
Abstract
Purpose
This field study aimed to guide the planning of iron amendments for phosphorus retention by investigating the long-term fate of iron added to two urban lakes (Plötzensee and Groß Glienicker See) in Berlin, Germany. The contributions of iron dosing to improve lake status as well as the relevance of competing processes for management success were evaluated.
Methods
Sediment stratigraphy, as well as occurrence of iron minerals, and fluxes between water and sediment were examined using geochemical analyses (i.e. element composition, sequential extraction, X-ray diffraction, and pore water analyses). A one-box lake model was used to relate these fluxes to monitoring data from the water column and to sediment inventories.
Results
In both lakes, the added iron was preserved in the sediment. Whereas phosphorus retention increased following the addition of iron to Groß Glienicker See, sulphur was retained by the excess iron in Plötzensee. This contrasting effect is attributed to significantly different sulphate reduction rates in two lakes (Wilcoxon rank sum test: W = 25, p = 0.008). According to the one-box model, sulphate reduction explained both the decrease in measured sulphate concentrations after iron application as well as the observed increase in sulphur deposition in the sediments.
Conclusion
Management interventions involving iron amendments to enhance phosphorus retention must consider the competing process of iron sulphide formation during the entire management plan period, and additional iron may need to be applied to account for this effect.
Similar content being viewed by others

1 Introduction
Iron (Fe) dosing is a logistically and financially feasible management tool for reducing phosphorus (P) concentrations in small- and medium-sized freshwater lakes (Orihel et al. 2016). The efficiency of the method is thought only to be applicable under aerobic conditions (Lürling et al. 2016; Jilbert et al. 2020), when Fe is precipitated as ferric particles that bind P. In the hypolimnion and at the sediment–water interface (SWI) of lakes and reservoirs, aerobic conditions do not usually occur throughout the year. Importantly, several studies indicate that anaerobic conditions decrease the effectiveness of Fe amendments because of Fe reduction and subsequent dissolution of Fe and P (Lürling et al. 2016; Jilbert et al. 2020). However, it has been shown that P can also be retained by Fe under anoxic conditions, e.g. through the formation of the ferrous mineral vivianite, Fe2+3[PO4]2 · 8 H2O (Berner 1981; Rothe et al. 2016). Vivianite formation in laboratory and field studies (Rothe et al. 2014; Heinrich et al. 2021) indicates that anoxic Fe reduction and subsequent dissolution of Fe and P are not the reasons for the failure of Fe amendments. Precipitation of vivianite was found to have a long-term effect on P retention via Fe dosing in Groß Glienicker See, an urban lake in Berlin, Germany (Kleeberg et al. 2013; Rothe et al. 2014).
Nevertheless, the effect of Fe amendments on P retention is often only short lived (Cooke et al. 1993; Immers et al. 2015). Indeed, several competing binding partners such as organic matter, sulphur (S), silicon, and carbonate can inhibit the coupling of P to ferrous Fe (Davison 1993; Hartikainen et al. 1996; Kleeberg et al. 2013). A key question is which sediment properties or processes at the SWI determine the long-term success of P retention by Fe amendments; according to a critical review by Wang and Jiang (2016) on the use of chemicals for in-lake P precipitation, detailed investigation of the biogeochemical behaviour of precipitants (e.g. Fe), target elements (i.e. P) and competing binding partners (e.g. carbonate, organic matter, and S) is still required. Field studies examining the sediment composition and processes at the SWI alongside the long-term fate of Fe dosing can provide insights into the relevance and mechanisms of the competing reactions. Except for a few well-investigated lakes such as Groß Glienicker See (Wolter 2010; Kleeberg et al. 2013; Rothe et al. 2014), long-term field studies of Fe and P interactions at the SWI exceeding a few years after Fe dosing remain lacking (Boers et al. 1994; Deppe and Benndorf 2002; Lürling et al. 2016).
The competitive effect of S binding to Fe has been frequently discussed (Ohle 1953; Caraco et al. 1993; Lamers et al. 2002; Rothe et al. 2016; Kelly Vargas and Qi 2019; Zak et al. 2021). Sulphide competing for Fe with P may originate either from decayed organic matter or the reduction of dissolved sulphate at the SWI. The origin of competing S is important, as the requirements for managing these sources may differ; however, field data on the underlying mechanisms of S outcompeting P for Fe binding sites remain scarce.
Competing reactions also need to be considered in the planning and management of Fe amendments (Immers et al. 2015). Generally, the planning and management of in-lake measures with the overall goal of increased P retention require multiple steps. According to Lürling et al. (2016) and Bakker et al. (2016), a system analysis needs to be carried out to reveal the biological structure as well as the main water and P flows in lakes. Specifically, the water balance and retention time, external P loading rate, P sedimentation rate, and P flux from the sediment need to be determined (Schauser et al. 2003; Lürling et al. 2016). Anthropogenic P sources often need to be reduced to achieve an effective reduction in P loadings (Kelly Vargas and Qi 2019). For the calculation of the required Fe dosage, Kleeberg et al. (2013) considered not only unavoidable external and internal P loadings but also the competing Fe demand of organic carbon (Corg) and sulphide. Field studies investigating the long-term fate of in-lake dosed Fe can help verify Fe dosage calculations and improve management procedures of Fe amendments.
Our study aims to understand the long-term fate of in-lake Fe dosing and, based on this knowledge, evaluate and improve the planning and management procedures for Fe amendments for P retention. In the two study lakes, Fe amendments were carried out 2 and 3 decades ago, respectively. Starting before these periods of Fe amendment up to the present, we investigate the sediment stratigraphy in the lakes, the occurrence of Fe minerals, and the fluxes between the water and sediment. This allows us to assess whether and how the Fe dosing has contributed to an improved lake status over long-term periods. Furthermore, we evaluate whether and which competing processes have determined the success of the management measures with respect to long-term P retention. Based on our analysis, we compare planning and managing approaches to Fe amendments and make recommendations for improvement.
2 Methods
2.1 Study sites
The lakes Groß Glienicker See and Plötzensee are dimictic subglacial channel lakes without surface inflows and outflows in Berlin, Germany. Groß Glienicker See is both larger and deeper than Plötzensee (Table 1). During summer stratification, the hypolimnion of Plötzensee begins at a depth of approximately 3–4 m (ILAT Berlin 2003). Therefore, the hypolimnetic area comprises roughly 50% of the lake (Senatsverwaltung für Stadtentwicklung Berlin 2002). In Groß Glienicker See, the metalimnion begins at a depth of approximately 6–7 m (Kleeberg et al. 2013), with approximately 60% of the lake area in the hypolimnion (Senatsverwaltung für Stadtentwicklung Berlin 2002). The catchment area of Groß Glienicker See comprises urban settlements, agricultural land, and forests, whereas the considerably smaller Plötzensee catchment encompasses parks, cemeteries, and allotment gardens. Both lakes are used for recreation (i.e. bathing and fishing).
The trophic state of both lakes deteriorated in the late twentieth century. In Groß Glienicker See, inputs of wastewater from leaking septic tanks and from the direct discharge of poorly treated wastewater from a military base were assumed the cause of the change (Wolter 2010). In Plötzensee, high inputs of P from bathers as well as the release of legacy P accumulated in the sediments were identified as the main reasons for eutrophication (ILAT Berlin 2003). Due to the eutrophic state of both lakes, Fe amendments were applied as part of a package of management interventions. Iron (500 g Fe m−2) was added to Groß Glienicker See in winter 1992/1993 as 50% solid ferric hydroxide and 50% dissolved ferric chloride (Wolter 2010). Furthermore, between 1992 and 1998, Groß Glienicker See was continuously aerated, and the external P load was bypassed and eliminated (Deneke and Mischke 1995; Wolter 2010). In comparison, 150 g Fe m−2 was added to Plötzensee in November 2000 by applying ferric hydroxide together with nitrate as an oxidising compound (ILAT Berlin 2003). Before the Fe amendment, the upper layers of sediment (approximately 60 000 m3) were removed from Plötzensee. The maximum water depth did not increase because of the dredging. Further, an external P-removal plant was operated for approximately 1 year. The sanitary facilities at the public baths were also modernised, and the shore was protected against leaf litter inputs and bank erosion using palisades.
After the management measures, the status of both lakes improved both in the short- and long-term. In the water column of Groß Glienicker See, total P and chlorophyll-a concentrations decreased, water transparency improved, and macrophytes began to dominate over phytoplankton (Wolter 2010; Kleeberg et al. 2013; Rothe et al. 2014). In Plötzensee, total P and chlorophyll-a concentrations also decreased, water transparency improved, and macrophytes began to establish (ILAT Berlin 2003; Enviteam & LimPlan 2018).
2.2 Sediment sampling and analyses
Sediment cores (60 mm in diameter) were retrieved from the deepest parts of both lakes (Groß Glienicker See: June 2020, Plötzensee: October 2019) using a gravity corer (Uwitec). After sampling, the cores were stored in the dark at 5 °C. The cores were subsequently cut into horizontal layers of either 10 or 20 mm. The fresh muddy dark brown to black sediments were homogenised by stirring, and the subsamples for sequential P extraction, dry weight (DW) determination, and loss on ignition (LOI) were taken before freezing and freeze-drying the remaining material. The dry samples were ground in an agate mortar.
