
Abstract
Transforming growth factor β-induced protein (TGFBIp), as an extracellular matrix protein, is expressed TGF-β in some types of cells. Experimental sepsis is mediated by expressed and released TGFBIp in primary human umbilical vein endothelial cells (HUVECs). Cornuside (CNS) is a bisiridoid glucoside compound found in the fruit of Cornus officinalis SIEB. et ZUCC. Based on the known functions of CNS, such as the immunomodulatory and anti-inflammatory activities, we tested whether TGFBIp-mediated septic responses were suppressed by CNS in human endothelial cells and mice and investigated the underlying anti-septic mechanisms of CNS. Data showed that the secretion of TGFBIp by lipopolysaccharide (LPS) and severe septic responses by TGFBIp were effectively inhibited by CNS. And, TGFBIp-mediated sepsis lethality and pulmonary injury were reduced by CNS. Therefore, the suppression of TGFBIp-mediated septic responses by CNS suggested that CNS may be used as a potential therapeutic agent for several vascular inflammatory diseases, with the inhibition of the TGFBIp signaling pathway as the mechanism of action.
Graphical abstract

Similar content being viewed by others
Introduction
Transforming growth factor β-induced protein (TGFBIp), as an extracellular matrix protein, contains the N-terminal secretion signal peptide, followed by a cysteine-rich domain, four internal homologous repeats (FAS1 domain), and a C-terminal Arg–Gly–Asp (RGD) motif [1]. Previous reports showed the biological functions of TGFBIp such as cell growth, cell differentiation, wound healing, tumorigenesis, and apoptosis [2,3,4]. We previously reported that TGFBIp is a promising therapeutic target for the treatment of sepsis [5, 6] and that the blood levels of acetylated 676th lysine TGFBIp (TGFBIp K676Ac) was elevated in patients with the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pneumonia [7]. In addition, TGFBIp acts as a lethal mediator in sepsis conditions in which serum TGFBIP levels are significantly elevated, and blocking TGFBIp even after the onset of infection has been shown to rescue mice from lethal [5, 6].
Cornuside (CNS, Fig. 1A), a secoiridoid glucoside, is found in the fruit of Cornus officinalis Sieb. et Zucc., which has been used as a traditional herbal medicine to treat inflammatory diseases and activate blood circulation. The crude extract of fruit of C. officinalis has various pharmacological actions, such as anti-neoplastic, anti-inflammatory, hepatoprotective, and anti-diabetic nephropathy [8, 9]. Studies also showed that the CNS inhibits the expression of cytokine-induced infectious and adhesion molecules in human endothelial cells and protects cultured murine cortical cells from damage due to oxygen–glucose deprivation [8, 9]. Based on previous potential functions of TGFBIp on the vascular inflammatory response [2, 5,6,7] and the known beneficial effects of the CNS [8, 9], we hypothesized that treatment with the CNS would be effective in inhibiting TGFBIp- or cecal ligation and puncture (CLP)-induced septic responses, respectively. To investigate the effects of the CNS on TGFBIp protein secretion and TGFBIp mRNA expression, human umbilical vein endothelial cells (HUVECs) were exposed to lipopolysaccharide (LPS) was used and to investigate the effects of CNS in TGFBIp-mediated septic responses, we assessed hyperpermeability, expression of cell adhesion molecules (CAMs), and adhesion and migration of leukocytes to the HUVECs. For the in vivo evaluation, after intravenous injection of TGFBIp into male C57BL/6 mice, the effects of CNS on leukocyte migration, mortality and lung injury were measured.

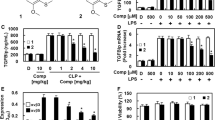
Structures of collismycin C (CNS) and its effects on the release of TGFBIp and expression of receptors. A Structures of CNS. B HUVECs were treated with the indicated concentrations of CNS for 6 h after stimulation with LPS (100 ng/ml, 1 h), and TGFBIp release was measured by ELISA. C Male C57BL/6 mice that underwent CLP were intravenously administered CNS at 0.08–0.8 mg/kg 12 h after CLP (n = 5). Mice were euthanized 24 h after CLP. Serum TGFBIp levels were measured by ELISA. D The same procedure as in (B) except that real-time qRT-PCR analysis was performed using specific primers for TGFBIp and actin, as described in the materials and methods section. E Confluent HUVECs were activated with LPS (100 ng/ml, 3 h), followed by incubation with CNS for 6 h. Expression of αv β3 (white column) and αv β5 (black column) was determined by cell-based ELISA. F Effect of CNS on cellular viability was measured by the MTT assay. D = 0.2% DMSO (vehicle control). Results are expressed as the means ± SD of at least 3 independent experiments (n = 5/group). *p < 0.05 relative to DMSO (E left), LPS (B, D, E right) or CLP (C), one-way ANOVA
Results
Effects of CNS on LPS- and CLP-mediated release of TGFBIp
Our previous study showed the stimulation of TGFBIp release by LPS in HUVECs and showed that 100 ng/ml LPS was sufficient to induce TGFBIp release [5, 6], which was consistent to in this study (Fig. 1B). To investigate the effect of CNS on LPS-mediated secretion of TGFBIp, HUVECs were stimulated with 100 ng/ml LPS for 1 h and then treated with increasing CNS concentration for 6 h. As shown in Fig. 1B, CNS inhibited the release of TGFBIp from HUVECs, and the optimal effective concentration was > 5 μM. Also, in the absence of LPS pretreatment, CNS had no effect on TGFBIp release (Fig. 1B). To confirm this effect in vivo, CLP-induced septic mice was applied. As shown in Fig. 1C, treatment with CNS resulted in marked inhibition of CLP-induced TGFBIp release. Because the mean circulating blood volume of mice was 72 ml/kg [10], the mean body weight of mice used in the study was 27 g, and the average blood volume was 2 ml in each mouse, the amount of injected CNS (0.08, 0.2, 0.4 or 0.8 mg/kg) yield a maximum concentration of 2, 5, 10, and 20 μM, respectively, in the peripheral blood.
