
Abstract
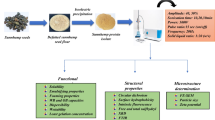
This study aimed to investigate the impacts of high-intensity ultrasound treatments on the physicochemical, thermal, and structural properties of faba bean protein isolates (FPI). Aqueous FPI suspensions were sonicated at 20 kHz for 1, 3, 5, and 10 min. Ultrasound treatment increased the solubility of FPI at all sonicated times, with maximum solubility (89.11%) observed at the 3-min treatment. Particle size (from 370.47 to 187.87 nm) and zeta potential (from − 40.83 to − 37.17 mV) reduction (in magnitude) by sonication were observed at all studied times. The usage of ultrasound has been found to have a more pronounced antibrowning impact compared to unsonication (higher L* and b*, lower a*, C*, H*, BI, and ΔE). With the increase in sonication treatment from 0 to 3 min, the Td decreased from 103.07 to 99.58 °C and ΔHd from 2.21 to 1.12 J/g, which were related to the changes in the secondary structure of proteins. Sonication changed the secondary structure of FPI, decreasing the α-helix, β-turn, and random coil content, and increasing the β-sheet for samples treated up to 3 min. Prolonged sonication increased the α-helices due to the extension of certain types of hydrogen bonds in the structure. The microstructural changes of FPI caused by ultrasound were further validated through observations made using a scanning electron microscope, which revealed the presence of smaller fragments. The present study indicates that the use of ultrasound has the potential to improve the functionality of FPI, hence potentially enhancing the efficacy of FPI-based food products.
Graphical abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Faba beans, also known as Vicia faba L., are a member of the Fabaceae family and are an excellent source of healthy proteins, energy, and fiber [1]. The native faba bean’s seeds have a protein content ranging from 24 to 35%, making it one of the most protein-rich crops, and their proteins are high in lysine. Other nutrients found in the seed include 60% carbohydrates and 25% dietary fiber. Compared to other crops, faba beans have a relatively low level of albumins (15–20%) and a large amount of globulins (57–60%) in their proteins [2].
Proteins, because of their composition and structure, provide a variety of diverse techno-functional properties in a variety of foods. These properties can be categorized into three main groups: (a) moisturizing properties, which depend directly on protein–water interactions, such as adhesion, dispersion, solubility, viscosity, and water retention capacity; (b) interfacial properties, which include emulsification and foaming; and (c) gelling properties, which are explained by protein–protein and protein-water interactions. These properties are susceptible to change when proteins are subjected to either chemical or physical treatments [3, 4].
Ultrasound, which is defined as sound waves with a frequency that surpasses the hearing limit of the human ear (20 kHz), has simple, cost-effective, energy-saving, and environmentally friendly advantages when compared to other physical approaches for protein modification [5]. Its operating mechanism is based on passing waves that generate zones of high and low pressure; the fluctuation in acoustic pressure is directly proportional to the amount of energy provided to the system. Ultrasound is categorized into two types: low intensity (1 W/cm2) with a frequency of 5–10 MHz and high intensity (10–1000 W/cm2) with a frequency of 20–100 kHz [6].
In recent years, many studies have reported on the effect of ultrasound treatment on the structure and functional properties of plant proteins. Researchers demonstrated that ultrasound treatment significantly altered the solubility, emulsification, and other functional properties of these proteins [4, 7,8,9,10,11,12]. The impact of high-intensity ultrasound treatment (20 kHz) on FPI was investigated by Martínez-Velasco et al. [13]. The researchers initially employed response surface methodology to examine the impact of amplitude and duration on various properties of the protein, including surface tension, solubility, particle size, and zeta potential (ζ-potential). Subsequently, an examination was conducted on the functional properties of FPI under optimized conditions. The results revealed that the application of ultrasound enhanced the foamability and adsorption dynamics at the air–water interface, while concurrently reducing the digestibility of faba bean protein. Alavi et al. [14] investigated the effect of alkaline shifting (the protein solution is first exposed to an extremely alkaline pH [pH 11] and then adjusted back to neutral [pH 7]), sonication (20 kHz, 10 or 20 min.) and their combination on functional modifications of faba bean protein isolate. They found that the foaming properties and emulsion stability of faba bean protein were stronger after being treated with ultrasound-assisted alkaline shifting treatment. The current studies related to the structural and functional features of faba bean protein are limited, resulting in the underutilization of faba bean proteins as a crop protein source. Interestingly, not a single report was found related to the influence of ultrasound treatment at different times on the functional, thermal, and structural properties of faba bean protein isolate. Hence, the objective of this study was to examine the impact of high-intensity ultrasound treatment at varying sonication times on the physicochemical, thermal, and structural characteristics of the faba bean protein isolate.
