
Abstract
Background
Alpelisib in combination with cetuximab showed synergistic anti-tumour activity in head and neck squamous cell carcinoma (HNSCC) models.
Objectives
The recommended phase 2 dose (RP2D) was determined in a phase 1b dose-escalation study. Phase 2 evaluated anti-tumour activity with a randomised part in cetuximab-naïve patients and a non-randomised part in cetuximab-resistant patients.
Patients and Methods
Alpelisib was administered in 28 d cycles as whole tablets, suspension from crushed tablets or suspension from dispersible tablets in patients with platinum-resistant, recurrent/metastatic HNSCC.
Results
The RP2D determined for alpelisib was 300 mg/d. Alpelisib–cetuximab achieved an overall response rate of 25% and 9.9% and disease control rate of 75% and 43.7% in phase 1b and phase 2 studies, respectively. Median progression-free survival (PFS) per central review was 86 d for combination treatment and 87 d for cetuximab monotherapy (unadjusted HR 1.12; 95% CI 0.69–1.82; P > 0.05). When adjusted for baseline covariates [sum of longest diameters from central data, haemoglobin and white blood cell (WBC), the results favoured combination treatment (adjusted HR 0.54; 95% CI 0.30–0.97; P = 0.039). PFS per investigator assessment resulted in an unadjusted HR of 0.76 (95% CI 0.49–1.19; P > 0.05) favouring combination treatment. The median PFS in cetuximab-resistant patients was 3.9 months.
Conclusions
The addition of alpelisib to cetuximab did not demonstrate a PFS benefit in cetuximab-naïve patients with advanced HNSCC. The alpelisib–cetuximab combination showed moderate activity in cetuximab-resistant patients, with a consistent safety profile.
Clinical Trial Registration
ClinicalTrials.gov NCT01602315; EudraCT 2011-006017-34.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Recommended phase 2 dose for alpelisib was 300 mg/d in combination with cetuximab. |
Safety profile of alpelisib-cetuximab was similar to previous alpelisib studies. |
Survival benefit in cetuximab-naive patients seen after adjustment of covariates. |
1 Introduction
Head and neck squamous cell carcinoma (HNSCC) represent ~91% of all head and neck cancers and usually has poor prognosis in advanced recurrent/metastatic stages. Despite the availability of conventional cytotoxic chemotherapy, immunotherapy or molecular-targeted agents, most patients with advanced disease have low median survival (6–15 months) [1, 2]. The choice of systemic therapy depends on several factors, including comorbidities, performance status, previous therapy and pathologic features such as programmed death-ligand-1 (PD-L1) expression status [3].
The overexpression of EGFR in ~90% of HNSCC represents a potential therapeutic target [4]. The anti-EGFR monoclonal antibody, cetuximab is the only approved targeted therapy with proven efficacy in treatment of advanced recurrent/metastatic HNSCC. However, several patients are refractory to EGFR inhibition by cetuximab due to intrinsic or acquired mechanisms of resistance, resulting in poor response rates (13%) [5]. The PI3K signalling pathway is commonly activated in HNSCC, promoting tumour growth and progression [6, 7], and is attributed to resistance to EGFR-directed therapy [8, 9]. The combination of a PI3K inhibitor (PI3Ki) with cetuximab could potentially overcome this resistance.
Alpelisib (BYL719), an oral α-isoform-specific PI3Ki, demonstrated anti-tumour efficacy in advanced solid tumours and is indicated in combination with fulvestrant for use in advanced hormone receptor-positive, HER2-negative breast cancer [10, 11]. Alpelisib potently inhibits PI3Kα isoforms, both wild type and mutant [6]. Simultaneous inhibition of PI3K and EGFR using alpelisib and cetuximab, respectively, leads to synergistic anti-tumour activity in HNSCC [12, 13] and inhibited most HNSCC tumour cell lines, irrespective of PIK3CA mutation [13]. Here, we report results from a phase 1b dose-escalation and phase 2 study of alpelisib–cetuximab in patients with recurrent/metastatic HNSCC who are resistant/ineligible/intolerant to platinum-based chemotherapy (ClinicalTrials.gov NCT01602315).
2 Materials and Methods
2.1 Study Design and Treatment
This was a multi-centre, open-label, phase 1b dose-escalation followed by a phase 2 randomised study in adult patients with platinum-resistant recurrent/metastatic HNSCC. Phase 1b included 3 treatment arms using different administration methods of alpelisib formulations administered once daily in 28 d cycles and weekly cetuximab 400 mg/m2 on day 1 and 250 mg/m2 weekly thereafter. The formulations tested included film-coated whole tablets (arm A), suspension prepared from crushed tablets in patients with swallowing dysfunction (arm B) and suspension from a dispersible tablet administered via G-tube (arm C). The primary objective of phase 1b part was to estimate the maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D) for the combination regimen and compare the single-dose exposure of a dispersible tablet versus alpelisib whole tablet in combination with cetuximab. The primary endpoint was the incidence of dose-limiting toxicity (DLT). MTD of the combination treatment was estimated based on the probability of DLT using a Bayesian logistic regression model (BLRM) [14].
