
Abstract
A new flavonoid compound, 4′-O-α-rhamnopyranosyl (1 → 2)-β-xylopyranoside of 7-O-methylapigenin (1) was isolated from the leaves of Trollius europaeus. The structure of this compound was elucidated by spectroscopic methods (UV, 1D NMR, 2D NMR and MS). Compound (1) was analyzed for antioxidant activity in DPPH assay.
Similar content being viewed by others
Introduction
The genus Trollius belonging to the Ranunculaceae family comprises species of perennial herbs that grow in a temperate climate. Trollius europaeus L. can be found in Europe as well as in western Siberia. This plant has traditionally been used for the treatment of scurvy due to the high content of vitamin C (Gruenwald et al. 2004).
In Chinese traditional medicine, the flowers of T. ledebouri and T. chinensis have been used in respiratory tract infections, e.g. colds, fever, pharyngitis, tonsillitis and bronchitis. The flowers of both species contain a number of flavonoids, mostly C-glycosylflavones. Extracts of T. ledebouri and T. chinensis have been reported to show antiviral and antimicrobial activity due to the presence of orientin and vitexin; additionally, T. ledebouri showed antioxidant and inflammatory activity (Zhou et al. 2005; Li et al. 2002; Cai et al. 2006; Wang et al. 2004).
Preliminary studies of T. europaeus leaves, which we were co-authored, reported the presence of flavonoids including C-glycoside of flavone and phenolic acids (Maciejewska-Rutkowska et al. 2007), whereas orientin 2″-O-β-xylopyranoside (adonivernith) was found in T. europeus flowers, whose seeds fed larvae Chiastocheta (Gallet et al. 2007).
A continuation of our study on flavonoids in T. europaeus leaves through a more extensive investigation led to isolation and structural identification of a new flavonoid compound, 4′-O-α-rhamnopyranosyl (1 → 2)-β-xylopyranoside of 7-O-methylapigenin.
The structure was determined by HR ESI–MS and ESI–MS and UV spectroscopic analyses, also 1D NMR and 2D NMR including COSY, HMQC and HMBC experiments.
The antioxidant activity of the compound was determined by the use of 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical scavenging method (Brand-Williams et al. 1995; Kang et al. 2010).
Experimental part
Chemicals
The reagents ethyl acetate, methanol, acetic acid, formic acid, n-propanol were purchased from POCH, Gliwice, Poland; aniline phthalate and diphenylboric acid 2-aminoethylester (Naturstoffreagenz A, NA) from ROTH, Germany; 1,1-diphenyl-2-picrylhydrazyl (DPPH) and butylated hydroxyanisole (BHA) from Sigma-Aldrich, USA.
General experimental procedures
The NMR spectra (1H NMR 400 MHz, 13C NMR 100 MHz) were recorded using a Bruker NMR Avance II 400 spectrometer in DMSO-d6 with trimethylsilane (TMS) as an internal standard.
The mass spectra were measured on two spectrometers: the HR ESI–MS on Thermo QExactive with Quadrupole-Orbitalp detection and ESI operating in a negative and positive ion mode at a spray voltage of 1.5 kV. Data were collected using Thermo QExactive Tune 2.4, and processed with 3 Thermo Xcalibur software. The spectrometer was calibrated using a dedicated calibration mixture. The mass measurement accuracy is a maximum of ±1 ppm and the ESI–MS mass spectra were measured on Waters/Micromass (Manchester, UK) ZQ mass spectrometer connected with a UV 996 Waters photodiode detector equipped with an electrospray interface operating in a negative and positive ion mode at an optimized sample cone voltage of 30 V (Figs. 1, 2, 3).

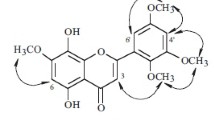
HMQC and HMBC correlation of 4′-O-α-rhamnopyranosyl (1 → 2)-β-xylopyranoside of 7-O-methylapigenin (400 MHz in DMSO)

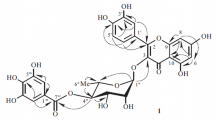
HMBC of 4′-O-α-rhamnopyranosyl (1 → 2)-β-xylopyranoside of 7-O-methylapigenin (1); correlation between H-1″′ and C-2″

DPPH radical scavenging activity of 4′-O-α-rhamnopyranosyl (1 → 2)-β-xylopyranoside of 7-O-methylapigenin (1)
The UV spectra were recorded on a UV/VIS Perkin Elmer Lambda 35 spectrophotometer.