Materials and methods
Materials
AGT Foods (SK, Canada) provided the FPI. According to the technical specification provided by the supplier, the FPI contained 90% protein, 6.8% moisture, 3.4% ash, 0.7% crude fiber, < 5.0 (mg/kg) gluten, and 2.5 (mg/kg) soy. Ultrapure water was used for all experiments. All chemicals and bovine serum albumin (BSA) were obtained from Sigma Aldrich (St. Louis, MO, USA).
Preparation of native protein solution
FPI powder was dissolved in ultrapure water to obtain a 5% (w/w) solution. The solution was continuously agitated at 300 rpm for two hours at room temperature (25 ± 2 °C) to achieve a homogeneous mixture. The pH of the solution was adjusted to 7 by using 0.1 N NaOH, then left at 4 °C overnight. The solutions were equilibrated at 25 °C for one hour the following day before further examination. Na-azide was used to prevent microbial growth in the solutions.
Ultrasound treatment of protein solutions
Sonication of the native FPI solution was undertaken following the method described by Martínez-Velasco et al. [13] and Jambrak et al. [15] with some modifications. The native FPI solution (50 mL) was sonicated in a 100 mL beaker using a sonicator (20 kHz, 250 W, Branson Sonifier SFX250, Danbury, CT, USA) equipped with a sonic horn (19 mm diameter) at an amplitude of 50% for 1 (U1), 3 (U3), 5 (U5), and 10 (U10) min. The durations of treatment were determined based on the significance of the effects identified during preliminary research in the laboratory. An ice bath was used during sonication to keep the solution temperature constant (24 ± 1 °C). After ultrasound treatment, unsonicated and sonicated FPI solutions were put into round aluminum plates (4 cm diameter, 2 cm height) and placed in a freezer at -78 °C for 24 h. Then, the frozen samples were lyophilized in a freeze dryer (CHRIST Alpha 1–4 LDPlus, Martin Christ, Germany) for 48 h. After freeze-drying, the powders were immediately transferred to plastic cups and stored in a desiccator at refrigeration temperature for further analysis.
Solubility determination
Solubility was determined using the method of Jambrak et al. [16] with some modifications. Lyophilized FPI powders (1% (w/w)) were dispersed in ultrapure water, and pH was adjusted by using 1N NaOH to 7. Suspensions were centrifuged at 5000 rpm for 10 min at 25 °C using a centrifuge (Eppendorf 5810R, Germany). The supernatant was collected to measure the protein content by the Biuret method using Bovine serum albumin as a standard. Absorbance at 540 nm was measured using a spectrophotometer (Jenway 6715, Bibby Scientific Limited, Beacon Road, Stone, Staffordshire, ST15 OSA, UK) [17]. Protein solubility (%) was calculated according to the following equation:
Zeta potential measurement
Lyophilized FPI powder was dissolved with milli-Q water (0.1%, w/v) and filtered through a 0.45 µ filter. Then, the pH of the samples was adjusted by using either 1N NaOH or 1N HCl to 7 [18]. The zeta potential values of the native and sonicated protein suspensions were measured using the Zetasizer Nano ZS instrument (Malvern Instruments Ltd, Malvern, Worcestershire, UK) and disposable capillary cells (DTS 1060).
Hydrodynamic diameter measurement
For measurements of the hydrodynamic diameters of the samples, lyophilized protein solutions (0.1%, w/v) were prepared via solubilization in ultrapure water. Then, the pH was adjusted to 7.0 using 1N NaOH or 1N HCl. The hydrodynamic diameter (Z-average diameter or particle size) measurement of the samples was performed using the Zetasizer Nano ZS instrument (Malvern Instruments Ltd, Malvern, Worcestershire, UK) using disposable plastic cuvettes (ZEN 0040) [18].
