
Abstract
Objectives
Isolation and culture of distinct primary endometrial cells are key to reliable in-vitro models to investigate the uterine immune response and optimse new disease interventions. Details on the isolation method and purity of distinct cell populations is lacking in currently available protocols leading to inconsistent results across laboratories.
Methods
Bovine endometrial tissue from non-pregnant bovine uteri were collected immediately post-mortem and separated using differential size filtering. Isolations (n = 15) yielded an average of 3.1 × 105 ± 0.7 × 105 epithelial cells and 1.88 × 106 ± 5.44 × 105 stromal fibroblasts per uterine horn. Following expansion in culture, the purity of cell populations was confirmed using morphology and positive staining for cytokeratin and vimentin which identifies epithelial and stromal fibroblast populations, respectively. Using PCR, cDNA from both cell populations was negative for CD45, a marker of immune cells.
Results
On challenge with a bacterial PAMP (LPS), epithelial and stromal fibroblasts showed a marked increase in the expression of the inflammatory mediators IL8, IL6, S100A8 and S100A9, with both cell populations displaying distinct expression profiles. Here we provide a detailed methodology on the culture of primary bovine endometrial epithelial and stromal cells and demonstrate these cells provide a physiologically relevant model for studies of endometrial inflammation and its regulation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The endometrium, the mucosal lining of the uterus, plays a complex role maintaining a homeostatic environment to allow for successful implantation and pregnancy while tolerating the local microbiome and also protecting against invading pathogens. The bovine endometrium is composed of a single layer of columnar epithelial cells which provide a barrier overlying the stromal matrix (Frandson et al. 2009). While the epithelial cells are in constant interaction with immunogenic material from the external environment and commensal microbiome, during parturition and the subsequent process of tissue remodelling the epithelial layer can be disrupted. This results in stromal fibroblast cells becoming exposed to microbial associated molecular patterns (MAMPs) and endogenous danger associated molecular patterns (DAMPs), thereby activating inflammatory pathways which can result in uterine disease if not resolved. Identifying the inflammatory pathways operating in and between endometrial cells is crucial to understanding the pathology behind uterine disease and the development of targeted therapeutics for local pathology.
In addition to their barrier function, epithelial cells are potent producers of a wide selection of host defence peptides (HDPs) including defensins, cathelicidins and whey acid proteins, as well as pro-inflammatory cytokines and chemokines such as IL-6 and IL-8 (Cronin et al. 2012; Davies et al. 2008; Narciandi et al. 2011; Whelehan et al. 2014). Stromal fibroblast cells also produce pro-inflammatory cytokines and play a role in defence against viral infection (Donofrio et al. 2007). Possession of such a wide repertoire of protective mechanisms demonstrates the pivotal roles epithelial and stromal fibroblast cells play in the early stages of the innate immune response before immune cell recruitment (MacKintosh et al. 2013). However detailed investigation into the innate immune properties of endometrial cells is hindered by difficulty in isolating and culturing these distinct cell populations.
While in-vivo models have classically been used to capture the overall complexity of immune responses, the visualisation of cell type-specific signalling pathways, particularly under controlled stimulation environments, poses a challenge. In addition, in-vivo modelling of bovine immune responses is expensive and restricted by the large inter-animal variation due to extraneous influences (nutrition, housing etc.). In order to examine the immune mechanisms operating within the endometrium, a model that allows us to easily manipulate and control experimental variables while providing quantitative readouts would be useful. Endometrial cell cultures, including the use of cell lines, explants and primary cell populations, have been developed (Borges et al. 2012; Fortier et al. 1988; Herath et al. 2006; Staggs et al. 1998). While cell lines provide a good basis for investigation, they come with limitations including the lack of inter-animal variation provided by primary cultures. In addition, the genetic manipulation require to immortalise a cell results in altered phenotype, function and responsiveness (Kaur and Dufour 2012). The culture of tissue explants provides a potential link between cellular studies and in-vivo models but is restricted by the limited viability of explants in-vitro. Primary cell cultures provide more realistic insight into immune mechanisms, retaining the physiology and genetic makeup of their cellular counterparts in-vivo and demonstrating a level of inter-animal variation which is more reflective of the whole population.