Phase 2, with open-label randomised and non-randomised parts, was initiated upon declaration of MTD or RP2D in arm A of phase 1b, regardless of the status of arms B and C. Per protocol, patients naïve to cetuximab were assigned to the open-label randomised part (scheme 1, arms I and II); those who had prior cetuximab were assigned to the non-randomised part (scheme 2, arm III) (Fig. 1). Patients who were resistant/ineligible/intolerant to platinum and cetuximab-naïve were randomised in a 2:1 ratio via interactive randomisation technology to alpelisib–cetuximab (arm I) or single-agent cetuximab (arm II). Those with disease progression, confirmed using central imaging review, upon treatment with single-agent cetuximab could switch to alpelisib–cetuximab (arm IIB). Patients received treatment until disease progression, unacceptable toxicity or withdrawal of informed consent, whichever occurred first.

Study design of phase 1b and 2 study of alpelisib in combination with cetuximab in recurrent or metastatic head and neck squamous cell carcinoma. MTD maximum tolerated dose; PD progressive disease; RP2D recommended phase 2 dose. *In phase 1b, arms A and B enrolled in parallel. Arm C began later after arms A and B have completed. ^Arms 1 and 2 of phase 2 portion of the trial are randomized in patients resistant to or intolerant/ineligible for platinum-based chemotherapy. **Arm 3 in phase 2 is non-randomised arm in patients with resistance to platinum and cetuximab treatment
The primary objective of the phase 2 study was to evaluate anti-tumour activity of alpelisib–cetuximab versus cetuximab alone in patients who were resistant/intolerant to platinum and cetuximab naïve (scheme 1, arms I and II) and evaluate the combination regimen in patients resistant to platinum and cetuximab (scheme 1, arm IIB and scheme 2, arm III).
2.2 Patients
Eligible patients were aged ≥ 18 years, with histologically/cytologically confirmed HNSCC regardless of PIK3CA mutation. Patients after prior treatment with an EGFR-targeted antibody for recurrent/metastatic disease were included in the phase 1b study. For phase 2, only patients without prior EGFR-targeted treatment were eligible for randomisation in arms I and II. The non-randomised part of phase 2 (arm III) included patients after disease progression on or within 9 months of treatment containing both a platinum agent and cetuximab in recurrent/metastatic setting. Eligible patients had ≥ 1 measurable/non-measurable lesion per Response Evaluation Criteria in Solid Tumours (RECIST version 1.1) and World Health Organization (WHO) performance status of ≤ 2 with adequate organ function. The supplementary text provides more details on patient eligibility criteria. The study was conducted in accordance with the principles of Good Clinical Practice and the Declaration of Helsinki, and in compliance with applicable data privacy protection laws and regulations. All patients provided written informed consent.
2.3 Study Assessments
Potential sites of tumour lesions were assessed by computed tomography or magnetic resonance imaging at screening within 21 d prior to the first dose of study treatment. Tumour assessments were performed after completion of 6 weeks of treatment (cycle 2 day 15), 12 weeks of treatment (cycle 4 day 1), 18 weeks of treatment (cycle 5 day 15) and every 8 weeks thereafter and at the end of study treatment. A blinded central imaging review was conducted. Overall response rate (ORR) was defined as the proportion of patients achieving a complete or partial response according to RECIST version 1.1. Disease control rate (DCR) was defined as the proportion of patients with a best overall response of complete response, partial response stable disease or either including non-complete response/non-progressive disease or not including. Tumour response by RECIST v1.1 was used to calculate progression-free survival (PFS, primary endpoint). The PFS by RECIST1.1 was assessed based on the blinded centrally reviewed tumour assessment data and additionally, median PFS was estimated by RECIST 1.1 based on central imaging review. Information about the patients’ survival status was recorded in the electronic case record form and the respective information was used to determine OS in phase 2.
2.4 Statistical Analysis
For the model to support MTD recommendations, ~12 patients were expected to be treated in the phase 1b dose-escalation part. In phase 2, assuming a 2:1 randomisation ratio, 76 events were needed to provide 85% power to detect a 0.575 hazard ratio (HR) in terms of progression-free survival (PFS) using a log-rank test at a one-sided significance level of 0.1. Allowing for 10% dropouts, 99 patients were planned to observe 76 events.