Color measurement
Color measurements of lyophilized FPI powders were evaluated based on the International Commission on Illumination (CIE) protocol using HunterLab ColorFlex (A60-1010-615 Model Colorimeter, HunterLab, Reston VA). The instrument was calibrated against standard black and white tiles (L* = 93.41, a* = -1.12, b* = 1.07) [19]. The parameters were measured where L* = lightness, + a* = redness, − a* = greenness, + b* = yellowness and –b* = blueness. The calculations for ΔE: total color difference, H*: hue, C*: chroma, and BI: browning index were made with the following equations [20]:
Thermal properties
The thermal characterization of lyophilized FPI powder was performed using Pyris Manager software and a Perkin Elmer differential scanning calorimeter (DSC 7, Perkin Elmer Corporation, Winter Street, Waltham, Massachusetts, 02451 USA). The instrument was calibrated using indium. Then, 15–20 mg of FPI suspensions (prepared from unsonicated and sonicated lyophilized proteins at 10% (w/w) concentration) were transferred into aluminum pans and hermetically sealed. The scanning temperature was raised at a 10 °C/min rate from 30 to 150 °C. An empty aluminum pan was used as a control [21].
Secondary structure analysis by FTIR
Secondary structure analysis was achieved using an FTIR spectrometer (FTIR 100, Perkin Elmer Incorporation, USA) equipped with a universal diamond ATR crystal. All spectra were measured against a background spectrum of air. The spectroscopy measurements were performed using the lyophilized sample in the wavenumber range of 4000 to 650 cm−1 at a resolution of 4 cm−1 and 32 scans [13]. After performing a second derivative analysis and Fourier self-deconvolution (FSD), the peaks in the amide-I region (1700–1600 cm–1) band were fitted using the equipment software. The following band assignments were made: the corresponding cm−1 values for the α-helix, β-sheet, β-turn, and random coil were 1650–1658, 1610–1640, 1660–1700, and 1640–1650, respectively. Based on their center, Gaussian peaks could be allocated to the appropriate structure, and the integral of each peak was divided by the total of all identified peaks to determine the percentage (%) of each structure [22].
Scanning electron microscopy
Morphological analysis of lyophilized FPI samples was performed using scanning electron microscopy (Thermo Fisher Scientific Apreo S, Waltham, Massachusetts, USA) at an accelerating voltage of 5.0 kV. Samples were located on circular aluminum stubs and coated with gold–palladium in an argon atmosphere. Digital images were taken at different magnifications [4].
Statistical analyses
All the analyses were performed in triplicate, and the average values and their standard deviations were utilized to express the results. Statistical analyses were performed via SPSS (Version 22, Polar Engineering and Consulting, Nikiski, USA). A one-way analysis of variance (ANOVA) along with Duncan’s multiple range test was used to test the differences between the FPI samples. Differences between sample mean values were significant when p < 0.05.
Results and discussion
Solubility
Protein solubility was viewed as the best indicator of protein functional properties, as increased protein solubility could result in improved protein qualities. High solubility is needed to utilize a protein as an emulsifier [18]. Figure 1 displays the solubility of unsonicated and ultrasound-treated faba bean protein isolates. When subjected to ultrasound treatment, the solubility of unsonicated FPI significantly (p < 0.05) increased from 1.15% to 62.08%, 89.11%, 81.56%, and 82.88% for U1, U3, U5, and U10 samples, respectively. The enhanced solubility of FPI following ultrasound treatment can be attributed to various factors. One such factor is the exposure of hydrophilic groups of amino acids within FPI to water, as evidenced by the disruption of internal interactions and conformational changes observed in the FTIR results (Table 3), particularly in the amide II region. Another contributing element to the enhancement in solubility could perhaps be attributed to the creation of soluble protein aggregates arising from initially insoluble protein aggregates [6]. As can be seen from the results, the solubility of FPI reached the maximum solubility (89.11%) at 3 min of sonication time. Afterwards, the increase in sonication slightly decreased the solubility of the FPI, which was still higher than the unsonicated FPI sample. One potential explanation is that the protein molecules undergo partial unfolding, leading to an enhanced interaction between the proteins and water molecules. The expansion of FPI molecules, leading to the exposure of hydrophobic groups, decreased their solubility [18]. Similarly, Nazari et al. [9] indicated that the millet protein isolate’s solubility increased after sonication in varying intensities and times (18.4, 29.58, and 73.95 W/cm2 for 5, 12.5, and 20 min, respectively).