To determine the molecular mechanism by which CNS inhibits LPS-mediated release of TGFBIp, we tested the effect of the CNS on the transcriptional regulation of TGFBIp by LPS in HUVECs. Therefore, real-time qRT-PCR was used to measure the effect of CNS on LPS-induced TGFBIp mRNA levels. As shown in Fig. 1D, LPS induced an increase in the expression levels of TGFBIp mRNA, and treatment with CNS resulted in decreased expression levels of LPS-induced TGFBIp mRNA. Next, we investigated the effects of CNS on the expression of the TGFBIp receptors integrins αv β3 and αv β5 in HUVECs. Data showed that LPS induced an increase in the expression level of TGFBIp mRNA, and treatment with CNS resulted in a decrease in the expression level of LPS-induced TGFBIp mRNA (Fig. 1D). Next, the effects of the CNS on TGFBIp receptors (integrins αv β3 and αv β5) expression were investigated. As shown in Fig. 1E, treatment with LPS increased the expression of αv β5 more than fourfold but not αv β3 in HUVECs, and treatment with CNS significantly suppressed expression of αv β5, indicating that the inhibitory effect of the CNS on the release of TGFBIp was mediated by the inhibition of the TGFBIp receptor (integrin αv β5). To evaluate CNS cytotoxicity, cell viability assays were performed on CNS-treated HUVECs for 6 h (Fig. 1F). At concentrations up to 100 μM, the CNS had no effect on cell viability (Fig. 1F).
Effect of CNS on TGFBIp-mediated vascular barrier disruption
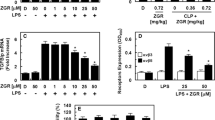
Permeability tests were performed to determine the effect of the CNS on the barrier integrity of HUVECs. Treatment with only 20 μM CNS did not alter barrier integrity (Fig. 2A). In contrast, treatment with CNS resulted in a dose-dependent decrease in TGFBIp-mediated barrier integrity (Fig. 2A). To confirm this vascular barrier protective effect of CNS in vivo, TGFBIp-mediated vascular permeability was evaluated in mice. As shown in Fig. 2B, treatment with CNS markedly inhibited TGFBIp-induced peritoneal leakage of dye. It is known that sepsis inducers such as high mobility group box 1 (HMGB1) and LPS induce an anti-inflammatory response by promoting phosphorylation of p38 mitogen-activated protein kinase (MAPK) [11,12,13,14,15]. Therefore, we investigated whether TGFBIp can promote phosphorylation of P38 MAPK and, if so, whether the CNS inhibits TGFBIp-induced activation of P38 MAPK in HUVECs. As shown in Fig. 2C, TGFBIp induced the activation of P38 MAPK, which was significantly inhibited by treatment by the CNS. These findings demonstrate the inhibition of TGFBIp-mediated endothelial destruction by CNS and maintenance of human endothelial barrier integrity in TGFBIp-treated mice.

Effects of CNS on TGFBIp-mediated permeability in vitro and in vivo. The effects of post-treatment with different concentrations of CNS for 6 h on the barrier disruptions caused by TGFBIp (A, 5 μg/ml, 6 h) were monitored by measuring the flux of Evans blue-bound albumin across HUVECs. B The effects of CNS at 0.08–0.8 mg/kg on TGFBIp-induced (0.1 mg/kg, iv.) vascular permeability in mice were examined by measuring the amount of Evans blue in peritoneal washings (expressed μg/mouse, n = 5/group). C HUVECs were activated with TGFBIp (5 μg/ml, 6 h), followed by treatment with different concentrations of CNS for 6 h. The effects of CNS on the TGFBIp-mediated expression of phospho-p38 were determined by ELISA. Results are expressed as the means ± SD of at least 3 independent experiments (n = 5/group). *p < 0.05 relative to TGFBIp, one-way ANOVA
Effects of CNS on TGFBIp-mediated cell adhesion molecules (CAMs) expression, neutrophil adhesion, and migration
Next, we tested whether CNS could inhibit the surface expression of vascular cell adhesion molecule (VCAM), intercellular adhesion molecule-1 (ICAM), and E-selectin induced by TGFBIp. Data showed that TGFBIp-induced upregulation of the surface expression of VCAM (Fig. 3A), ICAM (Fig. 3B), and E-selectin (Fig. 3C), which was inhibited by CNS. In addition, treatment with CNS resulted in downregulation of human neutrophil adsorption and subsequent migration through activated endothelial cells in a concentration-dependent manner (Figs. 3D, E). To confirm these effects in vivo, we investigated the TGFBIp-induced migration of leukocytes in mice. TGFBIp was found to stimulate migration of leukocytes into the peritoneal cavity of mice, and treatment with CNS markedly reduced the peritoneal leukocyte count (Fig. 3F).