The full analysis set (FAS) comprised all patients who received ≥ 1 dose of either alpelisib or cetuximab in phase 1b and phase 2 arm III and all randomised patients in phase 2 scheme 1, arms I and II. The safety analysis set comprised all patients who received ≥ 1 dose of alpelisib or standard cetuximab therapy and had ≥ 1 post-baseline safety assessment. Post hoc analysis was performed to explore how baseline characteristics and on-treatment variables may be associated with survival endpoints. Clinical baseline data and treatment were used to produce a parsimonious predictive model for PFS and overall survival (OS) using the partial least squares Cox model with cross-validation methodology. Statistical analyses describing Bayesian modelling and multivariate Cox regression modelling are shown in the supplementary information.
3 Results
3.1 Patients
In phase 1b, the FAS (N = 45) included patients without swallowing dysfunction (alpelisib 300 mg/d + cetuximab, n = 16; alpelisib 400 mg/d + cetuximab, n = 5); those with swallowing dysfunction were allocated to arm B (alpelisib 300 mg/d + cetuximab, n = 18) or C (alpelisib 300 mg/d + cetuximab, n = 6).
Of 106 cetuximab-naïve patients in the phase 2 randomised part, 71 received alpelisib + cetuximab and 35 received cetuximab monotherapy. Of these 35 patients, 16 (45.7%) switched to combination therapy (arm 2B) after disease progression; 19 did not (Table 1).
Patients in phase 1b and phase 2 discontinued the study treatment primarily due to disease progression or adverse events (AEs) (Table 2). Approximately, half of the patients in phase 1b and the non-randomised arm of phase 2 and 35% in randomised part of phase 2 had stage 3/4 disease at initial diagnosis. The demographic characteristics matched between treatment arms, except for ECOG status 0/1 and tumour lysis syndrome (TLS) risk (both higher in combination arm versus cetuximab arm) (Table 1). In the phase 2 part, PIK3CA alterations in archival tumour tissue by Foundation One next-generation sequencing was found in 8/44 samples and 5/20 samples analysed in the alpelisib + cetuximab and cetuximab monotherapy arms, respectively. Due to the low frequency of PIK3CA alterations in this patient population, a subset analysis by PIK3CA alteration was not deemed meaningful (Fig. 2).

a Best percentage change from baseline in sum of longest diameters and best overall response (full analysis set). Phase 1b. Phase 2 randomised part. Treatment arm—alpelisib 300 mg + cetuximab. Phase 2 randomised part. Treatment arm—cetuximab. n, number of patients with a baseline and ≥ 1 post-baseline assessment of target lesions based on investigator’s assessment. #Patients missing best percentage change from baseline are not included. b Waterfall plot of percent change from baseline in sum of longest diameters as per central radiology review by treatment (full analysis set). Phase 2 randomised part. n, number of patients with a baseline and ≥ 1 post-baseline assessment of target lesions based on central radiology review. CR complete response, PD progressive disease, PR partial response, SD stable disease, UNK unknown
Patients who did not receive ≥ 21 doses of alpelisib and ≥ 3 doses of cetuximab, i.e. 75% of the planned doses, during cycle 1 without experiencing a DLT were excluded from the dose-determining set (DDS) per protocol. Based on these criteria, 15 out of 45 patients have been excluded from the DDS. Two patients in the combination treatment arm died before the initiation of study treatment and were excluded from the safety set.
3.2 Safety
3.2.1 DLT
Of 30 patients, 9 had a DLT within 28 d of study initiation (1 on alpelisib 300 mg/d tablet, 2 on alpelisib 400 mg/d tablet, 4 on drinkable suspension and 2 on dispersible alpelisib). Per BLRM inference, alpelisib 300/350 mg/d whole-tablet cetuximab showed a 1.5% and 15.3% risk of overdosing for 300 and 350 mg/d, respectively. In patients on drinkable suspension (arm B) of alpelisib 300 mg/d, the posterior probability of the DLT rate lying within the targeted toxicity range (16–35%) for alpelisib 300 mg/d was 66.4%, with a 6.9% risk of overdosing (i.e. the posterior probability of a DLT rate of > 35%). In patients on dispersible tablets (arm C), the posterior probability of DLT rate lying within the targeted toxicity range (16–35%) for 300 mg alpelisib–cetuximab was 59.5%, with a higher risk of overdose (25.9%).