The determination of DW and LOI of the samples was performed gravimetrically (Sartorius research R 160 P, Sartorius) after drying and combusting a subsample of approximately 1 g at 105 °C and 450 °C (3 h), respectively. Organic carbon content (Corg) was estimated as 50% g g−1 of organic matter determined from LOI following Kleeberg et al. (2013).
For six selected samples from Groß Glienicker See and eight from Plötzensee, P fractions were distinguished according to Psenner et al. (1984) as modified by Hupfer et al. (1995). The fractions were characterised and named by the extracting agents. Loosely surface-adsorbed P was released using a N2 saturated 1 M ammonium chloride (A) solution, and redox-sensitive bound P was released using a 0.11 M bicarbonate/dithionite (BD) solution. Both fractions were determined after digestion of total P (TP) in potassium persulfate. The fraction dissolved by 1 M sodium hydroxide (NaOH) was further distinguished based on soluble reactive P (SRP) and non-reactive P (NRP), which was calculated as TP after digestion by potassium persulfate minus the SRP concentration. NaOH-SRP indicated P bound to, e.g. Fe oxides (Psenner et al. 1984) and vivianite (Rothe et al. 2015). NaOH-NRP was used as a measure of P released from organic material. P enclosed in carbonates was extracted with 0.5 M HCl and determined after digestion of TP, and finally, residual P was released by digestion of the remaining material. The TP content of the freeze-dried and ground subsamples was determined after wet digestion with one part H2O2 (30% V V−1) and one part H2SO4 (5 M) at 120 °C. P was measured in the solutions using the ammonium molybdate method (DIN EN ISO 6878).
The elemental content (Fe, S, and P) of all the sediment layers was determined by inductively coupled plasma optical emission spectrometry (ICP-OES, iCAP 7000series, Thermo Scientific) after aqua regia digestion (one part 37% V V−1 HCl and three parts 65% V V−1 HNO3) of the freeze-dried and ground samples in a high-pressure microwave oven (μPrep-A, MLS 169 GmbH). This method extracts a majority of Fe phases from the sediment, excluding Fe bound in silicates, which is not reactive in early diagenesis (Rothe et al. 2015). The selected subset elemental analyses of total S and Corg after vaporising inorganic carbon using 1 M HCl were carried out with a Vario EL (Elementar Analysensysteme GmbH). According to the applied methods, total S contained acid volatile sulphide, sulphate, and other sulphur species such as organic S. Dissolved sulphide might have evaporated during the sample preparation. In case all dissolved sulphide was lost, the S content decreased by approximately 6% in the upper sediment layer of Plötzensee.
All results are reported as single values for distinct sediment layers or as mean values and standard deviations for sediment sections.
X-ray diffraction (XRD) analyses were carried out on a D2Phaser (Bruker) equipped with a Cobalt X-ray tube and an SSD160 detector (active length = 12 mm) between 12 and 85° 2θ using a rotating sample holder (30 rotations min−1). The step size was 0.02° 2θ, and the measurement time was 2 s step−1. Measurements were carried out using a 1-mm fixed divergence slit, 2.5° primary and secondary soller collimator, a fixed knife edge (3 mm above the sample surface), and a Fe k-beta filter (0.5). Qualitative phase analysis was conducted using DIFFRAC.EVA V5.2 (Bruker) and the Crystallography Open Database (Grazulis et al. 2009). Qualitative analyses and quantitative estimations were carried out using the Rietveld refinement software Profex/BGMN (Bergmann et al. 1998; Doebelin and Kleeberg 2015). For quantitative estimations, one Fe-rich sample per lake was prepared by adding 10% g g−1 corundum (Baikowski) as an internal standard and subsequent hand-milling and intimate mixing with an agate mortar and pestle. The samples were then filled in the XRD holders using the top-loading technique. The quantitative scans were carried out on the same device between 5.7 and 120° 2θ using a rotating sample holder (10 rotations min−1). The step size was 0.02° 2θ, and the measurement time was 8 s step−1. The measurements were carried out using a 0.6-mm fixed divergence slit, 2.5° primary and 4.0° secondary soller collimator, a fixed knife edge (3 mm above the sample surface), and a Fe k-beta filter (0.5).
2.3 Pore water sampling and analyses
The pore water profiles at the SWI were obtained by in situ dialysis samplers (Hesslein 1976) with a vertical resolution of 10 mm and two neighboured lines of 58 chambers, respectively. The chambers, covered by a 0.2-µm polyethersulfone membrane (Supor®200, Pall Corporation), were filled with Millipore water (0.055 µS cm−1) and degassed with nitrogen for 24 h to remove oxygen. Subsequently, the samplers were installed at the deepest points of the lakes for more than 13 d during the summer stratification period.
The pore water samples were collected from the in situ dialysis samplers with syringes through the membrane. Sulphide samples (1 mL) were fixed in 1% g g−1 zinc acetate solution (200 µL) and measured photometric following the methylene blue method (Cline 1969). Sulphate was determined by ion chromatography (Metrohm) with detection via conductivity (DIN EN ISO 10304–1) in a sample aliquot of 1 mL. One subsample was fixed using hydrochloric acid immediately after sampling and was used to analyse the total dissolved Fe concentrations of the pore water via ICP-OES (iCAP 7000series, Thermo Scientific), SRP following the ammonium molybdate method (DIN EN ISO 6878) and nitrate (DIN EN ISO 13395), both using segmented flow analysis (Skalar Scan + + , Skalar Analytical B.V.). All samples were stored at 5 °C before analysis, and all measurements were performed in duplicate.
Reaction rates at and below the SWIs were calculated from pore water profiles following Wang et al. (2008) assuming only molecular diffusion (significance level = 0.05 with a minimum of three data points per reaction zone). All diffusion coefficients were retrieved from Schulz and Zabel (2006) for each lake’s median bottom temperature at the month of sampling. For Groß Glienicker See, temperature data from 2012 to 2014 with a 2-h resolution were used. For Plötzensee, monthly temperature measurements from 2010, 2012, 2015, 2018, and 2020 were available. For sulphate, the molecular diffusion coefficient was 6.79 ∙ 10−6 cm2 s−1 (10 °C) for Groß Glienicker See and 7.86 ∙ 10–6 cm2 s−1 (15 °C) for Plötzensee. For sulphide in Plötzensee, a molecular diffusion coefficient of 14.6 ∙ 10–6 cm2 s−1 (15 °C) was used for the species HS− (50% mol mol−1) and H2S (50% mol mol−1) assuming neutral pH at the SWI. The uncertainties of the reaction rates were calculated according to Wang et al. (2008) with a relative precision of 0.1 and 50 random concentration profiles.
The mean sulphate reduction rates (SRRs) and the standard deviations were calculated from five pore water profiles per lake, which were sampled after the Fe amendments and without any other active management measure, such as aeration. The pore water profiles were derived from September and June 2001, July and September 2002, and June 2020 for Plötzensee and from September 2004 and 2008, June and September 2009, and September 2020 for Groß Glienicker See. Three of the sulphate profiles are included in Kleeberg et al. (2013). For Plötzensee, the SRR before Fe addition was determined from the sulphate profiles of four dialysis samplers distributed over the lake in October 1998. Dissolved oxygen in the water column was measured by Multiparameter Water Quality Sonde (6600 V2, YSI) at the days of placing and retrieving the dialysis samplers in September 2020.
2.4 One-box model for annual in-lake sulphate concentrations after iron dosing
A one-box model designed to calculate P concentrations after lake restoration measures, such as P precipitation, under steady-state (Gächter and Imboden 1985) and non-steady-state conditions (Hupfer et al. 2016) was adapted to model the effect of Fe dosing on sulphate concentrations. The coefficients of the one-box model were retrieved from the literature (Table 1; water residence times, τ) and calculated from the sediment and pore water analyses. For the calculation, sulphate reduction was assumed to occur only at the SWI in the hypolimnion. The sulphate concentration of the external load, Sin, was determined by the steady-state condition after the Fe dosing. Finally, the net sulphate deposition per sulphate mass of the entire lake volume, σ, before the Fe amendments was required to match the steady-state condition before the management measure. The stratification factor, β, was approximated as 1, as the mean annual outflow concentration was assumed to equal the annual mean sulphate concentration of the lakes.
The one-box model was used to compute non-steady-state annual sulphate concentrations in the lakes after the Fe amendments by an increased net sulphate deposition in the sediment. The modelled sulphate concentrations were compared to measured sulphate concentrations, Slake, before and after Fe dosing reported by the Berlin Senate for the Environment.
The cumulative deposition of S from sulphate reduction was calculated from the results of the one-box model and, then, compared to the additional S stored in the sediment after the Fe amendment from the sediment stratigraphy. In addition, the theoretical consumption of Fe because of the formation of pyrite, FeS2 (i.e. two sulphide molecules bind one ferrous Fe cation), based on the modelled sulphate deposition was compared to the Fe surplus from the management measures; in those cases where all excess Fe was consumed, the net sulphate deposition was set back to zero.