Solubility values of FPI as a function of ultrasound treatment time (C: untreated, U1: 1 min treated FPI, U3: 3 min treated FPI, U5: 5 min treated FPI, U10: 10 min treated FPI). The values followed by different letters for each column significantly (p < 0.05) differ by Duncan’s multiple range test
Particle size
The particle size of unsonicated and ultrasound-treated FPI samples is shown in Fig. 2. The results showed that ultrasound treatment decreased the particle size of FPI from 370.47 to 288.93 nm (hydrodynamic diameter in nanometer) significantly (p < 0.05) within 1 min. When the sonication time was increased to 5 min, a further decrease in particle size to 196.50 nm was observed for sample U5 (Table 1). The highest reduction in particle size, from 370.47 to 187.87 nm, was observed in FPI sonicated for 10 min. It has been reported that large insoluble protein aggregates can be reduced in size and broken down into smaller particles by the cavitational turbulence and shear forces produced by the sonicator’s probe [18, 23]. A similar reduction in particle size after ultrasound treatment was obtained by Jambrak et al. [15] for soy protein, Arzeni et al. [23] for whey protein, Xiong et al. [24] for pea protein isolate, and Mir et al. [19] for album seed protein isolate. The particle size agrees well with the solubility of the samples (Fig. 1). It is clearly seen that when the particle size of the FPI decreased, solubility increased significantly (p < 0.05) after ultrasound treatment at 1 and 3 min.

Particle size values of FPI as a function of ultrasound treatment time (C: untreated, U1: 1 min treated FPI, U3: 3 min treated FPI, U5: 5 min treated FPI, U10: 10 min treated FPI). The values followed by different letters for each column significantly (p < 0.05) differ by Duncan’s multiple range test
Zeta potential
Zeta potential, which is a significant physical and chemical parameter that serves as an indicator of the stability of a crucial protein suspension solution, is influenced by the surface charge properties exhibited by protein particles in a solution system. It is determined by the presence of charged amino acids, which impact how proteins disperse and aggregate together [6]. In this study, zeta potential values of all the samples were negative (Fig. 3), which means the FPI contained more negatively charged amino acids than positively charged amino acids. The zeta potential values of FPI samples significantly (p < 0.05) reduced (in magnitude) from − 40.83 to − 37.60, − 36.03, − 37.00, and − 37.17 mV for 1, 3, 5, and 10 min treated, respectively (Table 1). The observed reduction in absolute zeta potential can mostly be attributed to alterations in noncovalent interactions brought about by sonic treatment. These interactions, namely electrostatic and hydrophobic forces, play a significant role in promoting protein aggregation [18]. However, there were no significant differences between ultrasound-treated samples after 3 min., which indicates that a short ultrasound treatment modifies protein charge, but a longer treatment time has no additional effect. Wang et al. [4] reported that the high-intensity ultrasound pretreatment decreased the net surface charge of chickpea protein isolate (CPI). This reduction indicates that the ionization degree of the surface groups in CPI was attenuated.

Zeta potential values of FPI as a function of ultrasound treatment time (C: untreated, U1: 1 min treated FPI, U3: 3 min treated FPI, U5: 5 min treated FPI, U10: 10 min treated FPI). The values followed by different letters for each column significantly (p < 0.05) differ by Duncan’s multiple range test
Color
The effects of ultrasound on color parameters (L*, a*, b*, C*, H*, ΔE and BI) of FPI powders are presented in Table 1. As can be seen from these data, the L* values of FPI samples significantly (p < 0.05) increased after applying ultrasound treatment for 1 and 3 min. However, an extension of the treatment to 5 or 10 min was associated with decreasing L* values. Thus, longer sonication resulted in a lightening of the FPI samples compared with the control sample. The a* values also showed a similar trend following ultrasound treatment. The a* values of FPI samples significantly (p < 0.05) decreased after ultrasound treatment until a specific time (3 min) and then increased by increasing sonication time. Ultrasound treated FPI samples exhibited significantly (p < 0.05) reduced b* values. Chrome (C ∗) is used to quantify the saturation, purity, or visual intensity of a color. It measures the extent to which a color deviates from grey towards a pure chromatic color. Consequently, higher values of Chrome (C ∗) correspond to greater color saturation [25]. The chroma values of sonicated FPI samples were significantly (p < 0.05) lower compared to the control. The hue angle, which is a reflection of tonality, increased in the sample that was sonicated for 1 min. but decreased with a further increase in the sonication time. The Browning Index (BI) is a metric that quantifies the degree of brown pigmentation and is considered a significant parameter in enzymatic or non-enzymatic browning processes [26]. In the present study, the unsonicated FPI sample showed the highest BI value. After ultrasound treatment, BI values of FPI samples reduced significantly (p < 0.05). During ultrasound treatment, all FPI samples exhibited a decrease in BI with the treatment time, indicating that ultrasound treatment effectively reduced non-enzymatic browning. ΔE also decreased with the application of ultrasound in FPI samples. The color differences in FPI samples are shown visually in Fig. 4. Based on the results of this investigation, it is possible to attribute the alterations in color observed after sonic treatment to the degradation of pigments. The physical and chemical degradation of biomolecules, such as the generation of radicals, can occur as a result of the cavitation phenomenon induced by ultrasound. The reduction in particle size may provide yet another explanation for the color change. Protein particles get smaller after undergoing ultrasound treatment and thus increase in number. This, in turn, contributes to the multiple scattering of light, resulting in the whiteness of the FPI sample [27]. Hence, sonication time is directly correlated with the variance in the color features of ultrasound-treated protein isolates. Sonication can positively or negatively impact the pigments found in food [28]. Zuniga-Salcedo et al. [8] isolated safflower protein isolate and exposed it to ultrasound treatment. They found no significant differences between the color of the control and ultrasound-treated protein solutions.