Effects of CNS on TGFBIp-mediated proinflammatory responses. A–C, HUVECs were stimulated with TGFBIp (5 μg/ml) for 6 h, followed by treatment with CNS for 6 h. TGFBIp-mediated A expression of VCAM (white column), ICAM (gray column), and E-selectin (black column) in HUVECs, B adherence of human neutrophils to HUVEC monolayers, and C migration of human neutrophils through HUVEC monolayers were analyzed. D The effects of post-treatment with CNS at 0.08–0.8 mg/kg on leukocyte migration into the peritoneal cavities of mice caused by TGFBIp (0.1 mg/kg, i.v.) were analyzed. Results are expressed as the means ± SD of at least 3 independent experiments (n = 5/group). *p < 0.05 relative to TGFBIp, one-way ANOVA
Effects of CNS on TGFBIp-stimulated activation of nuclear factor (NF)-κB and extracellular regulated kinase (ERK)
To understand the mechanism of inhibition of CNS on TGFBIp-mediated sepsis, the effect of CNS on TGFBIp-induced activation of NF-κB and ERK 1/2 was evaluated. HUVECs were activated with TGFBIp for 6 h, and then incubated with CNS for 6 h. TGFBIp enhanced the activation of NF-κB and ERK 1/2, and these increases were significantly reduced by treatment with CNS (Fig. 4A–C). The Raf-MAPK kinase (MEK)-ERK signaling cascade and downstream c-Jun-N-terminal kinase (JNK) activation have been previously characterized in response to the inflammatory response [16]. Therefore, we next measured the effect of CNS on TGFBIp-induced JNK, Raf, and MEK activation. The data show that TGFBIp enhanced the activation of JNK, Raf, and MEK, which was significantly reduced by treatment with CNS (Fig. 4D–F). Therefore, these results and Fig. 2C show that CNS inhibits the Raf-MEK-ERK-JNK signaling pathway after TGFBIp challenge.

Effects of CNS on TGFBIp-stimulated activation of NF-κB and ERK 1/2. A Confluent HUVECs were activated with TGFBIp (5 μg/ml) for 6 h, incubated with CNS for 6 h, and then analyzed by western blotting for TGFBIp-mediated activation of phospho-NF-κB p65, total NF-κB p65, phospho-ERK1/2, or total ERK1/2. The images are representative of 3 separate experiments conducted on different days with similar results. B The graph shows the densitometric intensities of phospho-NF-κB p65 and phospho-ERK1/2 normalized to total NF-κB and total ERK1/2 (n = 3 blots). C The same as (A) except that the activation of JNK, Raf, and MEK was measured by ELISA. The level of phosphorylation in each treatment was normalized to its total protein. Results are expressed as the means ± SD of at least 3 independent experiments (n = 3/group). *p < 0.05 relative to TGFBIp, one-way ANOVA (B-F)
Protective effect of CNS in TGFBIp-induced septic mortality
Based on the findings of the study described above, we hypothesized that treatment with CNS would reduce mortality in the TGFBIp-induced sepsis mouse model. To investigate whether CNS protects the TGFBIp-induced sepsis-induced lethality, CNS was administered to mice after TGBIp injection. 24 h after surgery, the mice showed signs of sepsis, shivering, hair bristles, and debilitating sepsis. In animals, administration of CNS (0.4 or 0.8 mg/kg) 12 h after TGFBIp injection did not prevent TGFBIp-induced death (data not shown). Therefore, the survival rate was 40–60% after two doses (12 h after TGFBIp injection, 50 h after TGFBIp injection) (p < 0.0001, Fig. 5A). The significant survival benefit achieved by CNS administration suggests that inhibiting TGFBIp release and TGFBIp-mediated inflammatory response may be a therapeutic strategy for the management of sepsis and septic shock.

Effects of CNS on lethality or pulmonary injury after TGFBIp injection. A Male C57BL/6 mice (n = 20) were administered CNS at 12 h and 50 h after TGFBIp (0.1 mg/kg, iv.) injection. Animal survival was monitored every 6 h after TGFBIp injection for 132 h. TGFBIp injection mice (filled circle) and sham mice (unfilled circle) were administered sterile saline (n = 20/group). The Kaplan–Meier survival analysis was used for determination of overall survival rates versus TGFBIp-injected mice. B The same procedure as in (A) except that mice were euthanized 96 h after TGFBIp injection. Histopathological scores of the lung tissue were recorded as described in the materials and methods section. Results are expressed as the means ± SD of at least 3 independent experiments (n = 5/group). *p < 0.05 relative to TGFBIp, one-way ANOVA. C Photomicrographs of lung tissues (H&E staining, ×200). Sham group (grade 1), TGFBIp group (grade 4), TGFBIp + CNS (0.4 mg/kg) (grade 2), and TGFBIp + CNS (0.8 mg/kg) (grade 1). Arrows indicate leukocyte infiltration. The illustrations are representative images from 3 independent experiments
Protective effect of CNS against TGFBIp-induced pulmonary injury
To confirm the protective effect of CNS against death by TGFBIp injection, the effect of the CNS on lung damage, which is one of the major organs damaged by sepsis, was measured. There were no significant differences between sham lung and sham lung + CNS mice in microscopic light observation (data not shown). In the TGFBIp-injected group, interstitial edema with massive infiltration of inflammatory cells into the interstitial and alveolar spaces was observed and the lung structures were severely damaged (Fig. 5B, C). These morphological changes were less pronounced in the TGFBIp + CNS group (Fig. 5B, C).