Column and thin layer chromatography
Column chromatography (CC) was performed using cellulose Whatman CF 11 (Whatman, Germany) and S-EtOAc-MeOH–H2O (100:6:20, upper layer) as a solvent system also using lipophilic Sephadex LH-20 (25–100 μm, Sigma-Aldrich, Germany) eluted by methanol. Thin layer chromatography (TLC) was performed on cellulose plates (Merck, Germany) using S1:CH3COOH–H2O (15:85), and S2: EtOAc-HCOOH–H2O (10:2:3) upper layer) and on silica gel 60 plates (Merck, Germany) using S3: EtOAc-HCOOH–CH3COOH–H2O (100:11:11:26) and S4: n-PrOH-EtOAc–H2O (7:2:1). Detection of flavonoids was achieved before and after spraying with 1 % NA in MeOH in UV (365 nm); sugar detection: aniline phthalate followed by heating at 100 °C for 5 min.
Plant material
The leaves of Trollius europaeus L. were collected in May 2007, from flowering plants growing in an experimental field of the Department of Botany, Poznań University of Life Sciences, Poland. The plant was identified by dr Wojciech Antkowiak (Department of Botany, Poznań University of Life Sciences). The voucher specimen is deposited at the Department of Pharmacognosy, Poznań University of Medical Sciences.
Extraction and isolation of flavonoid
500 g of air-dried powdered leaves of T. europaeus were percolated three times at room temperature with methanol for 24 h and then exhaustively extracted with a mixture of methanol–water (1:1) on a boiling water bath under reflux.
The obtained MeOH and MeOH:H2O extracts, after being monitored by TLC, were combined, concentrated under reduced pressure, and precipitated with hot water. The resulted precipitate was filtered off. The filtrate was extracted with chloroform and the chloroform fraction was discarded. The aqueous fraction was subjected to CC on cellulose Whatman CF 11. The column was eluted with S and the elution was monitored by TLC (S1, S2 and S3). Fractions of ca. 200–250 ml each were collected and the fractions which had similar constituents were combined. Fractions 9–15 containing compound 1 were rechromatographed on a column with Sephadex. The result was 55 mg yellow needles of compound 1.
4’-O-α-rhamnopyranosyl (1→2)-β-xylopyranoside of 7-O-methylapigenin (1)
Yellow crystals powder; UV λmax (H2O)nm: MeOH 324, 271; MeOH/NaOAc 325, 274; MeOH/NaOAc/H3BO3 326, 270; MeOH/NaOMe 369, 292; MeOH/AlCl3 378, 346, 300, 276; MeOH/AlCl3/HCl 378, 338, 299, 274; HR ESI–MS in negative ion mode m/z: 561.1358 (M – H)−, 283.2514 (A – H)−; in positive ion mode m/z 563.1749 (M + H)+ (Fig. 4); ESI–MS in negative ion mode m/z: 561 (M – H) −, 597 (M + Cl)−, 415 (M–H-rhamnose)−, 283 (A – H)−; in positive ion mode m/z: 563 (M + H)+, 585 (M + Na)+, 601 (M + K)+, 285 (A + H)+; 1H NMR and 13C NMR (Table 1).

The HR ESI–MS spectrum of 4′-O-α-rhamnopyranosyl (1 → 2)-β-xylopyranoside of 7-O-methylapigenin in a negative and positive ion mode
Total acid hydrolysis: 1 mg of compounds was refluxed with 2 ml of 2 % HCl in MeOH for 4 h. The reaction solutions were extracted with etyl acetate. The ethyl acetate extract containing aglycone was evaporated to dryness and the UV spectrum of the aglycone was analyzed. The aqueous fraction, after being neutralized and concentrated, was identified by comparison with standard sugars (TLC on silica gel 60 plates using S3 as a solvent system); spots were observed in VIS after spraying with aniline phthalate and heating at 110 °C for 10 min.
DPPH radical scavenging assay
The antioxidant activity was determined by the DPPH assay (Brand-Williams et al. 1995; Kang et al. 2010). The stable free radical DPPH was prepared at concentration of 6.0 × 10−5 mol/l in methanol. 0.2 ml of the solution of compound 1 in different concentrations was added to 3.9 ml of the DPPH solution. 1–20 μg of butylated hydroxyanisole (BHA) was used as a positive control. The absorbance of the solution was measured at λ = 515 nm 30 min after the addition of the DPPH radical. The scavenging activity was calculated as:
A–DPPH radical scavenging activity (%)
Ac–the absorbance of the DPPH radical
As–the absorbance of the sample
The IC10 (-the concentration of antioxidant which reduces the free radical DPPH about 10 %) was calculated from the concentration-effect linear regression analysis.