Photography of unsonicated and sonicated FPI samples (C: untreated, U1: 1 min treated FPI, U3: 3 min treated FPI, U5: 5 min treated FPI, U10: 10 min treated FPI)
Thermal properties
The thermal stability of FPI samples was investigated using DSC analysis. The thermal denaturation temperature (Td) is a parameter used to measure the temperature at which proteins undergo denaturation. ΔHd represents the enthalpy change associated with the denaturation process, indicating the amount of heat required for denaturation to occur [29]. All FPI samples showed a single broad endothermic peak at approximately 100 °C. Thermal properties like onset temperature (Tonset), denaturation temperature (Td), endset temperature (Tend), and denaturation enthalpy (ΔHd) of the FPI samples were determined from the curves and are tabulated in Table 2. The Td of the unsonicated FPI sample was 103.07 °C. After ultrasound treatment, the Td of the FPI samples was significantly (p < 0.05) reduced to 99.42 °C and 99.58 °C for the U1 and U3 samples, respectively. Such a reduction of Td could be linked to several structural and conformational changes that occur in FPI samples due to the sonication process which breaks links in the protein [19]. The FPI samples showed an increase in Td when the treatment period was further increased up to 10 min, and the values recorded for this parameter were 104.96 °C and 102.63 °C for U5 and U10, respectively. There is a possibility that the reformation of hydrophobic connections between protein structures is responsible for this rise [12]. In the study by Mir et al. [19], the application of high pressure was found to cause the development of aggregates. In the same way, extended durations of ultrasound treatment may also facilitate the development of protein aggregates, resulting in a decrease in the solubility of FPI (Fig. 1). According to Chandrapala et al. [12], the application of heat to globular proteins results in the disruption of some forces that contribute to the stability of tertiary and/or secondary protein structures. The forces at play encompass hydrogen connections between polar groups and interactions of non-polar groups, commonly referred to as hydrophobic interactions, mediated by the surrounding water molecules that provide a protective enclosure around hydrophobic groups. The process of heat denaturation also involves the participation of electrostatic bonds and Van der Waals interactions, albeit to a lower degree. Nevertheless, when subjected to extended heat treatment, the process undergoes a reversal, whereby heat intensifies the aggregation of proteins. Sonication can also induce the rupture of intramolecular protein forces through the application of shear forces, resulting in the formation of aggregates over extended periods of sonication, similar to the effects observed during heating.
ΔHd represents the amount of energy necessary to denature the protein. Similar to the changes in Td, ΔHd of the FPI samples significantly (p < 0.05) decreased from 2.21 J/g to 1.12 J/g up to 3 min (Table 2). This decrease in ΔHd, caused by ultrasound treatment, indicates that less energy was needed to unfold the sonicated FPI samples due to the breakdown of intramolecular interactions caused by the shear forces that formed cavitation bubbles [9]. Chandrapala et al. [21] reported similar trends for whey protein concentrate, and they correlated this result to the destruction of hydrophobic interactions caused by ultrasound treatment. The FPI samples showed an increase in ΔHd as the treatment time was further increased up to 10 min (Table 2). A slight increase in enthalpy due to a more extended sonication time might be correlated to a possible reformation of intra- and intermolecular hydrophobic bonds from the resulting disordered protein molecules [19].
Secondary structure changes
In this study, FTIR was used to correlate the proportions of α-helix, β-sheet, β-turn, and random coil or unordered groups to determine the secondary structure of FPI samples. The FTIR spectra of proteins have absorption bands that are amide groups. The Amide I band, which may be found between 1700 and 1600 cm−1, is widely regarded as the most reliable indicator of changes in secondary structure [13, 30]. The FTIR spectroscopy of unsonicated and ultrasound-treated FPI samples is shown in Fig. 5. The findings demonstrated that the application of ultrasound caused a shift in the peak positions of amide I (at a wavelength of 1700–1600 cm−1) and II (at a wavelength of 1530–1550 cm−1). Compared to the control value of 1638.25 cm−1, the values shifted to 1627.72, 1627.18, 1631.9, and 1632.44 cm−1 for U1, U3, U5, and U10 samples, respectively.