3.2.2 AE
Of 45 patients enrolled in phase 1b study, most patients (n = 44, 97.8%) experienced ≥ 1 any-grade AE, regardless of relationship to the study treatment (Table 3). The most commonly reported all-grade AEs were hyperglycaemia, hypomagnesaemia, stomatitis, diarrhoea, weight decrease, loss of appetite, rash and fatigue. The most common reason for discontinuation was AEs (15 patients [33%] in phase 1b and 18 [17%] in phase 2), with the most frequent being hyperglycaemia (9% of patients in phase 1b, 3% in phase 2). The incidence of AEs leading to study treatment discontinuation was higher in the randomized combination arm versus the cetuximab arm (15 patients [21.7%] versus 3 [8.6%]) in phase 2 (Table 4). The incidence of clinically notable AEs for phase 1b studies are presented in Table S1.
In phase 1b, all patients experienced AEs that were suspected to be related to study treatment. The most frequently reported AEs suspected to be related to study treatment included hyperglycaemia, hypomagnesaemia, diarrhoea and rash. The most frequently reported serious adverse events (SAEs) were pneumonia, hyperglycaemia and pyrexia. Grade 3/4 AEs were lower in treatment arm B (n = 12; 66.7%) compared with arm A (n = 13; 86.7%) and arm C (n = 5; 83.3%).
In phase 2, the incidence of any-grade AEs was higher (difference ≥ 10%) in the combination treatment arm compared with the cetuximab monotherapy arm. Patients experiencing grade 3/4 AEs and SAEs were higher in the combination treatment arm (87% and 58%, respectively) compared with the cetuximab monotherapy arm (57.1% and 42.9%, respectively). The most frequently reported AEs suspected to be study treatment related in the combination treatment arm were hyperglycaemia, stomatitis, diarrhoea, rash, decreased appetite, dermatitis acneiform, fatigue, weight decrease, hypomagnesaemia, paronychia, dry skin and nausea.
Eight on-treatment deaths were reported during phase 1b: disease progression (n = 3), AEs including pneumonia (n = 3), septic shock and TLS (n = 1 each); only TLS was considered treatment related. In phase 2, 15 on-treatment deaths (21.7%) were reported in the randomized combination arm and 8 (22.9%) in the cetuximab arm (4 patients on cetuximab monotherapy who did not cross over and 4 who switched to combination treatment); 3 deaths (10.3%) were reported in the non-randomised arm. Of the 15 patients who died in the combination treatment arm, 7 patients died due to the study indication or squamous cell carcinoma. Eight patients died due to AEs: pneumonia (n = 4), sepsis (n = 1), pulmonary haemorrhage (n = 1), respiratory failure (n = 1) and tumour haemorrhage (n = 1). Pneumonia in one patient was suspected to be related to study treatment. Of the four patients randomized to cetuximab monotherapy who died after the cross-over, two patients died due to study indication and two patients died due to AEs: one patient from sepsis and one patient from respiratory distress, neither event was suspected to be related to study treatment.
3.3 Efficacy
3.3.1 Response Rate
In the phase 1b study, the ORR [95% confidence interval (CI)] was 8.9% (2.5–21.2%), and the overall DCR (95% CI) was 51.1% (35.8–66.3%). Four patients (25%) treated with whole-tablet alpelisib 300 mg/d + cetuximab achieved partial response (PR) per RECIST version 1.1; 18 (40.0%) had stable disease (SD) (Table 5). No arm B patients who received an oral suspension of alpelisib crushed tablets and cetuximab achieved a partial response/complete response.
In the phase 2 randomised part, a trend indicating higher ORR and DCR in the combination arm was observed. The ORR (95% CI) was 9.9% (4.1–19.3%) in the combination arm and 5.7% (0.7–19.2%) in the cetuximab arm (Table 5). The DCR (95% CI), including CR, PR, SD or non-CR/non-PD, was 52.1% (39.9–64.1%) in the combination arm and 57.1% (39.4–73.7%) in the cetuximab arm. Of the nine patients in the cross-over part, four showed reductions in tumour burden; none achieved PR/CR (ORR 10.3%, 95% CI 2.2–27.4%; DCR 10.3%, 95% CI 20.7–57.7%).
Of the 23 patients who had tumour reduction in phase 1b, 11 had tumour shrinkage ≥ 30%. In the phase 2 part, tumour shrinkage was observed in 31 of 44 patients in the combination arm and in 9 of 21 patients in the cetuximab arm. There was a higher number of patients with tumour shrinkage ≥ 30% in the combination arm (12 of 44 patients) when compared with the cetuximab arm (2 of 21 patients (Fig. 2)).