Results and discussion
The flavonoid compound (1) obtained from T. europaeus was identified as 4′-O-α-rhamnopyranosyl (1 → 2)-β-xylopyranoside of 7-O-methylapigenin by interpretation of its ESI–MS, UV and NMR spectra.
The HR ESI–MS and ESI–MS analysis of compound 1 with the negative and positive ion mode indicated a mass of 562 and corresponded to the molecular formula of C27 H30O13.
The ESI–MS of 1 in the negative ion mode gave a deprotonated ion at m/z 561 [M – H]− and its adduct with chlorine at m/z 597 [M + Cl]−. The signal at m/z 563 corresponded to the protonated molecule [M + H]+, while ions at m/z 585 and m/z 601 corresponded to its adducts with sodium [M + Na]+ and potassium [M + K]+, respectively. The fragment ion at m/z 415 [M – H – 164]− indicated that the terminal sugar was a methylpentose moiety. The signals formed after the loss of the pentose moiety at m/z 285 corresponded to protonated [A + H]+ and at m/z 283 to deprotonated [A – H]− of aglycone, implying the molecular mass of aglycone m/z = 284 compatibility with methylapigenin (C16 H12 O5). The signals appearing in the ESI–MS spectrum were also observed in the HR ESI–MS spectrum. The ion peak of [M – H]+ was found at m/z 563.1749, from [M – H]− at 561.1358, whereas [A – H]− at 238.2514 (Fig. 4).
The UV spectrum of compound 1 in MeOH showed two characteristic bands at 271 nm and 324 nm, which suggested that the compound belonged to flavones. The shifts observed after addition of shift reagents (NaOMe, NaOAc, NaOAc/H3BO3, AlCl3, AlCl3/HCl) indicated a flavone structure with a free 5-hydroxyl group and the absence of a free OH group at C-7 and ortho- dihydroxy system at C-3′, 4′. The position of glycosidation was confirmed at C-4′ where the aglycone exhibited a bathochromic shift in band- I in NaOMe spectrum. The UV spectral data of aglycone in methanol after addition of NaOAc, NaOAc/H3BO3 showed that the compound did not have a 3′, 4′-ortho-hydroxylated group and OH at C-7.
The 1H NMR spectrum of compound 1 gave two doublets for the meta-coupled protons, at δ 6.34 ppm and at δ 6.81 ppm (each 1H, d, J = 2.1 Hz) assignable to H-6 and H-8, respectively. The values of both signals were shifted downfield, compared to apigenin, indicating that the OH group at C-7 could be substituted. The signal at δ 6.97 ppm (1H, s), which correlated to carbon at δ 103.84 ppm in HMQC spectrum was characteristic of the proton H-3. Signals of ring B protons were doublets (J = 8.97 Hz) and were observed at δ 8.07 ppm (H-2′ and H-6′) and at δ 7.13 ppm (H-3′ and H-5′). The downfield signal shifts of ortho protons (Δ + 0.15 ppm) and meta protons (Δ + 0.13 ppm), in comparison with apigenin, indicated the substitution of the hydroxyl group at C-4′. In the region of the anomeric protons of sugars there were two resonance signals. The doublet at 5.24 δ ppm appeared closer to the signals from the aglycone protons and was assigned to proton H-1″ xylose attached directly to the ring, while the doublet at δ 5.10 ppm was assigned to proton H-1″′ rhamnose. The values of coupling constants of anomeric protons (J = 7.25 and 1.20 Hz) confirmed the β-linkage of xylose and α-linkage of rhamnose, respectively. The doublet observed at δ 1.18 ppm (J = 6.3 Hz) was diagnostic for the methyl group of rhamnose.
The 13C NMR spectrum gave 27 carbon signals, which indicated the presence of 15 carbon signals due to the flavone skeleton and 12 carbon signals assigned to sugar moieties. In the anomeric carbon signals of sugar molecules two diagnostic signals were recorded at δ 100.67 ppm and 98.18 ppm from C-1″ xylose and C-1′′′ rhamnose. On the basis of the signal shifts in the downfield (Δ + 2.05 ppm) of C-2″ of xylose and in the upfield (Δ − 1.13 ppm, and Δ − 5.73 ppm, respectively) of the neighboring carbon atoms C-3″ and C-1″, compared with unsubstituted xylose, it was concluded that the molecule of rhamnose was attached to xylose at C-2″ (Harborne and Mabry 1982; Semmar et al. 2002).