FTIR spectra of FPI as a function of ultrasound treatment time (C: untreated, U1: 1 min treated FPI, U3: 3 min treated FPI, U5: 5 min treated FPI, U10: 10 min treated FPI)
The fitted peaks obtained subsequent to the resolving of the secondary molecular structure into its constituent-deconvolution of FPI samples are shown in Fig. 6. Table 3 illustrates that the unsonicated FPI sample contained 9.88% α-helix, 50.0% β-sheet, 26.90% β-turn, and 13.23% random coil, and this content was in agreement with the reports of Martínez-Velasco et al. [13] and Johnson et al. [31]. From the results, the α-helix content of FPI samples significantly (p < 0.05) decreased from 0 to 3 min and then increased from 3 to 10 min sonication. In the case of β-sheets, they increased from 0 to 3 min and then decreased slightly from 3 to 10 min ultrasound treatment. Compared to the unsonicated FPI sample, a rise in β-sheets in all the processed samples was observed. There was a reduction in the random coil and β-turn content after ultrasound treatment. A decrease in α-helix means less structured proteins. During the first sonication intervals (1 and 3 min), there was unfolding, leading to less structured proteins. As the sonication times increased (5 and 10 min), there was a restructuration of proteins by folding. Random coil and β-turns decrease in a continuous form due to proteins changing with ultrasound treatment. Proteins can refold but do not acquire the original structure of non-treated FPI [32]. Jin et al. [33] reported similar decreases in α-helix content and increasing β-sheet content for zein protein after ultrasound application. Gao et al. [34] worked on the effect of high-intensity ultrasound on pea protein isolate and reported that β-sheet proportion increased while β-turn decreased when the processing time was prolonged. However, other researchers have shown that different changes could happen in the secondary structure of proteins [9, 11, 18]. This implies that the effect of sonication on protein structure is dependent on protein type and sonication conditions.

Curve fitted of the amide I region of unsonicated and sonicated FPI (C: untreated, U1: 1 min treated FPI, U3: 3 min treated FPI, U5: 5 min treated FPI, U10: 10 min treated FPI)
Scanning electron microscopy
SEM was used to investigate the microstructure of lyophilized FPI samples to understand the effect of sonic treatment times on FPI. Figure 7 presents the microstructures of the unsonicated and treated FPI samples; the SEM images were obtained at a magnification of 150-fold and 2000-fold. As can be seen in Fig. 7, the unsonicated FPI appear to be distributed within a densely packed network and have a polydisperse protein size; whereas the FPI treated by ultrasound appear to be distributed into discrete entities, having a smaller and a slightly more uniform size in comparison to the unsonicated FPI. After sonic treatment, the larger sheets were broken into smaller irregular fragments, as seen in Fig. 7 (U1, U3, U5, and U10), consistent with the results in particle size described above. These could result from the micro-streaming, turbulent, and cavitational forces induced by the probe during sonic treatment, as Jiang et al. [22] suggested. The structure of the FPI sample which was sonicated for 3 min was more ordered than samples sonicated for extended periods. This result is in line with the FPI’s solubility results (Fig. 1). The unfolding of proteins might explain the relationship between the structure and solubility of FPI after ultrasound treatment. Sonication increases the surface area of proteins and thus increases the chances of protein-water contact [35].

SEM images of FPI as a function of ultrasound treatment time (C: untreated FPI at 150×, U1: 1 min treated FPI at 150×, U3: 3 min treated FPI at 150×, U5: 5 min treated FPI at 150×, U10: 10 min treated FPI at 150×: c: Untreated at 2000×, u1: 1 min treated FPI at 2000×, u3: 3 min treated FPI at 2000×, u5: 5 min treated FPI at 2000×, u10: 10 min treated FPI at 2000×)
Conclusions
This study showed that applying ultrasound treatment at varying times affected the functional and thermal properties, and secondary structure content of FPI. The protein solubility gradually enhanced as the sonication time increased, whereas excessive ultrasound caused a decline in solubility. The cavitation effect of ultrasound caused a reduction in particle size of FPI. This reduction in particle size caused more hydrophobic groups to be exposed on the surface thus decreasing the net surface charge. In terms of color outcomes, an excessive increase in ultrasound time caused the protein to darken. Both Td and ΔHd decreased with the 1 and 3 min ultrasound treatments but increased after that. The secondary structure content of FPI changed at lower sonicating times. With times higher than 5 min, an increase in α-helix and a subsequent decrease in β-sheet were observed. SEM revealed that the microstructure of ultrasound-treated FPI was altered, with smaller aggregates. These findings have the potential to contribute to the establishment of a theoretical framework concerning the functional properties of proteins, as well as their structural and thermal alterations in response to ultrasonic treatment. Nevertheless, additional investigation is necessary to ascertain the mechanisms by which this protein isolate operates inside food systems.