3 Results
3.1 Sediment compositions before and after iron amendments
3.1.1 Stratigraphy of iron, phosphorus, and sulphur
In the sedimentary stratigraphy of Groß Glienicker See, Fe content was 19 ± 3 mg g−1 DW at depths of 35–47 cm (N = 5, Fe-poor, Fig. 1) and increased to a maximum of 73 mg g−1 DW at 27–29 cm, indicating the Fe dosing event. The Fe content stabilised at 59 ± 3 mg g−1 DW between the sediment surface and a depth of 25 cm (N = 14, Fe-rich). The P content in the Groß Glienicker See sediment corresponded with Fe (Fig. 2) and increased from 1.5 ± 0.1 mg g−1 DW in the Fe-poor section to 5.0 ± 0.3 mg g−1 DW in the Fe-rich sediment layer (Fig. 1). In the Fe-rich part of the sediment, the largest P fractions were BD-TP with 27–45% g g−1 TP and NaOH-SRP with 34–43% g g−1 TP. BD-TP decreased and NaOH-SRP increased towards the deeper sediment layers. The fractions of NaOH-NRP and HCl-TP were between 6 and 14% g g−1 TP, and the A-TP and residual P fractions were < 3% g g−1 TP in all samples. In relation with the Fe content, the S content increased slightly (Fig. 2) from 16 ± 2 mg g−1 DW in the Fe-poor sediment section to 21 ± 3 mg g−1 DW in the Fe-rich layer (Fig. 1). Therefore, the molar S/Fe ratio decreased from 1.52 ± 0.04 (Fe-poor) to 0.62 ± 0.08 (Fe-rich). Corg increased continuously from 108 at the bottom to 174 mg g−1 DW at the top. The resulting molar Fe/Corg ratio was 0.079 ± 0.008 in the Fe-rich part of the sediment.

Fe and P content (line determined by ICP-OES after aqua regia dissolution, dots indicate TP after wet digestion and photometric analysis), and P-binding forms (BD-TP, redox-sensitive P; NaOH-SRP, redox-stable metal-bound P; NaOH-NRP, organic-bound P; HCl-TP, P bound in calcium carbonates and apatite) as well as S (line determined by ICP-OES after aqua regia dissolution and dots by an element analyser), the molar ratio of S/Fe, and Corg (line determined by 50% g g−1 of loss-on-ignition and dots determined by an element analyser after evaporation of inorganic carbon) in the sediments of Groß Glienicker See (top) and Plötzensee (bottom). According to the maximum Fe content in both sediments, the grey lines indicate the sedimentary layer from the respective year of Fe dosing

Relations of P and S content with Fe content (all determined by ICP-OES after aqua regia dissolution) in Plötzensee and Groß Glienicker See
In Plötzensee, the Fe content was 17 ± 1 mg g−1 DW at depths of 15–28 cm (N = 13, below Fe peak, Fig. 1) and peaked at 9–10 cm at 57 mg g−1 DW corresponding to the Fe amendment. At depths of 3–8 cm, the Fe content was 28 ± 2 mg g−1 DW (N = 5, above Fe peak) and then decreased to 21 mg g−1 DW at the sediment surface. The P content of the sediment did not follow the Fe profile (Fig. 2) but remained at 1.4 ± 0.1 mg g−1 DW between depths of 3 and 28 cm (N = 25) and then increased to 1.7 mg g−1 DW at the sediment surface (Fig. 1). The largest P fraction was bound as NaOH-NRP with 27–44% g g−1 TP followed by HCl-TP, NaOH-SRP, and BD-TP with 9–20% g g−1 TP. The A-TP and residual P fractions were < 8% g g−1 TP. The S content varied in proportion with the Fe profile (Fig. 2) and peaked at 61 mg g−1 DW together with the Fe peak (Fig. 1). Towards the sediment surface, the S content decreased to 29 mg g−1 DW. Consequently, the molar S/Fe-ratio increased slightly from 1.88 ± 0.08 (below Fe peak) to 2.19 ± 0.08 (above Fe peak). Corg decreased from 205 ± 23 mg g−1 DW below the Fe peak to 171 ± 7 mg g−1 DW between the Fe peak and the SWI. The molar Fe/Corg ratio was 0.074 ± 0.008 (N = 3) at the Fe peak in the sediment.
In summary, the Fe dosing interventions were clearly recorded in the sediments of both lakes. In Groß Glienicker See, the increase in Fe content was accompanied by an increase in P content (Fig. 2), especially in the BD-TP and NaOH-SRP fractions (Fig. 1), while this relation was not observed in Plötzensee (Fig. 2). In contrast, the increase in the Fe content in Plötzensee was associated with an increase in S content, whereas this was of minor importance in Groß Glienicker See (Fig. 2).
3.1.2 Iron-phosphorus and iron-sulphur mineral occurrence
The XRD analysis showed that the Groß Glienicker See sediments were mainly composed of quartz, calcite, white mica, and tri-trioctahedral chlorite (both layer silicates), vivianite, K-feldspar, plagioclase, and ankerite, Ca(Fe2+,Mg,Mn2+)(CO3)2 (in decreasing abundance for the 9–11-cm layer). Traces of pyrite, kaolinite, and actinolite were also detected. In the Plötzensee sediments, calcite, pyrite, quartz, and gypsum were detected (in decreasing abundance for the 9–10-cm layer) with additional traces of kaolinite, white mica, plagioclase, K-feldspar, and actinolite. The XRD patterns of the sediments from both lakes showed a significant amorphous hump.
Special attention was paid to the coupling of Fe with P, carbonate, and S. The diffractograms show vivianite, ankerite, and pyrite throughout the sediment layers of Groß Glienicker See after the Fe dosing (Fig. 3). The vivianite reflections (i.e. at 15° 2θ) increased towards the deeper sediment layers, indicating increasing amounts of vivianite. The intensity of the ankerite and pyrite reflections remained similar at all the analysed depths. Vivianite and ankerite were not detected in the sediment from Plötzensee, whereas pyrite was identified in all the samples (Fig. 3). The intensity of the pyrite reflections was approximately two times higher in the Fe-rich layers (9–10 cm and 10–11 cm, respectively) compared to the Fe-poor layers (5–6 cm and 13–14 cm, respectively).

X-ray diffraction (XRD) scans of three distinct layers from the Fe-rich part of the sediment from Groß Glienicker See (0-1, 9-11, and 19-21 cm) and two distinct layers each from the Fe-rich part (9-10 and 10-11 cm) and Fe-poor parts (5-6 and 13-14 cm) of the sediment from Plötzensee. Below, reference patterns of vivianite and pyrite are displayed, and their largest reflections are elongated into the sample scans. Other sample reflections were assigned to the minerals ankerite, calcite, chlorite, K-feldspar, gypsum, kaolinite, dioctahedral mica, plagioclase, and quartz. The y-axis displays squared intensities as arbitrary units (a.u.)
Consequently, in Groß Glienicker See, Fe was coupled directly to P as vivianite, S as pyrite, and carbonate as ankerite, whereas in Plötzensee, the only consequence of the Fe dosing was S-Fe coupling in pyrite.
3.2 Sulphide production and sulphate reduction after iron amendments
3.2.1 Sulphide sinks and sources
At and above the SWI of Plötzensee, the concentration of dissolved sulphide was 20 ± 1 mg L−1 and decreased with depth below the SWI (Fig. 4). According to the reaction rates determined following Wang et al. (2008), the source of dissolved sulphide was located at the SWI. A large portion of the produced sulphide migrated to a sink of dissolved sulphide between a depth of 3 and 12 cm, below which and above the SWI, sulphide reaction rates were considerably lower. In contrast, no dissolved sulphide was measured over the entire pore water profile of Groß Glienicker See.

Dissolved sulphide, total dissolved Fe, and soluble reactive phosphorus (SRP) concentrations at the sediment–water interfaces (SWIs) of Plötzensee and Groß Glienicker See during the summer stratification period in 2020. Sulphide was below the detection limit of 0.03 mg L−1 for Groß Glienicker See. For Plötzensee, the sulphide reaction rates were calculated according to Wang et al. (2008)
The relatively high sulphide concentrations at the SWI of Plötzensee were associated with low concentrations of dissolved Fe (Fig. 4). Comparatively, dissolved Fe was available at higher concentrations in Groß Glienicker See at the SWI down to a sediment depth of 15 cm. In both lakes, the SRP concentrations in the water column directly above the sediment surface were similar. At the SWI of Groß Glienicker See, the SRP concentration showed a narrow peak, and in the sediment of this lake, the SRP concentration was lower than in the Plötzensee sediment.
3.2.2 Sulphate reduction rates at the sediment–water interfaces
The SRRs, calculated following Wang et al. (2008), at the five sampling times after Fe dosing were significantly different between Groß Glienicker See and Plötzensee (Wilcoxon rank sum test: W = 25, p = 0.008, Fig. 5). Specifically, Groß Glienicker See had a lower SRR of 14 ± 9 g m−2 year−1 compared to 70 ± 35 g m−2 year−1 for Plötzensee. While the SRRs varied considerably in both lakes, their sulphate profiles at the SWIs did not overlap. Two years before Fe addition to Plötzensee, the SRR was 84 ± 6 g m−2 year−1 (N = 4). To our knowledge, before Fe addition, SRRs were not determined for Groß Glienicker See.
