DSDS17), (Cranfield University, 2017).
M. Zhang and R.P. Singh, Mat. Lett. 58, 408 (2004).
V. Yong and H.T. Hahn, Nanotechnology 15, 1338 (2004).
S. Lao, W. Ho, K. Ngyuen, J. Cheng, J.H. Koo, L. Pilato, and G. Wissler., SAMPE Fall Technical Conference-37th ISTC (2005).
J.H. Koo, L.A. Pilato, and G.E. Wissler, J. Spacecr. Rockets 44, 1250 (2007).
C. Chen, The Manufacture of Polymer Nanocomposite Materials Using Supercritical Carbon Dioxide (Doctoral dissertation, Virginia Tech., 2011).
Y. Chen, A.M. Kushner, G.A. Williams, and Z. Guan, Nat. Chem. 4, 467 (2012).
J. Tang, Y. Wang, H. Liu, Y. Xia, and B. Schneider, J. Appl. Polym. Sci. 90, 1053 (2003).
D. Ma, T.A. Hugener, R.W. Siegel, A. Christerson, E. Mårtensson, C. Önneby, and L.S. Schadler, Nanotechnology 16, 724 (2005).
Y. Chen, S. Zhou, H. Yang, and L. Wu, J. Appl. Polym. Sci. 95, 1032 (2005).
A. Usuki, M. Kawasumi, Y. Kojima, A. Okada, and T. Kurauchi, Jpn. J. Polym. Sci. Technol. 52, 440 (1995).
Z. Spitalsky, G. Tsoukleri, D. Tasis, C. Krontiras, S.N. Georga, and C. Galiotis, Nanotechnology 20, 405702 (2009).
M. Liu, T. Zhu, Z. Li, and Z. Liu, J. Phys. Chem. C 113, 9670–9675 (2009).
J.H. Koo, L.A. Pilato, G. Wissler, A. Lee, A. Abusafieh, and J. Weispfenning, International SAMPE Symposium and Exhibition (Proceedings), Society for the Advancement of Material and Process Engineering, vol. 50, p. 1935 (2005).
Y. Chen, S. Zhou, H. Yang, and L. Wu, J. Appl. Polym. Sci. 95, 1032 (2005).
S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst, and R.N. Muller, Chem. Rev. 108, 2064 (2008).
L.M. Bronstein, X. Huang, J. Retrum, M. Pink, B.D. Stein, and B. Dragnea, Chem. Mater. 19, 3624 (2007).
C. Bakkali-Hassani, D. Tunc, K. Roos, M. Planes, P. Lecomte, and S. Carlotti, Macromolecules 50, 175 (2016).
S. Wei, Q. Wang, J. Zhu, L. Sun, H. Lin, and Z. Guo, Nanoscale 3, 4474 (2011).
C. Wang, C. Xu, H. Zeng, and S. Sun, Adv. Mater. 21, 3045 (2009).
K.A. Al-Saad, M.A. Amr, D.T. Hadi, R.S. Arar, M.M. AL-Sulaiti, T.A. Abdulmalik, N.M. Alsahamary, and J.C. Kwak, Arab J. Nucl. Sci. Appl. 45, 335 (2012).
P.B. Shete, R.M. Patil, B.M. Tiwale, and S.H. Pawar, J. Magn. Magn. Mater. 377, 406 (2015).
G.K. Xu, W. Lu, X.Q. Feng, and S.W. Yu, Soft Matter 7, 4828 (2011).
N. Ngwuluka, Aaps Pharmscitech 11, 1603 (2010).
J. Pockett, Crystallinity in Linear Polyamides: A Study Using Melt Blending with Small-Molecule Diluents (Doctoral dissertation, University of South Australia, 2004).
Y.F. Shen, J. Tang, Z.H. Nie, Y.D. Wang, Y. Ren, and L. Zuo, Sep. Purif. Technol. 68, 312 (2009).
M. Mahdavi, M. Ahmad, M. Haron, F. Namvar, B. Nadi, and J. Amin, Molecules 18, 7533 (2013).
M.K. Verhoefen, G. Schäfer, S. Shastri, I. Weber, C. Glaubitz, W. Mäntele, and J. Wachtveitl, Biochim. Biophys. Acta Bioenerg. 1807, 1583 (2011).