Data Availability
The data used to support the findings of this study are included within the article.
References
R.S. Bhatty, G.I. Christison, Plant Foods Hum. Nutr. 34, 41–51 (1984). https://doi.org/10.1007/BF01095071
S.B. Dhull, M.K. Kidwai, R. Noor, P. Chawla, P.K. Rose, Legum Sci. (2021). https://doi.org/10.1002/leg3.129
J.E. Kinsella, N. Melachouris, Crit. Rev. Food Sci. Nutr. 7, 219–280 (1976). https://doi.org/10.1080/10408397609527208
Y. Wang, Y. Wang, K. Li, Y. Bai, B. Li, W. Xu, LWT 129, 109563 (2020). https://doi.org/10.1016/J.LWT.2020.109563
D.J. McClements, Trends Food Sci. Technol. 6, 293–299 (1995). https://doi.org/10.1016/S0924-2244(00)89139-6
M.M. Rahman, B.P. Lamsal, Compr. Rev. Food Sci. Food Saf. 20, 1457–1480 (2021). https://doi.org/10.1111/1541-4337.12709
O.A. Higuera-Barraza, C.L. Del Toro-Sanchez, S. Ruiz-Cruz, E. Márquez-Ríos, Ultrason. Sonochem. 31, 558–562 (2016). https://doi.org/10.1016/J.ULTSONCH.2016.02.007
M.R. Zuniga-Salcedo, J.A. Ulloa, P.U. Bautısta-Rosales, P. Rosas-Ulloa, J.C. Ramírez-Ramíez, Y. Silva-Carrilo, R. Gutıérrez-Leyva, C. Hernández, Ital. J. Food Sci. 31, 3 (2019). https://doi.org/10.14674/IJFS-1440
B. Nazari, M.A. Mohammadifar, S. Shojaee-Aliabadi, E. Feizollahi, L. Mirmoghtadaie, Ultrason. Sonochem. 41, 382–388 (2018). https://doi.org/10.1016/J.ULTSONCH.2017.10.002
H. Li, Y. Hu, X. Zhao, W. Wan, X. Du, B. Kong, X. Xia, LWT 137, 110403 (2021). https://doi.org/10.1016/J.LWT.2020.110403
H. Hu, J. Wu, E.C.Y. Li-Chan, L. Zhu, F. Zhang, X. Xu, G. Fan, L. Wang, X. Huang, S. Pan, Food Hydrocoll. 30, 647–655 (2013). https://doi.org/10.1016/J.FOODHYD.2012.08.001
B. Lo, S. Kasapis, A. Farahnaky, Food Hydrocoll. 124, 107345 (2022). https://doi.org/10.1016/J.FOODHYD.2021.107345
A. Martínez-Velasco, C. Lobato-Calleros, B.E. Hernández-Rodríguez, A. Román-Guerrero, J. Alvarez-Ramirez, E.J. Vernon-Carter, Ultrason. Sonochem. 44, 97–105 (2018). https://doi.org/10.1016/J.ULTSONCH.2018.02.007
F. Alavi, L. Chen, Z. Emam-Djomeh, Food Chem. 354(129494), 5 (2021). https://doi.org/10.1016/J.FOODCHEM.2021.129494
A.R. Jambrak, V. Lelas, T.J. Mason, G. Krešić, M. Badanjak, J. Food Eng. 93, 386–393 (2009). https://doi.org/10.1016/J.JFOODENG.2009.02.001
A.R. Jambrak, T.J. Mason, V. Lelas, Z. Herceg, I.L. Herceg, J. Food Eng. 86, 281–287 (2008). https://doi.org/10.1016/J.JFOODENG.2007.10.004
Z. Zhang, J.M. Regenstein, P. Zhou, Y. Yang, Ultrason. Sonochem. 34, 960–967 (2017). https://doi.org/10.1016/J.ULTSONCH.2016.08.008
L. Jiang, J. Wang, Y. Li, Z. Wang, J. Liang, R. Wang, Y. Chen, W. Ma, V. Qi, M. Zhang, Food Res. Int. 62, 595–601 (2014). https://doi.org/10.1016/J.FOODRES.2014.04.022
N.A. Mir, C.S. Riar, S. Singh, Food Hydrocoll 96, 433–441 (2019). https://doi.org/10.1016/J.FOODHYD.2019.05.052
D.E.S.H. Azab, R.I.M. Almoselhy, M.H. Mahmoud, J. Food Process. Preserv. 46, e17235 (2022). https://doi.org/10.1111/jfpp.17235