Discussion
In this study, we established the following hypotheses: Treatment with CNS will inhibit TGFBIp-induced sepsis response in human endothelial cells and CLP-induced sepsis in mice. Data showed that CNS inhibited LPS-induced secretion and mRNA expression of TGFBIp or CLP-induced TGFBIp secretion, inhibited TGFBIp-mediated hyperpermeability, p38, NF-κB, Raf-MEK-ERK-JNK signaling cascade activation, upregulation of CAM expression, leukocytes of HUVECs were inhibited. Finally, CNS was protected from TGFBIp-induced lethality and lung injury in vivo. All these results are consistent with our hypothesis.
The anti-septic functions of CNS on TGFBIp-mediated sepsis could be mediated by inhibition of TGFBIp release and inhibition of TGFBIp mRNA transcription (Fig. 1B–D), and TGFBIp-mediated hyperpermeability (Fig. 2A, B) via suppression of p38 activation (Fig. 2C). Noting that the LPS-induced transcriptional expression (Fig. 1D) and translational (Fig. 1B) levels of TGFBIp were repressed by CNS, the transcriptional stability of TGFBIp mRNA was maintained by LPS, which was repressed by CNS. Also, as shown in Fig. 1E, the LPS-induced expression of integrin αv β5 was suppressed by CNS. These results indicated that integrin αv β5 is an important regulator of the vascular endothelial leakage response in sepsis, suggesting that a function-blocking antibody against integrin αv β5 prevented the development of pulmonary vascular permeability and animal acute pulmonary disease models [6, 17, 18]. In addition, noting that high plasma concentrations of TGFBIp in sepsis patients correlate with the severity of sepsis [6], and pharmacological inhibition of TGFBIp improves survival in sepsis animal models [5], the prevention of CLP-induced release of TGFBIp by the CNS (Fig. 2) indicates the potential utility of the CNS in the treatment of vascular inflammatory diseases.
Several studies have reported that TGFBIp enhanced the expression of CAMs, such as VCAM, ICAM, and E-selectin, on the surfaces of human cells, thereby promoting adhesion and migration of leukocytes across the endothelium to sites of inflammation [2, 6, 19, 20]. And, previous studies have reported that TGFBIp enhances the expression of CAMs such as VCAM, ICAM, and E-selectin on the human cell surface, promoting leukocyte adhesion and migration across the endothelium to sites of inflammation [2, 6, 19, 20]. According to the current findings, TGFBIp-induced upregulation of the surface expression of VCAM, ICAM, and E-selectin, which was suppressed by CNS suggesting that the inhibitory effects of CNS on the expression of CAMs are mediated through attenuation of the TGFBIp signaling pathway by CNS. And, increased CAMs expression enhances binding of human neutrophils to TGFBIp-activated endothelial cells and subsequent migration. These results suggest that CNS not only inhibits the release of endotoxin-mediated TGFBIp from endothelial cells, but also downregulates the proinflammatory signaling effect caused by the release of TGFBIp, thereby inhibiting the expansion of inflammatory pathways by nuclear cytokines. Experiments on CAM have been extensively performed in vitro to study the regulation of interactions between leukocytes and endothelial cells [21, 22]. In this study, TGFBIp-induced levels of VCAM, ICAM, and E-selectin were down-regulated by CNS treatment, which was shown to inhibit adhesion and migration of leukocytes to the inflammatory endothelium.
Consistent with previous reports of interfering with TGFBIp, it is essential for leukocyte adhesion and migration through interaction with integrin αv β5 [23, 24]. In addition, the inhibitory effect of CNS on the interaction between leukocytes and endothelial cells is mediated by the suppression of the expression of CAMs such as VCAM, ICAM, and E-selection (Fig. 3). The underlying mechanism of the anti-inflammatory effects of CNS was downregulation activation of the inflammatory transcription factors NF-κB and Raf-MEK-ERK-JNK signaling cascades (Fig. 4). A possible major target of CNS in TGFBIp-mediated sepsis may be the interaction between released TGFBIp and its receptor (integrin αv β5). This is because the binding of the ligand TGFBIp to the receptor mediates a severe downstream vascular inflammatory response towards the leukocyte terminal [5, 6].
The present findings support the potential of CNS as a treatment for sepsis and septic shock. Although the herbal drug candidate has the advantage of being cheap in terms of price, the main limitation of this study was the inability to determine the pharmacokinetics of CNS. After intravenous injection by CNS, it undergoes excretion, which accounts for absorption, distribution, metabolism, and processing of drug compounds within the organism. All four of these criteria influence the level, kinetics, and overall performance of the drug. Therefore, further studies are needed to elucidate the pharmacokinetic properties of CNS in vivo.
Our results show that CNS inhibits TGFBIp receptor expression (integin αv β5) and TGFBIp-mediated barrier dysfunction through barrier integrity and inhibition of CAM expression. Moreover, CNS reduces human neutrophil adhesion and migration to HUVECs. This barrier protective effect of CNS was confirmed in a mouse model in which treatment with CNS resulted in a reduction in TGFBIp-induced mortality. Our findings show that CNS is worthy of consideration as a potential treatment for severe vascular inflammatory diseases such as sepsis and septic shock.