F. Yang, M. Zhao, B. Zheng, D. Xiao, L. Wu, and Y. Guo, J. Mater. Chem. 22, 25471 (2012).
C. Harrats and G. Groeninckx, Macromol. Rapid Commun. 29, 14 (2008).
M. Zhang, B.H. Guo, and J. Xu, Crystals 7, 4 (2016).
D. Tunc, H. Bouchekif, B. Améduri, C. Jérôme, P. Desbois, P. Lecomte, and S. Carlotti, Eur. Polym. J. 71, 575 (2015).
X.Y. Wang, D. Mertz, C. Blanco-Andujar, A. Bora, M. Ménard, F. Meyer, C. Giraudeau, and S. Bégin-Colin, RSC Adv. 6, 93784 (2016).
S.T. Navale, G.D. Khuspe, M.A. Chougule, and V.B. Patil, RSC Adv. 4, 27998 (2014).
R. Khandanlou, M. Ahmad, K. Shameli, E. Saki, and K. Kalantari, Int. J. Mol. Sci. 15, 18466 (2014).
A.S. Perera, S. Zhang, S. Homer-Vanniasinkam, M.O. Coppens, and M. Edirisinghe, ACS Appl. Mater. Interfaces 10, 15524 (2018).
C. Scott, “Nylon-6 information and properties”, http://www.polymerprocessing.com/polymers/PA6.html. Accessed 20 February 2018.
I. Goodman and A.H. Kehayoglou, Polym. J. 19, 321 (1983).
P. Rittigstein and J.M. Torkelson, J. Polym. Sci. Part B 44, 2935 (2006).
F. Rybnikář and P.H. Geil, J. Appl. Polym. Sci. 49, 1175 (1993).
S. Pashaei, M.M. Avval, and A.A. Syed, Chem. Ind. Chem. Eng. Q. 17, 141 (2011).
B. Wunderlich, Macromol. Phys. 3, 69 (2003).
K. Khodabakhshi, Anionic Polymarisation of Caprolactam: An Approach to Optimising the Polymerisation Condition to be Used in the Jetting Process (Doctoral dissertation, Khosrow Khodabakhshi, 2011).
L.P. Ramirez and K. Landfester, Macromol. Chem. Phys. 204, 22 (2003).
L. Ricco, S. Russo, G. Orefice, and F. Riva, Macromolecules 32, 7726 (1999).
S. Kalia, S. Kango, A. Kumar, Y. Haldorai, B. Kumari, and R. Kumar, Colloid Polym. Sci. 292, 2025 (2014).
M. Doi and S.F. Edwards, The theory of polymer dynamics, vol. 73 (Oxford: Oxford University Press, 1988), pp. 140–176.
L.A. Feigin and D.I. Svergun, Structure analysis by small-angle x-ray and neutron scattering, vol. 1 (New York: Plenum, 1987), pp. 14–15.
J. Teixeira, J. Appl. Crystallogr. 21, 781 (1988).
G. Porod, in Small angle X-ray Scattering, ed. by O. Glatter, O. Kratky (London: Academic Press, 1982), p. 17.
K. Hino, R. Shingai, T. Morita, K. Toku, T. Hirano, H. Yoshikawa, H. Nakano, and N. Nishi, Chem. Phys. Lett. 460, 173 (2008).
P. Vassili, A. Evandro, N. Mario, A. Craievich, and I. Polikarpov, SAXSMoW 2.0: Online Calculator of the Molecular Weight of Proteins in Dilute Solution from Experimental SAXS Data Measured on a Relative Scale. http://saxs.ifsc.usp.br/. Accessed 22 April 2018.
A. Dow, Various Applications of Magnets—Induction Coil Testing on Self-Healing Polymer Nanocomposites and ITER Tokamak. (Thesis repository, Robert Gordon University, 2018).
Acknowledgements
Ranjeetkumar thanks Robert Gordon University for Ph.D. financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gupta, R., Pancholi, K., De Sa, R. et al. Effect of Oleic Acid Coating of Iron Oxide Nanoparticles on Properties of Magnetic Polyamide-6 Nanocomposite. JOM 71, 3119–3128 (2019). https://doi.org/10.1007/s11837-019-03622-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11837-019-03622-5