In the HMBC spectrum of compound 1 correlations observed among H-2″ and C-1″’ confirmed a connection 1 → 2 between sugar molecules. The sequence of sugar moieties was also determined by the correlation between H-2 of the xylosyl group and the anomeric proton of rhamnosyl moiety in the 1H -1H COSY spectrum. In the HMBC spectrum, the anomeric proton of the xylosyl group was correlated with C-4′ (δ 5.22 at 162.32 ppm) of the aglycone. The carbon signal from the methyl group of rhamnose occurred at δ 18.53. 1H-NMR spectrum also exhibited one singlet for the methoxyl group (δ = 3.86, 3H, s). Evidence for the position of the methoxyl group at C-7 was provided by HMBC spectral data. H-3 showed cross peaks with C-1′, C-4, C-10 and H-8 showed HMBC correlation with C-10, C-6 and C-9, also H-6 to C-10, C-8 and C-5, confirming the positions of these hydrogens (Table 1; Figs. 1, 2).
The results of the chromatographic analysis of the hydrolyzate of compound 1 confirmed the presence of two sugars corresponding to xylose and rhamnose.
Thus, the structure of 1 was recognized as 4′-O-α-rhamnopyranosyl (1 → 2)-β-xylopyranoside of 7-O-methylapigenin, which is a new natural compound.
The antioxidant potency of compound 1 has been shown in Fig. 3. The IC10 % value of the compound was 3.19 mg/ml. BHA showed similar antioxidant strength at a dose of 2.19 μg/ml.
Author contribution statement
Ewa Witkowska-Banaszczak collected the material to be tested, isolated the flavonoid, and performed the analysis of identification and antioxidant activity test. She prepared the documentation, interpreted and described the results. Wiesława Bylka was involved in preparing the paper. She interpreted and adjusted the results.
References
Brand-Williams W, Cuvelier ME, Berset C (1995) Use of a free radical method to evaluate antioxidant activity. Lebensmittel-Wissenschaft Und Technology 28:25–30
Cai SQ, Wang R, Yang X, Shang M, Ma C, Shoyama Y (2006) Antiviral flavonoid—type C–glycosides from the flowers of Trollius chinensis. Chem Biodivers 3:343–348
Gallet C, Ibanez S, Zinger L, Ravel FR, Trierweiler M, Jeacomine I, Despres L (2007) Plant chemical defense included by a seed—eating pollinator mutualist. J Chem Ecol 33(11):2078–2089
Gruenwald J, Brendler T, Jaenicke MD (2004) PDR for herbal medicines. Thomson PDR, Montvale, pp 387–388
Harborne JB, Mabry TJ (1982) The flavonoids. Advances in research. Chapman and Hall, London
Kang WY, Li CF, Liu YX (2010) Antioxidant phenolic compounds and flavonoids of Mitragyna rotundifolia (Roxb.) Kuntze in vitro. Med Chem Res 19:1222–1232
Li YL, Ma SCh, Yang YT, Ye SM, But PPH (2002) Antiviral activities of flavonoids and organic acid from Trollius chinensis Bunge. J Ethnopharm 79:365–368
Maciejewska-Rutkowska I, Antkowiak W, Jagodziński A, Bylka W, Witkowska-Banaszczak E (2007) Chemical composition and morphology of basal leaves of Trollius europaeus L. and T. altissimus Crantz (Ranunculaceae). Pol J Environ Stud 16:595–605
Semmar N, Fenet B, Gluchoff-Fiasson K, Comte G, Jay M (2002) New flavonol tetraglycosides from Astragalus caprinus. Chem Pharm Bull 50:981–984
Wang RF, Yang XW, Ma CM, Liu HY, Shang MY, Zhang QY, Cai SQ, Park JH (2004) Trollioside, a new compound from the flowers of Trollius chinensis. J Asian Nat Prod Res 6:139–144
Zhou X, Peng J, Fan G, Wu Y (2005) Isolation and purification of flavonoid glycosides from Trollius ledebourii using high-speed counter-current chromatography by stepwise increasing the flow-rate of the mobile phase. J Chrom 1092:216–221
Acknowledgments
Thank you Wojciech Antkowiak (PhD, Department of Botany, Poznań University of Life Sciences) for providing plant material for research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. V. Jorrin-Novo.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Witkowska-Banaszczak, E., Bylka, W. New flavonoid compound of Trollius europaeus . Acta Physiol Plant 37, 99 (2015). https://doi.org/10.1007/s11738-015-1838-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11738-015-1838-3