Directly above the SWI of Plötzensee, oxygen was between 0.0 and 0.2 mg L−1 and between 0.3 and 0.6 mg L−1 in Groß Glienicker See. In Plötzensee, nitrate was below the detection limit of 0.01 mg N L−1 over the whole pore water profile in September 2020. In 2001, nitrate reduction occurred at the SWI of Plötzensee as a result of nitrate dosing together with Fe in 2000. In 2002, nitrate was again below 0.06 mg N L−1, which at the time was the detection limit. In the dialysis sampler retrieved from Groß Glienicker See in 2020, nitrate was between 0.03 and 0.06 mg N L−1 without a trend. In 2004, 2008, and 2009, nitrate at the SWI of Groß Glienicker See was below the detection limit of 0.06 mg N L−1.
3.3 In-lake sulphate concentrations and sedimentary sulphur retention after Fe amendments
3.3.1 Sulphate concentrations in the water column
Shortly after the Fe dosing to Groß Glienicker See in 1992, the sulphate concentration (0.5 m below water surface) began to decrease from 81 ± 1 mg L−1 (N = 41) and stabilised at 40 ± 1 mg L−1 (N = 123) after 2005 (Fig. 6). Similarly, in Plötzensee, the sulphate concentration decreased from 143 ± 13 mg L−1 (N = 46) after the Fe dosing (Fig. 6), although after a minimum sulphate concentration of 102 ± 8 mg L−1 in 2006 and 2007 (N = 25), concentrations increased again to 140 ± 4 mg L−1 (N = 5) in 2020.

Sulphate profiles and resulting sulphate reduction rates (SRR) at the sediment–water interfaces (SWIs) of Groß Glienicker See and Plötzensee at five different sampling times. The SRRs in the two lakes were significantly different (Wilcoxon rank sum test: p = 0.008, W = 25)

Annual mean sulphate concentrations and their standard deviations in the water column (0.5-m depth) of Groß Glienicker See (N = 7–13) and Plötzensee (N = 5–13) in comparison to the sulphate concentrations calculated using a one-box model (Gächter and Imboden 1985) assuming maximum sulphate deposition in the sediments after Fe dosing
3.3.2 Sedimentary sulphur sinks via excess iron
The one-box model adopted in this study relates sulphate concentrations to the quantities of S deposited in the sediments after the Fe amendments (see Table 2 for model coefficients). It was assumed that after the Fe dosing, all sulphide from net sulphate reduction was retained by the excess Fe. Therefore, the net sulphate deposition per sulphate mass in the entire volume of each lake, σ, after the Fe dosing was calculated based on the SRRs (Fig. 5) and the steady-state sulphate concentrations, Slake, after the Fe dosing (Fig. 6).
In Groß Glienicker See, the modelled sulphate concentrations decreased after the model coefficient σ increased (Fig. 6). According to the sediment core retrieved in 2020 from Groß Glienicker See, a surplus of 80 t of S was stored in the sediment after the Fe dosing. This amount was calculated by considering a background content of 17 mg S g−1 DW (Fig. 1) below the Fe peak as well as the hypolimnetic area available for sulphate reduction. The additional S deposition after the Fe amendment and up to 2020 calculated by the one-box model corresponded to 94% g g−1 of the additional S inventory in the sediment. In Groß Glienicker See, the Fe dosing of 500 g Fe m−2 over the entire lake area resulted in a total Fe mass of 340 t in the lake. According to the one-box model, pyrite formation after sulphate reduction had consumed 19% g g−1 of the excess Fe by 2020.
The modelled sulphate concentrations in Plötzensee also decreased after the model coefficient σ increased (Fig. 6). In this lake, the amount of 150 g Fe m−2 accounted for a total Fe mass of 12 t over the entire lake area. According to the one-box model, the amount of excess Fe was completely consumed by pyrite formation by 2013. Therefore, the net sulphate deposition, σ, was reset to the pre Fe-dosing value from 2014 onwards, after which the modelled sulphate concentrations increased again (Fig. 6). According to the sediment core retrieved from Plötzensee in 2019, a surplus of 13 t S was stored in the sediment after the Fe dosing. This amount was calculated by considering a background content of 20 mg S g−1 DW (Fig. 1) below the Fe peak as well as the hypolimnetic area available for sulphate reduction (Table 1). The additional S deposition after the Fe amendment corresponded to 99% g g−1 of the additional S inventory in the sediment.
4 Discussion
4.1 Sulphur competition with phosphorus for dosed iron: field scale evidences
In both of the studied lakes, the sediment layers originating from past Fe applications were identified from their increased Fe content in comparison to lower sediment layers (Fig. 1). Correspondingly, we were able to distinguish the compositions of the sediments before and after Fe dosing. In Groß Glienicker See, the current Fe content remains considerably higher than before the Fe dosing, whereas in Plötzensee, the Fe content has fallen back to the pre-amendment level.
In the Groß Glienicker See sediment, Fe amendment has enabled the long-term retention of P (Kleeberg et al. 2013). Among other P-binding forms, such as BD-TP, vivianite couples P to Fe under anoxic conditions and significantly increases P retention following Fe dosing (Rothe et al. 2014) which exemplifies that microbial Fe reduction does not prevent long-term P retention by Fe. This has contributed to the low P concentrations in the lake and, thus, improvement of its trophic state. Based on the high share of Fe-related P-binding forms (BD-TP and NaOH-SRP) and by the presence of vivianite, the more recent data show that the high degree of P retention by Fe remains active in Groß Glienicker See (Figs. 1 and 3). In contrast, in Plötzensee, the Fe dosing did not increase P retention (Figs. 1 and. 2). In particular, Fe-related P-binding forms (BD-TP, NaOH-SRP, and vivianite) in the sedimentary record are low or absent and did not increase after the period of management intervention. Overall, the Fe amendment successfully retained P in Groß Glienicker See, whereas long-term P-Fe binding failed in Plötzensee.
Pyrite was present in the sediments obtained from both lakes, and, therefore, S is relevant as a competitor for the Fe-driven long-term retention of P in both cases (Fig. 3). In Groß Glienicker See, vivianite and pyrite both consume Fe, whereas in Plötzensee, Fe is bound in pyrite but not vivianite. According to the XRD analyses, considerably more pyrite is present in the Plötzensee sediment layers containing excess Fe. Also, the strongly increasing relation of total S content with Fe content in Plötzensee in comparison to Groß Glienicker See (Fig. 2) shows that the competition of S with P for excess Fe was of greater importance in Plötzensee. Furthermore, the molar S/Fe ratio decreased in the Groß Glienicker See sediment after the period of Fe dosing but slightly increased in the Fe-rich layers of Plötzensee (Fig. 1). According to Rothe et al. (2015), S/Fe molar ratios can be interpreted as the Fe availability after sulphidisation. Therefore, after the Fe amendment in Groß Glienicker See, P retention was not inhibited by sulphidisation, but S was bound to Fe (in pyrite) and P was bound to Fe (e.g. as vivianite). In Plötzensee, the S/Fe ratio shows that Fe availability did not increase after the Fe amendment, but, rather, sulphidisation consumed the excess Fe. Whether the increase in the S/Fe molar ratio towards the SWI in Plötzensee is a result of the Fe dosing is unclear; however, the addition of redox-sensitive Fe hydroxide might facilitate S-Fe binding relative to less-reactive Fe compounds reaching the lake via natural pathways. In all, based on evidence from both lakes, S has been the relevant competitor determining the long-term success of P-Fe binding after the Fe amendments.
As a competing binding partner to Fe, carbonate (i.e. ankerite) must be considered in the sediment of Groß Glienicker See (Fig. 3); however, the formation of ankerite did not prevent the coupling of Fe and P as BD-TP and in vivianite, and these Fe binding forms coexist. The amount of vivianite increased with depth, but ankerite remained relatively constant suggesting no replacement of vivianite by ankerite during diagenesis. Corg might also be relevant, which may have bound Fe via complexation. However, the similar Corg content in the sediments of both lakes (Fig. 1) and the similar Fe/Corg molar ratios in the Fe-rich sediment layers do not indicate that this was a key factor determining the long-term success of P-Fe coupling in these two lakes. Furthermore, the so-called cryptic cycling of Fe, Corg, and S might have an important influence on the final binding forms of these elements and, therefore, affect the formation of stable P-Fe binding forms (Hansel et al. 2015; Kappler and Bryce 2017). Nevertheless, the long-term stable Fe binding forms remain relevant for the calculation of Fe dosages.
Overall, the sedimentary stratigraphy and element correlations as well as the XRD results indicate that S, as a competitive binding element, determined the relative success (Groß Glienicker See) and failure (Plötzensee) of long-term P-Fe coupling following Fe amendment. Importantly, our results show that pyrite formation competes with P-Fe coupling and has the potential to outcompete long-term P-Fe binding.
4.2 Sulphur competition with phosphorus for dosed iron: mechanisms
Pyrite is formed by S and Fe, both in their reduced species form. Therefore, sulphide competes with long-term P-Fe coupling under anoxic conditions. Pyrite formation in sediment immobilises Fe, and consequently, pyrite formation limits the precipitation of P by Fe(II) under anoxic conditions and prohibits P adsorption to Fe(III) under oxic conditions in the water column and at the SWI as immobile Fe cannot accumulate in an oxic SWI (Gächter and Müller 2003; Lehtoranta et al. 2009). Furthermore, laboratory studies have shown that sulphide releases P from both Fe(III)-P (i.e. BD-TP) and Fe(II)-P (i.e. vivianite), whereas P release from reduced Fe(II)-P is higher at identical sulphide inputs (Wilfert et al. 2019).