Experimental
Reagents
CNS, Evans blue, and crystal violet were obtained from Sigma (St. Louis, MO). Vybrant DiD (used at 5 μM) was obtained from Invitrogen (Carlsbad, CA). TGFBIp protein was purified as described previously [6].
Cell culture
Primary HUVECs were obtained from Cambrex Bio Science (Charles City, IA) and maintained as described previously [25,26,27]. All experiments were performed with HUVECs in passages 3–5. Human neutrophils were freshly isolated from whole blood (15 ml) obtained by venipuncture from 5 healthy volunteers and maintained as previously described [28, 29]. The study protocol (KNUH 2019-01-010) was approved by the IRB of Kyungpook National University Hospitals (Daegu, Republic of Korea).
Animals and husbandry
Male C57BL/6 mice (6–7 weeks old, weight 27 g) were purchased from Orient Bio (Seongnam, Korea) and used after 12 days of adaptation. Animals were housed five per polycarbonate cage under temperature (20–25 °C) and humidity (40–45%), 12 h 12 h light:dark cycle. Animals received normal rodent pellet food and water ad libitum during acclimatization. Mice were euthanized by CO2 anesthesia after cervical dislocation [30]. All animals were treated in accordance with the Guidelines for the Care and Use of Laboratory Animals issued by Kyungpook National University (IRB No. KNU 2016–54).
CLP
In this study, CLP-induced septic mouse model was used because it is more similar to human sepsis than LPS-induced endotoxemia, were used [31]. For sepsis induction, male mice were first anesthetized in a breathing room with 2% isoflurane (Poran, JW Pharmaceutical, Korea) in oxygen delivered through a small rodent gas anesthesia machine (RC2, Vetequip, Pledon, CA), and then through a face mask. They were able to breathe spontaneously during the operation. CLP-induced sepsis model was prepared as previously described [28, 29]. This protocol was approved by the Animal Care Committee at Kyungpook National University prior to conducting the study (IRB No. KNU 2016–54).
Cell viability assay
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma) was used as an indicator of cell viability as previously described [26, 28]. Cells were grown to a density of 5 × 103 cells/well in 96-well plates. After 24 h, the cells were washed with fresh medium and treated with CNS. After 6 h, the cells were washed and incubated for 4 h after adding 100 μl of MTT (1 mg/ml). Finally, DMSO (150 µL) was added to dissolve the amount of the resulting formazan salt. Adjust the optical density (OD) to 540 nm using a microplate reader (Tecan Austria GmbH, Austria).
Enzyme-linked immunosorbent assay (ELISA) for TGFBIp
TGFBIp concentrations in cell culture media or mouse serum were determined by competitive ELISA, as described previously [2, 6]. 96-well plastic plate microtiter plates (Corning, NY) were coated with TGFBIp protein in 20 mM carbonate–bicarbonate buffer (pH 9.6) with 0.02% sodium azide overnight at 4 °C. Plates were rinsed three times with phosphate buffered saline (PBS)-0.05% Tween20 (PBS-T) and then kept at 4 °C. The lyophilized culture medium was pre-injected with anti-TGFBIp antibody (PBS-T 1:2000 dilution) in 96-well plastic circular microtiter plates for 90 min at 37 °C. Pre-incubated samples were transferred to pre-coated plates and incubated for 30 min at room temperature. Plates were rinsed 3 times with PBS-T and then incubated for 90 min with peroxidase-conjugated anti-rabbit IgG antibody (PBS-T, diluted 1:2000 min from Amersham Pharmacya Biotech) at room temperature. After rinsing the plate 3 times with PBS-T, it was incubated for 60 min at room temperature in the dark with 200 μl substrate solution (100 μg/ml o-phenylenediamine, 0.003% H2O2). After the reaction was stopped with 50 μl of 8 N H2SO4, the absorbance was measured at 490 nm.
In vitro permeability assay
Permeability was quantified via spectrophotometric measurements of Evans blue-bound albumin flux across functional HUVEC monolayers using a two-sphere chamber model modified as previously described [28, 29]. Briefly, HUVECs were plated (5 × 104 cells/well) in Transwells with a pore size of 3 μm and a diameter of 12 mm for 3 days. The confluent monolayers were treated with TGFBIp (5 μg/ml for 6 h) followed by incubation with CNS for 6 h.
Expression of CAMs and receptors
Expression of VCAM, ICAM, and E-selectin on HUVECs was determined by whole-cell ELISA, as described previously [28, 29]. Binding monolayers of HUVECs were treated with TGFBIp (5 μg/ml) for 6 h, followed by CNS treatment for 6 h. After removing the medium and washing the cells with PBS, they were fixed with 50 μl of 1% paraformaldehyde at room temperature for 15 min. After washing, 100 μl of murine anti-human monoclonal antibodies (VCAM, ICAM and E-selectin); Temecula, CA; 1:50 each) was applied. After 1 h (37 °C, 5% CO2), cells were washed 3 times and then 100 μl of 1:2000 peroxidase-conjugated anti-mouse IgG antibody (Sigma) was administered for 1 h. The cells were washed again 3 times and developed using O-phenylenediamine substrate (Sigma). Chromaticity analysis was performed by measuring absorbance at 490 nm. All measurements were performed in 3 wells. The same experimental procedure was used to monitor the cell surface expression of αv β3 and αv β5 using specific antibodies obtained from EMD Millipore (MA).