J. Chandrapala, B. Zisu, M. Palmer, S. Kentish, M. Ashokkumar, Ultrason. Sonochem. 18, 951–957 (2011). https://doi.org/10.1016/J.ULTSONCH.2010.12.016
C. Zhao, Z. Chu, Z. Miao, J. Liu, J. Liu, X. Xu, Y. Wu, B. Qi, J. Yan, Food Biosci. 39, 100827 (2021). https://doi.org/10.1016/J.FBIO.2020.100827
C. Arzeni, K. Martínez, P. Zema, A. Arias, O.E. Pérez, A.M.R. Pilosof, J. Food Eng. 108, 463–472 (2012). https://doi.org/10.1016/J.JFOODENG.2011.08.018
T. Xiong, W. Xiong, M. Ge, J. Xia, B. Li, Y. Chen, Food Res. Int. 109, 260–267 (2018). https://doi.org/10.1016/J.FOODRES.2018.04.044
M.N. Islam, M. Zhang, B. Adhikari, C. Xinfeng, B.G. Xu, Int. J. Refrig. 42, 121–133 (2014). https://doi.org/10.1016/j.ijrefrig.2014.02.012
B.K. Tiwari, K. Muthukumarappan, C.P. O’Donnell, P.J. Cullen, LWT 41, 1876–1883 (2008). https://doi.org/10.1016/j.lwt.2007.11.0164
X. Bi, Y. Hemar, M.O. Balaban, X. Liao, Ultrason. Sonochem. 27, 567–575 (2015). https://doi.org/10.1016/J.ULTSONCH.2015.04.011
Y. Zou, A. Jiang, Food Sci. Technol. 36, 111–115 (2016). https://doi.org/10.1590/1678-457X.0061
P. Kaushik, K. Dowling, S. McKnight, C.J. Barrow, B. Wang, B. Adhikari, Food Chem. 197, 212–220 (2016). https://doi.org/10.1016/J.FOODCHEM.2015.09.106
J. Kong, S. Yu, Acta Biochim. Biophys. Sin. (Shanghai) 39, 549–559 (2007). https://doi.org/10.1111/j.1745-7270.2007.00320.x
J.B. Johnson, K. Walsh, M. Naiker, Legum Sci. 2(3), e40 (2020). https://doi.org/10.1002/LEG3.40
Y. Meng, Z. Liang, C. Zhang, S. Hao, H. Han, P. Du, A. Li, H. Shao, C. Li, L. Liu, LWT 152, 112272 (2021). https://doi.org/10.1016/J.LWT.2021.112272
J. Jin, H. Ma, B. Wang, A.E.G.A. Yagoub, K. Wang, R. He, C. Zhou, Ultrason. Sonochem. 30, 44–51 (2016). https://doi.org/10.1016/J.ULTSONCH.2015.11.021
K. Gao, F. Zha, Z. Yang, J. Rao, B. Chen, Food Hydrocoll. 125, 107409 (2022). https://doi.org/10.1016/J.FOODHYD.2021.107409
R. Tian, J. Feng, G. Huang, B. Tian, Y. Zhang, L. Jiang, X. Sui, Ultrason. Sonochem. 68, 105202 (2020). https://doi.org/10.1016/J.ULTSONCH.2020.105202
Acknowledgements
The author acknowledges AGT Food (SK, Canada) for supplying faba bean protein isolate. Thanks to the Center for Science and Technology Studies and Research of Iskenderun Technical University for providing structural analyses of samples.
Funding
Open access funding provided by the Scientific and Technological Research Council of Türkiye (TÜBİTAK).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author has declared no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Adal, E. Modification of faba bean protein isolate by high-intensity ultrasound treatment: screening of physicochemical, thermal, and structural properties. Food Measure 18, 2438–2449 (2024). https://doi.org/10.1007/s11694-024-02379-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11694-024-02379-z