In Plötzensee, significant amounts of dissolved sulphide were produced at the SWI. The sulphide migrated both towards the water column and deeper within the sediment. At the time of pore water sampling in June 2020, assuming a steady-state situation, a major portion of the dissolved sulphide was trapped in a reactive layer at a sediment depth corresponding with the current depth of the Fe peak in the sediment solids. This shows that sulphide is still being immobilised by the Fe-rich layer originating from the Fe amendment in 2000. However, the flux of sulphide towards the water column might peak during those seasons when less sulphide is accumulated in the water column. In Groß Glienicker See, although pyrite was identified in the upper sediment layers, no dissolved sulphide was detected at the SWI.
A central question is why the long-term P-Fe coupling in Plötzensee was substantially diminished because of the competing S but not in Groß Glienicker See. Apparently, it refers to varying sulphide formation in both lakes. In addition to the contrasting sulphide profiles at the SWI (Fig. 4), the SRRs of the two lakes are significantly different (Fig. 5); Groß Glienicker See has a lower SRR than Plötzensee. The SRRs of both lakes have been previously calculated following alternative approaches, which is consistent with the differences we observed (Kleeberg 1998; 2013). In addition to sulphate reduction, mineralisation of settling organic matter can be a source of sulphide (Urban 1994; Zhao et al. 2019).
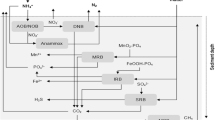
In Plötzensee and Groß Glienicker See, the monitored sulphate concentrations in the water column decreased after the Fe applications. The sediment stratigraphy and XRD analyses demonstrate increased S retention because of the availability of additional Fe. In a relatively Fe-poor lake, the SRR might be as equally high as in a lake after Fe dosing. However, reduced sulphide would not be retained in the sediment by Fe but instead migrate upwards to the water column and be oxidised back to sulphate. Consequently, we predicted that Fe dosing alters the S deposition rates and sulphate concentrations in the water column. Specifically, we applied the one-box model to test the hypothesis that the entire amount of sulphide produced by sulphate reduction is deposited in the sediment because of the availability of excess Fe. The determining factor in the model was the mean SRR of each lake. Accordingly, the decreasing sulphate concentrations in the water column following the Fe dosings explain the increased S deposition in the sediments due to sulphate reduction. Therefore, sulphate reduction was the main mechanism determining the competition between long-term S-Fe and P-Fe coupling. In Plötzensee, the high SRR and the resulting high S deposition rate after the Fe dosing potentially consumed all of the excess Fe (Fig. 7: High sulphate reduction rate). In contrast, in Groß Glienicker See, the lower SRR together with the higher Fe dosage has kept the share of the excess Fe consumed by S at a low level (Fig. 7: Low sulphate reduction rate).

Process scheme illustrating long-term effects of Fe dosing at low or high sulphate reduction rate. Brown indicates processes deliberately and successfully managed. Grey refers to possible interactions between excess Fe and sulphate reduction or sulphide production that might stimulate ( +) or decline ( −) the immobilisation of Fe by S after Fe dosing
Several controlling factors of SRRs in lakes have been suggested and discussed such as availability of other electron acceptors, availability of electron donors (mainly organic matter), and sulphate availability (Holmer and Storkholm 2001). In both Groß Glienicker See and Plötzensee, the alternative electron acceptors nitrate and oxygen were not relevant as electron acceptors at the SWIs at the times of pore water sampling, except during the aeration phases in Groß Glienicker See and directly after the nitrate addition in Plötzensee together with the Fe dosage. Moreover, increasing redox potentials by addition of other electron acceptors through oxygenation (e.g. artificial aeration) or addition of nitrate has been shown to shift the sulphate-reduction zone rather than suppress sulphate reduction (Zou et al. 2017; Fuchs et al. 2018; Cao et al. 2019). Organic matter has been shown to limit SRRs in marine and limnic sediments (Lehtoranta et al. 2008; Chen et al. 2016). Plötzensee and Groß Glienicker See are both rich in organic matter compared to the above cited studies. Nevertheless, a limitation of the SRR by the supply of organic matter cannot be excluded, since the quality of organic matter and not the quantity could limit the SRR. In addition, sulphate concentration limited the SRR at the laboratory scale in riverine sediments (Zak et al. 2006), and wild rice lake sediment (Myrbo et al. 2017) and sustained high supplies of sulphate have been suggested to deplete a high proportion of Fe in freshwater systems due to sulphate reduction (Smolders et al. 1995; 2001; 2006). In Plötzensee and Groß Glienicker See, the different SRRs might reflect the different sulphate concentrations in the water columns. Thus, the sulphate concentrations in Groß Glienicker See are moderate among 58% out of 456 natural lakes in Germany (2014–2020), whereas the concentrations in Plötzensee fall in the highest 24% of the monitored German lakes (Zak et al. 2021).
Further, the addition of excess Fe as a management measure might interact with sulphate reduction and change the SRR (Fig. 7). Amorphous ferric hydroxide in particular may increase the redox potential (Vandieken et al. 2014), which in comparison makes sulphate reduction thermodynamically less favourable and might, therefore, inhibit Fe immobilisation by S. Chemical reduction of ferric Fe consumes sulphide (Lehtoranta et al. 2008), which in effect might decrease Fe immobilisation by S and, further, has been supposed to block microbial Fe reduction (Wu et al. 2019). In addition, the immobilisation of Fe by S might be decreased by Fe sulphide precipitates that inhibit sulphate reducers by forming a barrier between the bacteria and the reactants in the pore water (Koschorreck 2008). On the other hand, it has been observed that Fe amendment stimulated the utilisation of sulphate. Hamdan and Salam (2021) supposed dissolved Fe acted as an electron mediator for sulphate reduction. Another possible mechanism causing an increased SRR and, therefore, faster Fe immobilisation by S is that excess Fe binds sulphide which might prevent product inhibition of sulphate reduction by toxic sulphide (Koschorreck 2008). The possible interactions via organic matter produced and consumed during the reduction of excess Fe and sulphate reduction are manifold: Generally, sulphate reducers compete with Fe-reducing bacteria for electron donors (Koschorreck 2008) possibly excluding microbial sulphate reduction (Chapelle and Lovley 1992). On the other hand, microbial Fe reduction and microbial sulphate reduction have often been shown to co-occur in the presence of poorly crystalline Fe oxides (Finke et al. 2007) as usually applied in Fe amendments. As excess Fe can stimulate the production of volatile fatty acids (Laufer et al. 2016; Yang et al. 2019), which in turn have been identified as favourable substrates of microbial sulphate reduction (Llobet-Brossa et al. 2002; Finke et al. 2007), Fe dosing might even increase SRR and Fe immobilisation by S. However, volatile fatty acids are consumed also by Fe reducing bacteria (Kappler et al. 2021), and microbial sulphate reduction can be driven by other electron donors (e.g. hydrogen, short alcohols, longer alcohols, fatty acids as well as aromatic and aliphatic hydrocarbons) (Finke et al. 2007). Edenborn and Brickett (2001) explained an abnormal pattern of microbial sulphate reduction by the adaptation of the sulphate reducers in the experiments to lactate instead of other more common electron donors. Such adaptations of sulphate reducing bacteria to specific electron donors and the large variety of possible electron donors suggest that sulphate reducers in lake sediments are able to adapt to electron donors depending on their availability that in turn depends on the processes occurring in the specific lake. At elevated concentrations, volatile fatty acids have been argued to even inhibit sulphate reducers (Koschorreck 2008). Moreover, successful Fe amendment decreases primary production and production of organic matter (Fig. 7). This might limit sulphate reduction by overall electron donor availability.
From the field-scale evidence together with previous studies (Lehtoranta et al. 2008; Heinrich et al. 2021), a conceptual understanding of how microbial reduction of sulphate and Fe determine P retention in sediments after Fe amendments can be derived (Table 3): At low SRR and low microbial Fe reduction, Fe(oxi)hydroxides persist in the sediment and bind P on the long term (Lehtoranta et al. 2008). At low SRR and relatively high microbial Fe reduction, vivianite binds P on the long term (Heinrich et al. 2021). Eventually, the burial of Fe(oxi)hydroxides and the formation of vivianite can be observed in parallel as in Groß Glienicker See. At high SRR, sulphide may either reduce Fe(oxi)hydroxides or dissolve vivianite (Wilfert et al. 2019) and immobilise Fe. As a result, P is not bound to Fe on the long term. In all, the extent of Fe reduction determines how P is bound to Fe on the long-term. In addition, the intensity of sulphate reduction determines whether P is retained by Fe on the long-term.
4.3 Management implications for long-term phosphorus retention by iron dosing
Sulphur to Fe coupling after Fe amendment can contribute to the improvement of lake status by preventing the toxic effects of free sulphide (Smolders et al. 1995; Zak et al. 2021). In Plötzensee, the retention of sulphide might have contributed to the improved water transparency and the development of macrophytes following Fe amendment in 2000. In addition, re-oxidation of mobile sulphide can consume oxygen from the water column at high rates (Holmer and Storkholm 2001; Berg et al. 2019). In Groß Glienicker See, this is apparently prevented by the surplus Fe in the sediment which prevents a sulphide flux to the water column, whereas, in Plötzensee, oxygen depletion because of high sulphide production is a possible threat for the lake ecosystem (Fig. 7).