3.3.2 PFS
Based on central review data, the mean of posterior distribution HR of PFS between combination treatment and cetuximab monotherapy estimated using the Bayesian Cox proportional-hazards model was 0.991 (95% credible interval, 0.643–1.529). The posterior probability of an HR of > 1 was 48.4%. As the posterior summaries did not meet the predefined criteria for success (interval probabilities: substantial efficacy 0.550%, moderate efficacy 5%, slight efficacy 46.06%, no efficacy 48.39%), combination therapy was not declared superior to cetuximab monotherapy. Median PFS (95% CI) per central review was 86 d (73–135 d) in the combination arm and 87 d (49–133 d) in the cetuximab arm. Median PFS for the 16 patients who crossed over from cetuximab to combination treatment was 43 d (95% CI 27.0–88.0 d). The median of the posterior distribution of PFS in cetuximab-resistant patients was 3.896, and the posterior probability of the median PFS being > 2.5 months was 99.69%, indicating moderate efficacy in the non-randomised arm.
The Cox regression model (frequentist approach) showed no significant difference between the combination and cetuximab arms for PFS (unadjusted HR 1.12; 95% CI 0.69–1.82) (Fig. 3). In the model adjusted for selected baseline covariates (sum of the longest diameters [SLD]) from central review/data, haemoglobin and white blood cell (WBC), the adjusted HR was 0.54, indicating a significant difference in favour of the combination arm (adjusted HR 0.54; 95% CI 0.30–0.97).

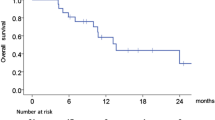
a Kaplan–Meier plot of progression-free survival as per central review for phase 2 randomised part (full analysis set). b Overall survival—phase 2 randomised part (full analysis set). CI confidence interval, HGB haemoglobin, SLD (C) sum of the longest diameters from central review/data, SLD (L) sum of the longest diameters from investigator/local data, WBC white blood cell
The analysis of PFS based on investigational site assessment resulted in an unadjusted HR of 0.76, trending in favour of the combination arm [not significant (NS)]. In the model adjusted for selected baseline covariates, the HR reduced to 0.64 in favour of the combination arm (non-significant).
3.3.3 Overall Survival
The median OS (95% CI) was 173 d (142–249) in the combination arm and 263d (181–444) in the cetuximab arm. Unadjusted Cox proportional-hazards model data represented a non-statistically significant trend of higher death risk in the combination arm (unadjusted HR 1.28; 95% CI 0.80–2.05). However, the model adjusted for selected baseline covariates resulted in an adjusted HR of 1.00, suggesting that the initial trend observed in the unadjusted analysis might be attributed to unbalanced baseline covariates across the two groups (Fig. 3). The pharmacokinetics and biomarker data are shown in the supplementary information (Supplementary Fig. S1, Table S2).
4 Discussion
This phase 1b/2 study aimed to evaluate the MTD, preliminary efficacy and safety of alpelisib, an α-isoform-specific PI3Ki, in combination with cetuximab in adult patients with recurrent/metastatic HNSCC who were resistant/ineligible/intolerant to platinum-based chemotherapy. In the first-in-human trial of alpelisib in patients with PIK3CA-mutant advanced solid tumours, disease control was achieved in 13/19 patients with PIK3CA-mutant HNSCC [15]. Based on these findings and the strong synergy between cetuximab and alpelisib in pre-clinical HNSCC models, the current study was designed.
Phase 1b included patients who received prior treatment with an EGFR-targeted antibody for recurrent/metastatic disease. The RP2D of alpelisib was determined as 300 mg/d (as whole-tablet or drinkable suspension) in combination with cetuximab.
Alpelisib displays a predictable pharmacokinetic profile; systemic exposure appears dose-proportional within the tested range [16]. We found no significant difference in the pharmacokinetic profiles, despite administering two formulations (tablet and dispersible tablet) in three service forms (oral tablet, crushed tablet suspension and dispersible solution/suspension) and two routes (orally versus intragastric); hence, the disintegration of alpelisib tablet in the stomach is unlikely to be a rate-limiting step in the absorption process.
The alpelisib–cetuximab combination safety results were consistent with previous safety data from the respective single agents and aligned with the safety profile of alpelisib in other combination trials, [5, 14, 15, 17]. Additionally, the safety profile in patients with HNSCC was similar in phases 1b and 2 irrespective of prior cetuximab treatment. The most frequently reported AE, hyperglycaemia, is considered an on-target effect of PI3K inhibition, given the role of the PI3K pathway in glucose homeostasis. The hyperglycaemia (onset during the first few weeks of treatment) was generally manageable with anti-hyperglycaemic agents and dose adjustments. The skin-related events, including rash, dermatitis acneiform and dry skin, are also considered a class effect of PI3K/mTOR inhibitors. Gastrointestinal events were managed with concomitant medications and dose adjustments. Higher incidences of asthenia/fatigue, nausea and rash observed with the combination regimen versus single agents might result from overlapping toxicities of the EGFR and PI3K inhibitors. The death rates were comparable between the alpelisib–cetuximab versus the cetuximab arms and appear not unusual given the relapsed refractory population (patients ≥ 2 prior treatment regimens: 40.8% and 31.4%, respectively).