In vitro migration assays
Migration tests were performed in transwell plates with a diameter of 6.5 mm and a filter pore size of 8 μm. HUVECs (6 × 104) were cultured for 3 days to obtain fused endothelial monolayers. Before adding human neutrophils on top, the cell monolayer was treated with TGFBIp (5 μg/ml for 6 h) and then treated with CNS for 6 h. Cells on the top of the filter were aspirated and non-migrating cells were removed using a cotton swab. Human neutrophils at the bottom of the filter were fixed with 8% glutaraldehyde and stained with 0.25% crystal violet in 20% methanol (w/v). Each experiment was repeated in duplicate wells, and nine randomly selected high-power microscope fields (HPF, 200×) were counted within each well and expressed as a migration index.
Cell–cell adhesion assay
Adherence of human neutrophils to endothelial cells was evaluated by fluorescent labeling of human neutrophils as previously described [28, 29]. Briefly, purified human neutrophils (1.5 × 106 cells/ml, 200 μl /well) were labeled with Vybrant DiD dye and then added to washed and stimulated HUVECs. HUVEC monolayers were treated with TGFBIp (5 μg/ml) for 6 h followed by treatment with CNS for 6 h.
In vivo permeability and leukocyte migration assay
For the in vivo study, male mice were first anesthetized in a breathing room with 2% isoflurane (Forane; JW Pharmaceutical, Korea) in oxygen delivered through a small rodent gas anesthesia machine (RC2; Vetequip, Prendon, CA), and then anesthetized through a face mask. Mice were treated with TGFBIp (0.1 mg/kg, i.v.) for 6 h followed by CNS for 6 h.
For the in vivo permeability assay, each mouse was intravenously injected with a 1% Evans blue dye solution in normal saline. After 30 min, the mice were euthanized, and the peritoneal exudate was collected by washing the cavities with 5 ml of normal saline and centrifuging the supernatant at 200×g for 10 min. The upper peak absorptance was measured to be 650 nm. Vascular permeability is expressed as µg of dye/mouse measured using a standard curve as previously described [28, 29].
For evaluation of leukocyte migration [28, 29], mice were euthanized after 6 h and peritoneal cavities were washed with 5 ml of normal saline. A sample (20 μL) of the collected peritoneal fluid was mixed with 0.38 ml of Turk's solution (3% acetic acid crystal purple 0.01%), and the white blood cell count was measured under a light microscope.
Hematoxylin and eosin staining and histopathologic examination
Mice were administered with TGFBIp (0.1 mg/kg, iv.) followed by treatment with CNS at 12 h and 50 h (n = 5). Mice were euthanized 96 h after TGFBIp injection. To analyze phenotypic changes in mouse lungs [26, 28, 29], lung specimens were removed from each mouse, washed three times in PBS (pH 7.4) to remove residual blood, and 4% in PBS (pH 7.4). It was fixed in formaldehyde solution (Junsei, Tokyo, Japan) at 4 °C for 20 h. After fixation, the sample was dehydrated with a series of ethanol concentrations, embedded in paraffin, sectioned to a thickness of 4 μm, and placed on a slide. The slides were deparaffinized in an oven at 60 °C to rehydrate and stained with hematoxylin. To limit excessive staining, slides were rapidly dipped in 0.3 % acid alcohol three times and then counter-painted with eosin (Sigma). A series of ethanol and xylene were used to remove excess stain and coverlips were placed on slides. Light microscopic analysis of lung specimens was performed by blind observation to evaluate previously defined lung structures, tissue edema and inflammatory cell infiltration [32]. The results were classified into 4 grades, grade 1 indicating normal histopathology, grade 2 minimal neutrophil infiltration, grade 3 moderate neutrophil infiltration, angioedema formation, partial destruction of lung structures, grade 4 high-density neutrophil infiltration, in the form of abscession, and complete destruction of lung structures.
ELISA for phosphorylated p38 MAPK, JNK, Raf, and MEK
The activity of phosphorylated p38 MAPK (Cell Signaling Technology, Danvers, MA), JNK (R&D System, Inc., Minneapolis, MN), Raf, and MEK (RayBiotech, Norcross, GA) were quantified in accordance with the manufacturer’s instructions using a commercially available ELISA kit. The values were measured using an ELISA plate reader (Tecan, Austria GmbH, Austria).
Preparation of nuclear extracts and western blotting for NF-κB and ERK 1/2
The cells were harvested rapidly by sedimentation, and nuclear and cytoplasmic extracts were prepared on ice, as previously described [33]. Briefly, the cells were harvested and were washed with 1 ml of buffer A (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 19 mM KCl) for 5 min at 600×g. Subsequently, the cells were resuspended in buffer A, centrifuged at 600×g for 3 min, resuspended in 30 μl of buffer B (20 mM HEPES, pH 7.9, 25% glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA), rotated for 30 min at 4 °C, and centrifuged at 13,000×g for 20 min. The supernatant was the nuclear extract. The nuclear extracts were analyzed for protein content by the Bradford assay.