Importantly, the competition of S needs to be considered when planning Fe amendments that aim to sustain long-term internal P precipitation. According to Wang and Jiang (2016), no definitive recommendations for Fe dosages are available; dosages have previously been determined based on Fe/P molar ratios or laboratory tests (Quaak et al. 1993; Hansen et al. 2003; Gołdyn et al. 2014; Wang and Jiang 2016) but these approaches do not consider lake-specific competitive effects over timescales of a few decades. To our knowledge, the only planning approach available for Fe dosing that considers competing reactions, such as S-Fe coupling, was suggested by Kleeberg et al. (2013); however, in this approach, sulphide originating from sulphate reduction is assumed to bind with Fe over the course of just 1 year. In contrast, dissolved sulphate and free sulphide are both mobile species at the SWI. Indeed, the evidence for Plötzensee demonstrates that dissolved sulphide reaches the excess Fe layer even when buried 10 cm below the sediment surface. As a result, sulphide from sulphate reduction can continue to consume Fe year on year until the excess Fe is used up. Fe amendment planning must, therefore, consider sulphate reduction as a competing process over the entire management period until the P-Fe compounds are buried in unreactive sediment layers.
A challenge for the application of this finding is that Fe dosing might affect the SRR, the sulphide production at the SWI and the resulting immobilisation of Fe by S after Fe dosing (Fig. 7). As the resulting effect of the manifold interactions between excess Fe and SRR remains unclear, it is impossible to calculate the Fe dosage before the management intervention. For Plötzensee, the SRR in October 2 years before Fe addition was in the range of the SRRs determined after Fe dosing, which gives no indication of a systematic change of the SRR after Fe dosing in this lake. However, due to the distribution of the data, a possible effect may be overlooked. Until possible effects of excess Fe on SRR are better understood, the problem can be addressed by using the SRR determined before Fe dosing as an initial estimate, checking the SRR after the Fe amendment, and adjusting the Fe dosage if necessary.
To evaluate whether the adopted Fe dosing considering sulphate reduction as a competing process over the entire management period would still be realistic, we recalculated Fe dosages for Groß Glienicker See and Plötzensee based on Kleeberg et al. (2013) and, additionally, considered long-term (i.e. 30-year period) Fe consumption by pyrite formation (i.e. two sulphide molecules immobilise one ferrous Fe cation) resulting from sulphate reduction in the hypolimnion. Recent literature was also used to calculate the excess Fe consumed by organic matter. For example, Herndon et al. (2017) found more Fe (19.8 ± 3.3% g g−1 of total Fe) bound to organic matter in the mineral layers of arctic tundra soils than in organic layers, and based on 11 different marine sediments, Barber et al. (2017) concluded that up to 18.1% g g−1 of total Fe was complexed by organic matter. Thus, we suggest providing 20% g g−1 of the excess Fe for competitive binding to Corg, although further work is needed to verify this. The competitive effects of ankerite formation and whether ankerite might contribute to P-Fe coupling via adsorption cannot yet be determined. Based on our calculations, the recalculated Fe dosage for Plötzensee is 521 g Fe m−2, of which 71% g g−1 is supplied for S-Fe coupling, 20% g g−1 is supplied for Corg-Fe binding, and 9% g g−1 is supplied for P-Fe binding based on external and internal P loadings (Gunkel and Pachur 1994). The calculated Fe dosage is substantially higher than the 150 g Fe m−2 adopted in 2000 but only slightly exceeds the range of typical Fe treatments. Typical Fe dosages targeting P-Fe coupling range between 30 and 500 g Fe m−2, which can be applied in single or repeated treatments (Smolders et al. 2001; Wolter 2010). For Groß Glienicker See, our recalculations yield a total dose amount of 559 g Fe m−2 (33% g g−1 for S-Fe, 20% g g−1 for Corg-Fe, and 47% g g−1 for P-Fe), which is only 12% g g−1 higher than the 500 g Fe m−2 dose used in 1992 but considerably higher than the 242 g Fe m−2 calculated by Kleeberg et al. (2013). These calculations demonstrate that considering Fe consumption by sulphide after sulphate reduction will increase the required Fe dosages and associated costs.
A more sustainable and cost-effective approach would be to integrate the management of SRRs with long-term Fe treatments. For example, if the SRR in Plötzensee was decreased by 50%, the Fe dosage over the same period would be reduced by 44% g g−1. For Groß Glienicker See, a 50% reduction in the SRR would reduce the Fe dosage by 20% g g−1. Practically, SRRs might be decreased through additional management measures targeting the control variables of sulphate reduction. As discussed in the previous section, these are the availability of both sulphate and organic matter. For successful management of SRRs, it is not only decisive whether the change in a variable reliably reduces the sulphate reduction rate, but also whether the variable itself can be effectively controlled. Minimising organic matter in a lake (i.e. primary productivity) is usually part of the goal of Fe dosing. If high sulphate concentrations could impede long-term P retention by excess Fe, they could be most efficiently reduced by managing sulphate loads, which in urban and industrial areas often derive from anthropogenic sources including mining, wastewater treatment plants, and war debris (Zak et al. 2021). In all, how SRRs can be effectively controlled in lakes is not currently well understood, requiring further research to inform the development of more sustainable and cost-effective Fe treatments for P retention.
5 Conclusions
Detailed investigations of chemical sediment composition coupled with one-box lake modelling at the example of two urban lakes enabled to evaluate the long-term success of P retention following Fe amendments alongside the competing effect of S-Fe coupling. Sulphate reduction was identified as a relevant factor controlling the long-term efficacy of Fe amendments with respect to P retention. Over a period of several decades, pyrite formation involving sulphide originating from a continuously high SRR was attributed to the consumption of a large part of the excess Fe. On the one hand, this process can retain sulphide in lake sediments and prevent the negative impacts of this toxic compound. However, Fe amendments aiming to retain P must account for S-Fe coupling over the entire management plan period. One approach is to provide additional Fe to account for S-Fe binding during each year of the management period. Alternatively, additional measures to decrease SRR could be adopted as a more sustainable approach to lake management.
Data availability
The datasets used and/or analysed within the current study are available from the corresponding author upon reasonable request.
Code availability
Not applicable.
Change history
18 August 2022
Missing Open Access funding information has been added in the Funding Note.
References
Bakker ES, Van Donk E, Immers AK (2016) Lake restoration by in-lake iron addition: a synopsis of iron impact on aquatic organisms and shallow lake ecosystems. Aquat Ecol 50:121–135. https://doi.org/10.1007/s10452-015-9552-1
Barber A, Brandes J, Leri A, Lalonde K, Balind K, Wirick S, Wang J, Gelinas Y (2017) Preservation of organic matter in marine sediments by inner-sphere interactions with reactive iron. Sci Rep 7:366. https://doi.org/10.1038/s41598-017-00494-0
Berg JS, Pjevac P, Sommer T, Buckner CRT, Philippi M, Hach PF, Liebeke M, Holtappels M, Danza F, Tonolla M, Sengupta A, Schubert CJ, Milucka J, Kuypers MMM (2019) Dark aerobic sulfide oxidation by anoxygenic phototrophs in anoxic waters. Environ Microbiol 21:1611–1626. https://doi.org/10.1111/1462-2920.14543
Bergmann J, P. Friedel P, Kleeberg R (1998) BGMN – a new fundamental parameter based Rietveld program for laboratory X-ray sources, its use in quantitative analysis and structure investigations. Commission on Powder Diffraction Newsletter, IUCr 20:5–8
Berner RA (1981) A new geochemical classification of sedimentary environments. J Sediment Res 51:359–365. https://doi.org/10.1306/212f7c7f-2b24-11d7-8648000102c1865d
Boers P, Vanderdoes J, Quaak M, Vandervlug J (1994) Phosphorus fixation with iron(III)-chloride - a new method to combat internal phosphorus loading in shallow lakes. Arch Hydrobiol 129:339–351