In this study, both whole-tablet and drinkable suspension of alpelisib with cetuximab showed good tolerability and comparable pharmacokinetics, and hence included in phase 2 to evaluate anti-tumour efficacy. In the primary analysis, the alpelisib–cetuximab combination did not demonstrate PFS benefit by central review versus cetuximab alone (Bayesian method; HR, 0.99). In cetuximab-naïve patients, no significant trend for unadjusted PFS in favour of the combination was observed by central review, but significant improvement of PFS was shown while adjusting for baseline covariates including WBC, haemoglobin and SLD. PFS based on investigator assessment showed a trend to favour the combination treatment arm (unadjusted HR, 0.76; 95% CI 0.49–1.19). In cetuximab-resistant patients, moderate anti-tumour activity was observed with true median PFS of 3.9 months. Some patients with platinum-refractory disease had confirmed PRs (ORR, ~10% with the combination).
Interestingly, these responses were observed in tumours harbouring multiple underlying molecular-activation mechanisms. However, combination treatment failed to demonstrate any improvement in OS. OS results similar to PFS were obtained when the model was adjusted for baseline covariates (adjusted HR, 1.00), suggesting that the initial trend observed in the unadjusted analysis could be explained by unbalanced baseline covariates across the two groups [17].
Treatment with PI3Ki leads to upregulation of receptor tyrosine kinase (RTKs) including EGFR, that, in turn, can limit the antitumor effects of PI3Ki [18, 19]. Hence, PI3K inhibitors used as single agents might not elicit durable responses, and thus provides the rationale to test combinations of PI3Ki with agents that block the activity of RTKs. Combinations of alpelisib and EGFR inhibitor have shown synergistic effect in HNSCC cell lines, specifically decreased the mTOR activity. Additionally, in an in vitro study, the combination of cetuximab and alpelisib induced over 50% growth inhibition in 85% of the HNSCC lines tested regardless of PIK3CA mutational status, suggesting that this combination is both broadly synergistic and efficacious [20]. The pan-PI3Ki buparlisib–cetuximab combination also exerted a synergistic antiproliferative effect in vitro in HNSCC. Clinical studies have evaluated the combination of buparlisib–cetuximab and copanlisib–cetuximab [21,22,23]. PI3Ki PX-866 with cetuximab showed a reduction of tumours in human xenograft models [24], but clinical data have not shown improvement in PFS or OS [25, 26].
At the time the study was conducted (first patient first visit November 2012), platinum-based chemotherapy was the standard first-line treatment for inoperable recurrent or metastatic HNSCC. Immune checkpoint inhibitors, anti-programmed cell death-1 therapy and Pi3K blockade together induced a synergistic effect in HNSCC animal models [27]. Immunotherapies have now shown major advantages in subsets of HNSCC patients, as their responses are durable with longer follow-up [28,29,30]. Immune checkpoint inhibitors are now approved for treatment of recurrent/metastatic HNSCC as first line and in platinum refractory disease [31], representing the new standard of care. Clinical studies evaluating the efficacy of PI3Ki/immune checkpoint inhibitors in combination therapy remain to be explored.
5 Conclusions
The addition of alpelisib to cetuximab did not demonstrate a PFS benefit in cetuximab-naïve patients with advanced HNSCC. The alpelisib–cetuximab combination showed moderate activity in cetuximab-resistant patients with a consistent safety profile. While no superiority of alpelisib + cetuximab combination could be demonstrated, the value of a PI3Kα inhibitor such as alpelisib in the era of immune checkpoint inhibitors might warrant further investigation, particularly in PIK3CA-mutant HNSCC.
References
Argiris A, Karamouzis MV, Raben D, Ferris RL. Head and neck cancer. Lancet. 2008;371(9625):1695–709.
Malone E, Siu LL. Precision medicine in head and neck cancer: myth or reality? Clin Med Insights Oncol. 2018;12:1179554918779581.
National Comprehensive Cancer Network clinical practice guideline in oncology (NCCN guideline®). Head and neck cancers. Version 1.2018—Feb 15, 2018. https://oncolife.com.ua/doc/nccn/Head_and_Neck_Cancers.pdf. Accessed 3 June 2022.
Harari PM. Epidermal growth factor receptor inhibition strategies in oncology. Endocr Relat Cancer. 2004;11(4):689–708.