For western blotting, nuclear extracts were separated by electrophoresis in polyacrylamide gels of different percentages, depending on the size of the protein of interest. The gels were transferred to polyvinylidene difluoride (PVDF) membranes via semidry electrophoretic transfer at 20 mA for 2 h. The PVDF membranes were blocked for 2 h at room temperature in 5% bovine serum albumin (BSA), incubated overnight at 4 °C with a 1:500 dilution of primary antibody in Tris-buffered saline/Tween 20 (TBS-T) containing 5% BSA, and then incubated with a 1:5000 dilution of secondary antibody in TBS-T containing 1% BSA at room temperature for 1 h. After incubation, the membranes were washed 3 times with TBS-T solution, incubated with western blot enhanced chemiluminescence (ECL) detection reagents (Amersham, Piscataway, NJ), and exposed to Xomat AR films (Eastman Kodak, Rochester, NY). For western blotting of NF-kB and ERK 1/2, anti-phospho-ERK1/2, anti-total ERK1/2, anti-NF-κB p65, and anti-total NF-κB p65 antibodies (Santa Cruz) were used. Densitometry analysis was performed using the ImageJ Gel Analysis tool (NIH, Bethesda, MD).
Statistical analyses
All experiments were independently performed a minimum of 3 times. Values were expressed as the mean ± standard deviation (SD). The statistical significance of differences between test groups was evaluated by SPSS for Windows, version 16.0 (SPSS, Chicago, IL). Statistical relevance was determined by one-way analysis of variance (ANOVA) and Tukey’s post-test. Values of p < 0.05 were considered to indicate statistical significance. The survival outcomes of TGFBIp injection were assessed using Kaplan–Meier analysis.
References
Skonier J, Neubauer M, Madisen L, Bennett K, Plowman GD, Purchio AF (1992) cDNA cloning and sequence analysis of β ig-h3, a novel gene induced in a human adenocarcinoma cell line after treatment with transforming growth factor-β. DNA Cell Biol 11:511–522. https://doi.org/10.1089/dna.1992.11.511
Bae JS, Lee SH, Kim JE et al (2002) β ig-h3 supports keratinocyte adhesion, migration, and proliferation through α3β1 integrin. Biochem Biophys Res Commun 294:940–948. https://doi.org/10.1016/S0006-291X(02)00576-4
Skonier J, Bennett K, Rothwell V et al (1994) β ig-h3: A transforming growth factor-β-responsive gene encoding a secreted protein that inhibits cell attachment in vitro and suppresses the growth of CHO cells in nude mice. DNA Cell Biol 13:571–584. https://doi.org/10.1089/dna.1994.13.571
Kim JE, Kim SJ, Jeong HW et al (2003) RGD peptides released from β ig-h3, a TGF-β-induced cell-adhesive molecule, mediate apoptosis. Oncogene 22:2045–2053. https://doi.org/10.1038/sj.onc.1206269
Bae JS, Lee W, Son HN, Lee YM, Kim IS (2014) Anti-transforming growth factor β-induced protein antibody ameliorates vascular barrier dysfunction and improves survival in sepsis. Acta Physiol (Oxf) 212:306–315. https://doi.org/10.1111/apha.12398
Bae JS, Lee W, Nam JO, Kim JE, Kim SW, Kim IS (2014) Transforming growth factor β-induced protein promotes severe vascular inflammatory responses. Am J Respir Crit Care Med 189:779–786. https://doi.org/10.1164/rccm.201311-2033OC
Park HH, Kim HN, Kim H et al (2020) Acetylated K676 TGFBIp as a severity diagnostic blood biomarker for SARS-CoV-2 pneumonia. Sci Adv 6:eabc1564. https://doi.org/10.1126/sciadv.abc1564
Kang DG, Moon MK, Lee AS, Kwon TO, Kim JS, Lee HS (2007) Cornuside suppresses cytokine-induced proinflammatory and adhesion molecules in the human umbilical vein endothelial cells. Biol Pharm Bull 30:1796–1799. https://doi.org/10.1248/bpb.30.1796
Jiang WL, Chen XG, Zhu HB, Hou J, Tian JW (2009) Cornuside attenuates apoptosis and ameliorates mitochondrial energy metabolism in rat cortical neurons. Pharmacology 84:162–170. https://doi.org/10.1159/000235621
Diehl KH, Hull R, Morton D et al (2001) A good practice guide to the administration of substances and removal of blood, including routes and volumes. J Appl Toxicol 21:15–23. https://doi.org/10.1002/jat.727
Qin YH, Dai SM, Tang GS et al (2009) HMGB1 enhances the proinflammatory activity of lipopolysaccharide by promoting the phosphorylation of MAPK p38 through receptor for advanced glycation end products. J Immunol 183:6244–6250. https://doi.org/10.4049/jimmunol.0900390
Sun C, Liang C, Ren Y et al (2009) Advanced glycation end products depress function of endothelial progenitor cells via p38 and ERK 1/2 mitogen-activated protein kinase pathways. Basic Res Cardiol 104:42–49. https://doi.org/10.1007/s00395-008-0738-8
Mittelstadt PR, Salvador JM, Fornace AJ Jr, Ashwell JD (2005) Activating p38 MAPK: new tricks for an old kinase. Cell Cycle 4:1189–1192. https://doi.org/10.4161/cc.4.9.2043