Cao J, Zhang L, Hong J, Sun J, Jiang F (2019) Different ferric dosing strategies could result in different control mechanisms of sulfide and methane production in sediments of gravity sewers. Water Res 164:114914. https://doi.org/10.1016/j.watres.2019.114914
Caraco NF, Cole JJ, Likens GE (1993) Sulfate control of phosphorus availability in lakes. Hydrobiol 253:275–280. https://doi.org/10.1007/BF00050748
Chapelle FH, Lovley DR (1992) Competitive exclusion of sulfate reduction by Fe(lll)-Reducing bacteria: a mechanism for producing discrete zones of high-iron ground water. Groundwater 30:29–36. https://doi.org/10.1111/j.1745-6584.1992.tb00808.x
Chen M, Sun HQ, Jiang HL (2016) The addition of FeOOH binds phosphate in organic matter-rich sediments. Chem and Ecol 32:432–445. https://doi.org/10.1080/02757540.2016.1150455
Cline JD (1969) Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol Oceanogr 14:454–458. https://doi.org/10.4319/lo.1969.14.3.0454
Cooke GD, Welch EB, Martin AB, Fulmer DG, Hyde JB, Schrieve GD (1993) Effectiveness of Al, Ca, and Fe salts for control of internal phosphorus loading in shallow and deep lakes. Hydrobiol 253:323–335. https://doi.org/10.1007/BF00050758
Davison W (1993) Iron and manganese in lakes. Earth-Sci Rev 34:119–163. https://doi.org/10.1016/0012-8252(93)90029-7
Deneke R, Mischke U (1995) Welche Bedeutung haben Planktonuntersuchungen im Rahmen der Seentherapie? Fallbeispiel: Kombinierte hypolimnische Belüftung und Phosphatfällung im Groß-Glienicker See (Berlin). In: Jaeger D, Koschel R (eds) Verfahren zur Sanierung und Renaturierung stehender Gewässer. Gustav Fischer Verlag, Stuttgard, pp 225–238
Deppe T, Benndorf J (2002) Phosphorus reduction in a shallow hypereutrophic reservoir by in-lake dosage of ferrous iron. Water Res 36:4525–4534. https://doi.org/10.1016/S0043-1354(02)00193-8
Doebelin N, Kleeberg R (2015) Profex: a graphical user interface for the Rietveld refinement program BGMN. J Appl Crystallogr 48:1573–1580. https://doi.org/10.1107/S1600576715014685
Edenborn HM, Brickett LA (2001) Bacteria in gel probes: comparison of the activity of immobilized sulfate-reducing bacteria with in situ sulfate reduction in a wetland sediment. J Microbiol Method 46:51–62. https://doi.org/10.1016/S0167-7012(01)00261-5
Enviteam & LimPlan (2018) Limnologisches Monitoring am Plötzensee 2018 und Entwicklung des Sees nach den Restaurierungsmaßnahmen im Jahre 2000, Berlin
Finke N, Vandieken V, Jørgensen BB (2007) Acetate, lactate, propionate, and isobutyrate as electron donors for iron and sulfate reduction in Arctic marine sediments, Svalbard. FEMS Microbiol Ecol 59:10–22. https://doi.org/10.1111/j.1574-6941.2006.00214.x
Fuchs E, Funes A, Saar K, Reitzel K, Jensen HS (2018) Evaluation of dried amorphous ferric hydroxide CFH-12 (R) as agent for binding bioavailable phosphorus in lake sediments. Sci Total Environ 628–629:990–996. https://doi.org/10.1016/j.scitotenv.2018.02.059
Gächter R, Imboden D (1985) Lake Restoration. In: Stumm W (ed) Chemical Processes in Lakes. J. Wiley & Sons, New York, pp 365–388
Gächter R, Müller B (2003) Why the phosphorus retention of lakes does not necessarily depend on the oxygen supply to their sediment surface. Limnol Oceanogr 48:929–933. https://doi.org/10.4319/lo.2003.48.2.0929
Gołdyn R, Podsiadłowski S, Dondajewska R, Kozak A (2014) The sustainable restoration of lakes—towards the challenges of the Water Framework Directive. Ecohydrol Hydrobiol 14:68–74. https://doi.org/10.1016/j.ecohyd.2013.12.001
Grazulis S, Chateigner D, Downs RT, Yokochi AF, Quiros M, Lutterotti L, Manakova E, Butkus J, Moeck P, Le Bail A (2009) Crystallography Open Database - an open-access collection of crystal structures. J Appl Crystallogr 42:726–729. https://doi.org/10.1107/S0021889809016690
Gunkel G, Pachur H-J (1994) Limnologisch-geomorphologische Untersuchungen im Rahmen der angestrebten Sanierung des Plötzensees/Bezirk Wedding, Gutachterliche Bewertung. Senatsverwaltung für Stadtentwicklung und Umweltschutz Berlin, Berlin
Hamdan HZ, Salam DA (2021) Ferric iron stimulation in marine SMFCs: impact on the microbial structure evolution in contaminated sediments with low and high molecular weight PAHs. J Environ Manage 280:111636. https://doi.org/10.1016/j.jenvman.2020.111636
Hansel CM, Ferdelman TG, Tebo BM (2015) Cryptic cross-linkages among biogeochemical cycles: novel insights from reactive intermediates. Elements 11:409–414. https://doi.org/10.2113/gselements.11.6.409
Hansen J, Reitzel K, Jensen HS, Andersen FØ (2003) Effects of aluminum, iron, oxygen and nitrate additions on phosphorus release from the sediment of a Danish softwater lake. Hydrobiol 492:139–149. https://doi.org/10.1023/A:1024826131327
Hartikainen H, Pitkänen M, Kairesalo T, Tuominen L (1996) Co-occurrence and potential chemical competition of phosphorus and silicon in lake sediment. Water Res 30:2472–2478. https://doi.org/10.1016/0043-1354(96)00139-X
Heinrich L, Rothe M, Braun B, Hupfer M (2021) Transformation of redox-sensitive to redox-stable iron-bound phosphorus in anoxic lake sediments under laboratory conditions. Water Res 189:116609. https://doi.org/10.1016/j.watres.2020.116609
Herndon E, AlBashaireh A, Singer D, Chowdhury TR, Gu BH, Graham D (2017) Influence of iron redox cycling on organo-mineral associations in Arctic tundra soil. Geochim Cosmochim Acta 207:210–231. https://doi.org/10.1016/j.gca.2017.02.034
Hesslein RH (1976) An in situ sampler for close interval pore water studies. Limnol Oceanogr 21:912–914. https://doi.org/10.4319/lo.1976.21.6.0912
Holmer M, Storkholm P (2001) Sulphate reduction and sulphur cycling in lake sediments: a review. Freshw Biol 46:431–451. https://doi.org/10.1046/j.1365-2427.2001.00687.x
Hupfer M, Gächter R, Giovanoli R (1995) Transformation of phosphorus species in settling seston and during early sediment diagenesis. Aquat Sci 57:305–324. https://doi.org/10.1007/BF00878395
Hupfer M, Reitzel K, Kleeberg A, Lewandowski J (2016) Long-term efficiency of lake restoration by chemical phosphorus precipitation: scenario analysis with a phosphorus balance model. Water Res 97:153–161. https://doi.org/10.1016/j.watres.2015.06.052
ILAT Berlin (2003) Der Plötzensee - Limnologische Untersuchungn vor, während und nach den Sanierungsmaßnahmen, Berliner Betrieb für Zentrale Gesundheitliche Aufgaben, Institut für Lebensmittel, Arzneimittel und Tierseuchen Berlin, Fachbereich Umwelt- und Gesundheitsschutz, Berlin
Immers AK, Bakker ES, Van Donk E, Ter Heerdt GNJ, Geurts JJM, Declerck SAJ (2015) Fighting internal phosphorus loading: an evaluation of the large scale application of gradual Fe-addition to a shallow peat lake. Ecol Eng 83:78–89. https://doi.org/10.1016/j.ecoleng.2015.05.034
Jilbert T, Couture R-M, Huser BJ, Salonen K (2020) Preface: restoration of eutrophic lakes: current practices and future challenges. Hydrobiol 847:4343–4357. https://doi.org/10.1007/s10750-020-04457-x
Kappler A, Bryce C (2017) Cryptic biogeochemical cycles: unravelling hidden redox reactions. Environ Microbiol 19:842–846. https://doi.org/10.1111/1462-2920.13687
Kappler A, Bryce C, Mansor M, Lueder U, Byrne JM, Swanner ED (2021) An evolving view on biogeochemical cycling of iron. Nat Rev Microbiol 19:360–374. https://doi.org/10.1038/s41579-020-00502-7
Kelly Vargas KG, Qi Z (2019) P immobilizing materials for lake internal loading control: a review towards future developments. Crit Rev Environ Sci Technol 49:518–552. https://doi.org/10.1080/10643389.2018.1551300
Kleeberg A (1998) The Quantification of sulfate reduction in sulfate-rich freshwater lakes - a means for predicting the eutrophication process of acidic mining lakes? Water, Air, and Soil Pollut 108:365–374. https://doi.org/10.1023/A:1005194404417
Kleeberg A, Herzog C, Hupfer M (2013) Redox sensitivity of iron in phosphorus binding does not impede lake restoration. Water Res 47:1491–1502. https://doi.org/10.1016/j.watres.2012.12.014
Koschorreck M (2008) Microbial sulphate reduction at a low pH. FEMS Microbiol Ecol 64:329–342. https://doi.org/10.1111/j.1574-6941.2008.00482.x
Lamers LPM, Falla SJ, Samborska EM, van Dulken LAR, van Hengstum G, Roelofs JGM (2002) Factors controlling the extent of eutrophication and toxicity in sulfate-polluted freshwater wetlands. Limnol Oceanogr 47:585–593. https://doi.org/10.4319/lo.2002.47.2.0585
Laufer K, Byrne JM, Glombitza C, Schmidt C, Jørgensen BB, Kappler A (2016) Anaerobic microbial Fe(II) oxidation and Fe(III) reduction in coastal marine sediments controlled by organic carbon content. Environ Microbiol 18:3159–3174. https://doi.org/10.1111/1462-2920.13387