Vermorken JB, Trigo J, Hitt R, Koralewski P, Diaz-Rubio E, Rolland F, et al. Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. J Clin Oncol. 2007;25(16):2171–7.
Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, et al. Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014;13(5):1117–29.
Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–44.
Huang WC, Hung MC. Induction of Akt activity by chemotherapy confers acquired resistance. J Formos Med Assoc. 2009;108(3):180–94.
Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011;13(6):224.
Andre F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med. 2019;380(20):1929–40.
Juric D, Janku F, Rodon J, Burris HA, Mayer IA, Schuler M, et al. Alpelisib plus fulvestrant in PIK3CA-altered and PIK3CA-wild-type estrogen receptor-positive advanced breast cancer: a phase 1b clinical trial. JAMA Oncol. 2019;5(2): e184475.
Elkabets M, Pazarentzos E, Juric D, Sheng Q, Pelossof RA, Brook S, et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell. 2015;27(4):533–46.
Sheng Q, Wang H, Das R, Chen Y, Liang J, Cao A, et al. Abstract 4261: targeting HER3 and PI3K in head and neck squamous cancer cells. Can Res. 2013;73(8 Supplement):4261.
Dunn L, Riaz N, Baxi SS, McBride SM, Sherman EJ, Michel L, et al. Phase Ib study of cetuximab + BYL719 + IMRT in stage III-IVB head and neck squamous cell carcinoma (HNSCC). J Clin Oncol. 2017;35(15):6081.
Juric D, Rodon J, Tabernero J, Janku F, Burris HA, Schellens JHM, et al. Phosphatidylinositol 3-kinase alpha-selective inhibition with alpelisib (BYL719) in PIK3CA-altered solid tumors: results from the first-in-human study. J Clin Oncol. 2018;36(13):1291–9.
Gonzalez-Angulo AM, Juric D, Argilés G, Schellens JH, Burris HA, Berlin J, et al. Safety, pharmacokinetics, and preliminary activity of the α-specific PI3K inhibitor BYL719: results from the first-in-human study. J Clin Oncol. 2013;31(15):2531.
Razak ARA, Ahn M-J, Yen C-J, Solomon BJ, Lee S-H, Wang H-M, et al. Phase lb/ll study of the PI3Kα inhibitor BYL719 in combination with cetuximab in recurrent/metastatic squamous cell cancer of the head and neck (SCCHN). J Clin Oncol. 2014;32(15):6044.
Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71.
Tao JJ, Castel P, Radosevic-Robin N, Elkabets M, Auricchio N, Aceto N, et al. Antagonism of EGFR and HER3 enhances the response to inhibitors of the PI3K-Akt pathway in triple-negative breast cancer. Sci Signal. 2014;7:29.
Sheng Q, Wang H, Das R, Chen Y, Liang J, Cao A, et al. Targeting HER3 and PI3K in head and neck squamous cancer cells. Cancer Res. 2013;73(8):4261.
Bozec A, Ebran N, Radosevic-Robin N, Chamorey E, Yahia HB, Marcie S, et al. Combination of phosphotidylinositol-3-kinase targeting with cetuximab and irradiation: a preclinical study on an orthotopic xenograft model of head and neck cancer. Head Neck. 2017;39(1):151–9.
Lattanzio L, Tonissi F, Monteverde M, Vivenza D, Russi E, Milano G, et al. Treatment effect of buparlisib, cetuximab and irradiation in wild-type or PI3KCA-mutated head and neck cancer cell lines. Invest New Drugs. 2015;33(2):310–20.
Clinicaltrials.gov [NCT01816984]. PI3K Inhibitor BKM120 and Cetuximab in treating patients with recurrent or metastatic head and neck cancer. Available from: https://clinicaltrials.gov/ct2/show/NCT01816984. Accessed 18 Sept 2020.
Keysar SB, Astling DP, Anderson RT, Vogler BW, Bowles DW, Morton JJ, et al. A patient tumor transplant model of squamous cell cancer identifies PI3K inhibitors as candidate therapeutics in defined molecular bins. Mol Oncol. 2013;7(4):776–90.
Jimeno A, Bauman JE, Weissman C, Adkins D, Schnadig I, Beauregard P, et al. A randomized, phase 2 trial of docetaxel with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic head and neck squamous cell cancer. Oral Oncol. 2015;51(4):383–8.
Jimeno A, Shirai K, Choi M, Laskin J, Kochenderfer M, Spira A, et al. A randomized, phase II trial of cetuximab with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic head and neck squamous cell cancer. Ann Oncol. 2015;26(3):556–61.
Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375(19):1856–67.