Walton KL, Holt L, Sartor RB (2009) Lipopolysaccharide activates innate immune responses in murine intestinal myofibroblasts through multiple signaling pathways. Am J Physiol Gastrointest Liver Physiol 296:G601–G611. https://doi.org/10.1152/ajpgi.00022.2008
Bae JS (2012) Role of high mobility group box 1 in inflammatory disease: focus on sepsis. Arch Pharm Res 35:1511–1523. https://doi.org/10.1007/s12272-012-0901-5
Lu N, Malemud CJ (2019) Extracellular signal-regulated kinase: a regulator of cell growth, inflammation, chondrocyte and bone cell receptor-mediated gene expression. Int J Mol Sci. https://doi.org/10.3390/ijms20153792
Su G, Atakilit A, Li JT et al (2013) Effective treatment of mouse sepsis with an inhibitory antibody targeting integrin αvβ5. Crit Care Med 41:546–553. https://doi.org/10.1097/CCM.0b013e3182711b1e
Su G, Hodnett M, Wu N et al (2007) Integrin αvβ5 regulates lung vascular permeability and pulmonary endothelial barrier function. Am J Respir Cell Mol Biol 36:377–386. https://doi.org/10.1165/rcmb.2006-0238OC
Lee SH, Bae JS, Park SH et al (2003) Expression of TGF-β-induced matrix protein β ig-h3 is up-regulated in the diabetic rat kidney and human proximal tubular epithelial cells treated with high glucose. Kidney Int 64:1012–1021. https://doi.org/10.1046/j.1523-1755.2003.00158.x
Park SW, Bae JS, Kim KS et al (2004) β ig-h3 promotes renal proximal tubular epithelial cell adhesion, migration and proliferation through the interaction with α3β1 integrin. Exp Mol Med 36:211–219. https://doi.org/10.1038/emm.2004.29
Lin WN, Luo SF, Wu CB, Lin CC, Yang CM (2008) Lipopolysaccharide induces VCAM-1 expression and neutrophil adhesion to human tracheal smooth muscle cells: involvement of Src/EGFR/PI3-K/Akt pathway. Toxicol Appl Pharmacol 228:256–268. https://doi.org/10.1016/j.taap.2007.11.026
Ruiz-Torres MP, Perez-Rivero G, Rodriguez-Puyol M, Rodriguez-Puyol D, Diez-Marques ML (2006) The leukocyte-endothelial cell interactions are modulated by extracellular matrix proteins. Cell Physiol Biochem 17:221–232. https://doi.org/10.1159/000094127
Kim JE, Jeong HW, Nam JO et al (2002) Identification of motifs in the fasciclin domains of the transforming growth factor-β-induced matrix protein β ig-h3 that interact with the αv β5 integrin. J Biol Chem 277:46159–46165. https://doi.org/10.1074/jbc.M207055200
Lee BH, Bae JS, Park RW, Kim JE, Park JY, Kim IS (2006) β ig-h3 triggers signaling pathways mediating adhesion and migration of vascular smooth muscle cells through αv β5 integrin. Exp Mol Med 38:153–161. https://doi.org/10.1038/emm.2006.19
Jeong SY, Kim M, Park EK, Kim J-S, Hahn D, Bae J-S (2020) Inhibitory functions of novel compounds from dioscorea batatas decne Peel on HMGB1-mediated septic responses. Biotechnol Bioproc Eng 25:1–8. https://doi.org/10.1007/s12257-019-0382-1
Lee W, Ku SK, Kim JE, Cho GE, Song GY, Bae JS (2019) Pulmonary protective functions of rare ginsenoside Rg4 on particulate matter-induced inflammatory responses. Biotechnol Bioproc Eng 24:445–453. https://doi.org/10.1007/s12257-019-0096-4
Lee I-C, Ryu C-W, Bae J-S (2020) Novel herbal medicine C-KOK suppresses the inflammatory gene iNOS via the inhibition of p-STAT-1 and NF-κB. Biotechnol Bioproc Eng 25:536–542. https://doi.org/10.1007/s12257-020-0126-2
Kim JE, Lee W, Yang S et al (2019) Suppressive effects of rare ginsenosides, Rk1 and Rg5, on HMGB1-mediated septic responses. Food Chem Toxicol 124:45–53. https://doi.org/10.1016/j.fct.2018.11.057
Lee IC, Bae JS (2019) Pelargonidin protects against renal injury in a mouse model of sepsis. J Med Food 22:57–61. https://doi.org/10.1089/jmf.2018.4230
Carbone L, Carbone ET, Yi EM et al (2012) Assessing cervical dislocation as a humane euthanasia method in mice. J Am Assoc Lab Anim Sci 51:352–356
Buras JA, Holzmann B, Sitkovsky M (2005) Animal models of sepsis: setting the stage. Nat Rev Drug Discov 4:854–865. https://doi.org/10.1038/nrd1854
Ozdulger A, Cinel I, Koksel O et al (2003) The protective effect of N-acetylcysteine on apoptotic lung injury in cecal ligation and puncture-induced sepsis model. Shock 19:366–372. https://doi.org/10.1097/00024382-200304000-00012
Mackman N, Brand K, Edgington TS (1991) Lipopolysaccharide-mediated transcriptional activation of the human tissue factor gene in THP-1 monocytic cells requires both activator protein 1 and nuclear factor κB binding sites. J Exp Med 174:1517–1526. https://doi.org/10.1084/jem.174.6.1517
Acknowledgements
This study was financially supported by a grant of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI15C0001) and by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2020R1A2C1004131).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ryu, S.H., Kim, C., Kim, N. et al. Inhibitory functions of cornuside on TGFBIp-mediated septic responses. J Nat Med 76, 451–461 (2022). https://doi.org/10.1007/s11418-021-01601-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11418-021-01601-2