Lehtoranta J, Ekholm P, Pitkänen H (2008) Eutrophication-driven sediment microbial processes can explain the regional variation in phosphorus concentrations between Baltic Sea sub-basins. J Mar Sys 74:495–504. https://doi.org/10.1016/j.jmarsys.2008.04.001
Lehtoranta J, Ekholm P, Pitkänen H (2009) Coastal eutrophication thresholds: a matter of sediment microbial processes. Ambio 38:303–308. https://doi.org/10.1579/09-a-656.1
Llobet-Brossa E, Rabus R, Böttcher M, E., Könneke M, Finke N, Schramm A, Meyer R, L., Grötzschel S, Rosselló-Mora R, Amann R (2002) Community structure and activity of sulfate-reducing bacteria in an intertidal surface sediment: a multi-method approach. Aquat Microb Ecol 29:211–226. https://doi.org/10.3354/ame029211
Lürling M, Mackay E, Reitzel K, Spears BM (2016) Editorial - A critical perspective on geo-engineering for eutrophication management in lakes. Water Res 97:1–10. https://doi.org/10.1016/j.watres.2016.03.035
Myrbo A, Swain EB, Engstrom DR, Wasik JC, Brenner J, Shore MD, Peters EB, Blaha G (2017) Sulfide generated by sulfate reduction is a primary controller of the occurrence of wild rice (Zizania palustris) in Shallow Aquatic Ecosystems. J Geophys Res-Biogeosci 122:2736–2753. https://doi.org/10.1002/2017jg003787
Ohle W (1953) Der Vorgang rasanter Seenalterung in Holstein. Naturwissenschaften 40:153–162
Orihel DM, Schindler DW, Ballard NC, Wilson LR, Vinebrooke RD (2016) Experimental iron amendment suppresses toxic cyanobacteria in a hypereutrophic lake. Ecol Appl 26:1517–1534. https://doi.org/10.1890/15-1928
Psenner R, Pucsko R, Sager M (1984) Fractionation of organic and inorganic phosphorus compounds in lake sediments, an attempt to characterize ecologically important fractions. Arch Hydrobiol 70:111–155
Quaak M, van der Does J, Boers P, van der Vlugt J (1993) A new technique to reduce internal phosphorus loading by in-lake phosphate fixation in shallow lakes. Hydrobiol 253:337–344. https://doi.org/10.1007/BF00050759
Rothe M, Frederichs T, Eder M, Kleeberg A, Hupfer M (2014) Evidence for vivianite formation and its contribution to long-term phosphorus retention in a recent lake sediment: a novel analytical approach. Biogeosci 11:5169–5180. https://doi.org/10.5194/bg-11-5169-2014
Rothe M, Kleeberg A, Grüneberg B, Friese K, Perez-Mayo M, Hupfer M (2015) Sedimentary sulphur:iron ratio indicates vivianite occurrence: a study from two contrasting freshwater systems. PLoS ONE 10:e0143737. https://doi.org/10.1371/journal.pone.0143737
Rothe M, Kleeberg A, Hupfer M (2016) The occurrence, identification and environmental relevance of vivianite in waterlogged soils and aquatic sediments. Earth-Sci Rev 158:51–64. https://doi.org/10.1016/j.earscirev.2016.04.008
Schauser I, Lewandowski J, Hupfer M (2003) Decision support for the selection of an appropriate in-lake measure to influence the phosphorus retention in sediments. Water Res 37:801–812. https://doi.org/10.1016/S0043-1354(02)00439-6
Schulz H, Zabel M (2006) Marine Geochemistry. Springer, Berlin Heidelberg
Senatsverwaltung für Stadtentwicklung Berlin (2002) Gewässeratlas von Berlin, Berlin
Smolders AJP, Nijboer RC, Roelofs JGM (1995) Prevention of sulphide accumulation and phosphate mobilization by the addition of iron(II) chloride to a reduced sediment: an enclosure experiment. Freshw Biol 34:559–568. https://doi.org/10.1111/j.1365-2427.1995.tb00913.x
Smolders AJP, Lamers LPM, Moonen M, Zwaga K, Roelofs JGM (2001) Controlling phosphate release from phosphate-enriched sediments by adding various iron compounds. Biogeochem 54:219–228. https://doi.org/10.1023/a:1010660401527
Smolders AJP, Lamers LPM, Lucassen ECHET, Van der Velde G, Roelofs JGM (2006) Internal eutrophication: how it works and what to do about it - a review. Chem Ecol 22:93–111. https://doi.org/10.1080/02757540600579730
Urban NR (1994) Retention of Sulfur in Lake Sediments. In: Baker LA (ed) Environmental Chemistry of Lakes and Reservoirs. American Chemical Society, pp 323–369
Vandieken V, Finke N, Thamdrup B (2014) Hydrogen, acetate, and lactate as electron donors for microbial manganese reduction in a manganese-rich coastal marine sediment. FEMS Microbiol Ecol 87:733–745. https://doi.org/10.1111/1574-6941.12259
Wang C, Jiang H-L (2016) Chemicals used for in situ immobilization to reduce the internal phosphorus loading from lake sediments for eutrophication control. Crit Rev Environ Sci Technol 46:947–997. https://doi.org/10.1080/10643389.2016.1200330
Wang G, Spivack AJ, Rutherford S, Manor U, D’Hondt S (2008) Quantification of co-occurring reaction rates in deep subseafloor sediments. Geochim Cosmochim Acta 72:3479–3488. https://doi.org/10.1016/j.gca.2008.04.024
Wilfert P, Meerdink J, Degaga B, Temmink H, Korving L, Witkamp G-J, Goubitz K, Loosdrecht MCM (2019) Sulfide induced phosphate release from iron phosphates and its potential for phosphate recovery. Water Res 171:115389. https://doi.org/10.1016/j.watres.2019.115389
Wolter K-D (2010) Restoration of eutrophic lakes by phosphorus precipitation, with a case study on Lake Gross-Glienicker. In: Eiseltová M (ed) Restoration of Lakes, Streams, Floodplains, and Bogs in Europe: Principles and Case Studies. Springer, Dordrecht, pp 85–99
Wu S, Zhao Y, Chen Y, Dong X, Wang M, Wang G (2019) Sulfur cycling in freshwater sediments: a cryptic driving force of iron deposition and phosphorus mobilization. Sci Total Environ 657:1294–1303. https://doi.org/10.1016/j.scitotenv.2018.12.161
Yang H, Liu J, Hu P, Zou L, Li Y-Y (2019) Carbon source and phosphorus recovery from iron-enhanced primary sludge via anaerobic fermentation and sulfate reduction: Performance and future application. Bioresour Technol 294:122174. https://doi.org/10.1016/j.biortech.2019.122174
Zak D, Kleeberg A, Hupfer M (2006) Sulphate-mediated phosphorus mobilization in riverine sediments at increasing sulphate concentration, River Spree, NE Germany. Biogeochem 80:109–119. https://doi.org/10.1007/s10533-006-0003-x
Zak D, Hupfer M, Cabezas A, Jurasinski G, Audet J, Kleeberg A, McInnes R, Kristiansen SM, Petersen RJ, Liu H, Goldhammer T (2021) Sulphate in freshwater ecosystems: a review of sources, biogeochemical cycles, ecotoxicological effects and bioremediation. Earth-Sci Rev 212:103446. https://doi.org/10.1016/j.earscirev.2020.103446
Zhao Y, Zhang Z, Wang G, Li X, Ma J, Chen S, Deng H, Annalisa O-H (2019) High sulfide production induced by algae decomposition and its potential stimulation to phosphorus mobility in sediment. Sci Total Environ 650:163–172. https://doi.org/10.1016/j.scitotenv.2018.09.010
Zou YC, Grace MR, Roberts KL, Yu XF (2017) Thin ferrihydrite sediment capping sequestrates phosphorus experiencing redox conditions in a shallow temperate lacustrine wetland. Chemosphere 185:673–680. https://doi.org/10.1016/j.chemosphere.2017.07.052
Acknowledgements
We are thankful to current and former members of the former IGB department of Chemical Analytics and Biogeochemistry for support in fieldwork and chemical analyses, for contributing earlier pore water and monitoring data and for discussions on previous versions of the manuscript. In particular, we thank F. Auer, C. Herzog, S. Jordan, A. Kleeberg, M. Lau, C. Levertz, A. Lüder, T. Rossoll, C. Schmalsch, E. Schütte, G. Siegert, N. Welteke, and D. Zak. The sulphate concentrations in Groß Glienicker See and Plötzensee were provided by A. Köhler (Senatsverwaltung für Umwelt, Verkehr und Klimaschutz, Berlin), K.-D. Wolter (Systeminstitut Aqua Terra e.V.), and A. Kleeberg (Landeslabor Berlin-Brandenburg). A. Köhler also provided previous reports on the management measures at both lakes. We would like to thank Editage (www.editage.com) for English language editing.
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was funded by the German Research Foundation (DFG) as part of the Research Training Group “Urban Water Interfaces (UWI)” (GRK 2032/2, Project F3: “Controlling of phosphorus fluxes in urban systems: Analogous processes in limnic sediments and sewage sludges”).
Author information
Authors and Affiliations
Contributions
LH and MH conceived and designed the analyses; LH collected and evaluated the data; LH and JD conducted and evaluated the XRD analyses; LH wrote the manuscript. All authors read, edited, and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Responsible editor: Shiming Ding
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Heinrich, L., Dietel, J. & Hupfer, M. Sulphate reduction determines the long-term effect of iron amendments on phosphorus retention in lake sediments. J Soils Sediments 22, 316–333 (2022). https://doi.org/10.1007/s11368-021-03099-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11368-021-03099-3