Massarelli E, William W, Johnson F, Kies M, Ferrarotto R, Guo M, et al. Combining immune checkpoint blockade and tumor-specific vaccine for patients with incurable human papillomavirus 16-related cancer: < phase 2 clinical trial. JAMA Oncol. 2019;5:67–73.
Mehra R, Seiwert TY, Gupta S, Weiss J, Gluck I, Eder JP, et al. Efficacy and safety of pembrolizumab in recurrent/metastatic head and neck squamous cell carcinoma: pooled analyses after long-term follow-up in KEYNOTE-012. Br J Cancer. 2018;119:153–9.
Gettinger S, Horn L, Jackman D, Spigel D, Antonia S, Hellmann M, et al. Five-year follow-up of nivolumab in previously treated advanced non-small-cell lung cancer: results from the CA209-003 study. J Clin Oncol. 2018;36:1675–84.
Botticelli A, Cirillo A, Strigari L, Valentini F, Cerbelli B, Scagnoli S, et al. Anti-PD-1 and Anti-PD-L1 in head and neck cancer: a network meta-analysis. Front Immunol. 2021;12: 705096.
Acknowledgements
We thank the patients who participated in this trial and their families, as well as the staff members at individual trial centres who provided support. Editorial assistance with the preparation of this manuscript was provided by Lakshmi Kasthurirangan and Avinash Yerramsetti, both of Novartis Healthcare Pvt Ltd.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by Novartis Pharmaceuticals AG.
Conflicts of interest
Dr. Abdul Razak reports grants from Novartis, during the conduct of the study. Dr. Wang has nothing to disclose. Dr. Chang has nothing to disclose. Dr. Ahn has nothing to disclose. Dr. Munster has nothing to disclose. Dr. Blumenschein, Jr reports personal fees from Abbvie, personal fees from Adicet, personal fees from Amgen, personal fees from Ariad, grants and personal fees from Bayer, personal fees from Clovis Oncology, grants from Adaptimmune, grants from Elelixis, grants from GlaxoSmithKline, grants from Immatics, grants from Immunocore, grants from Incyte, grants from Kite Pharma, grants from Macrogenics, grants from Torque, grants and personal fees from AstraZenaca, grants and personal fees from Bristol-Myers Squibb, grants and personal fees from Celgene, grants and personal fees from Genetech, grants and personal fees from MedImmune, grants and personal fees from Merck, grants and personal fees from Novartis, grants and personal fees from Roche, grants and personal fees from Xcovery, personal fees from Virogin Biotech, personal fees from Johnson & Johnson/Janssen, personal fees from Maverick Therapeutics, outside the submitted work. Dr. Solomon has nothing to disclose. Dr. Lim has nothing to disclose. Dr. Hong has nothing to disclose. Dr. Pfister reports grants and non-financial support from Novartis, during the conduct of the study; personal fees from Boehringer Ingelheim, outside the submitted work. Dr. Saba reports grants and personal fees from Merck, GSK, BMS and Rakuten, outside the submitted work. Dr. Lee reports personal fees from AstraZeneca/MedImmune, personal fees from Roche, grants and personal fees from Merck, personal fees from Novartis, outside the submitted work. Dr. van Herpen has nothing to disclose. Dr. Quadt is a paid employee of Novartis Pharma AG (Study Sponsor). Dr. Bootle is a paid employee of Novartis Pharma AG (Study Sponsor). Dr. Blumenstein is a paid employee Novartis Institute for Biomedical Research (Study Sponsor). Demanse paid employee of Novartis Pharma AG (Study Sponsor). Dr. Delord has nothing to disclose.
Authors’ contributions
ARA-R, CQ, DB and DD developed the study design. DD collected data. All authors critically reviewed and approved final draft and are accountable for accuracy and integrity.
Data availability
The datasets that support the findings of this study are available from the corresponding (Dr Razak) upon reasonable request.
Ethical Approval
Independent Review Board / Ethics Committee reviewed and approved all study materials prior to initiating patient recruitment and data collection at the participating Institutions.
Consent to participate
Written Informed Consent was obtained from all study participants prior to initiation any study activities.
Consent to publish
All authors provided consent for publication.
Additional information
Previous presentations: Razak AR et al. Phase 1b/2 study of the PI3Kα inhibitor BYL719 in combination with cetuximab in recurrent/metastatic squamous cell carcinoma of head and neck. Journal of Clinical Oncology 32, 15 suppl (May 20, 2014) 6044–6044.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Razak, A.R.A., Wang, HM., Chang, JY. et al. A Phase 1b/2 Study of Alpelisib in Combination with Cetuximab in Patients with Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma. Targ Oncol 18, 853–868 (2023). https://doi.org/10.1007/s11523-023-00997-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-023-00997-z