Primary bovine endometrial cell isolation and culture has been widely reported in past literature (Fortier et al. 1988; Herath et al. 2006), although these reports often lack crucial details allowing for their methods to be replicated. Here we detail a protocol for the isolation, culture and characterisation of pure, distinct populations of endometrial epithelial and stromal fibroblast cells, aiming to provide a useful model for the investigation of endometrial cell-mediated inflammation.
Methods
Cell isolation and culture
Bovine female reproductive tracts were collected at a local abattoir within 15 min of slaughter. Collected tracts were identified as being free from disease and visible infection. Only tracts in the early luteal stage of oestrous (as identified by the presence of a stage 1 corpus luteum on one ovary) were used. Details of age and breed were also recorded at the time of collection.
The external surface of the tract was washed in 70% industrial methylated spirits (IMS) and the uterine horn ipsilateral to the corpus luteum was opened longitudinally with sterile scissors. The exposed endometrium was washed in PBS (Gibco, Paisley, UK) supplemented with 50 IU/ml penicillin, 50 IU/ml streptomycin (Gibco) and 2.5 g/ml amphotericin B (Sigma-Aldrich, Poole, UK). The endometrial surface was dissected using a sterile scissors and forceps. Dissection was performed at the abattoir with the tissue harvested into a 50 ml tube containing 20 ml of RPMI (Gibco) supplemented with antibiotics as above. Samples were transported to the laboratory at room temperature within 90 min of collection.
On arrival at the laboratory, tissue was washed twice in room temperature HBSS (Gibco, Madison, WI, UK) supplemented with antibiotics. The tissue was then chopped into fine pieces (approximately 1-3 mm3 in size) using a sharp scissors and scalpels. The tissue was then incubated in HBSS with antibiotics at 37 °C for 10 mins in order to slowly return the tissue temperature to 37 °C. HBSS was removed before the addition of 20 ml digestive solution (pre-warmed to 37 °C). Digestive solution (100 ml) consisted of 375 BAEE units of trypsin-EDTA (Sigma-Aldrich), 50 mg collagenase II (Sigma-Aldrich), 100 mg bovine serum albumin (BSA)(Sigma-Aldrich) and 10 mg DNase I (Sigma-Aldrich) made up to 100 ml with HBSS and 0.2 μm sterile filtered (Sartorius, Göttingen, Germany). Samples incubated with digestive solution were placed in a 37 °C shaking incubator at 150 rpm for 1 h.
The resulting mixture was filtered through a 70 μm nylon mesh cell strainer (Becton, Dickinson, Flintshire, UK) to remove any cell debris with the filtrate being collected in a tube containing HBSS with 10% FBS (Gibco). This solution was then passed through a 40 μm cell strainer (Pluristrainer, Pluiselect, Leipzig, Germany) to isolate the stromal cells. As the epithelial cells were too large to pass through the 40 μm cell strainer they remained on the strainer. The epithelial cells could then be isolated by turning the 40 μm cell strainer upside down and washing the epithelial cells into a clean 50 ml tube using 10 ml of HBSS supplemented with 10% FBS. Cells were then pelleted by centrifugation at 700 x g for 10 min. Red blood cells were lysed by re-suspending the pellet in 1 ml sterile water (Gibco) before addition of 4 ml of complete growth media. This consisted of RPMI 1640 supplemented with antibiotics as above, 10% FBS and 1X insulin, transferring, selenium supplement with ethanolamine (ITS-X) (Gibco).
Cells were pelleted as before and re-suspended in complete growth media. Cells were counted using a haemocytometer with trypan blue (Sigma-Aldrich, Poole, UK) staining to distinguish live from dead cells. Cell counts were adjusted to 3 × 105 cells/ml and 8 × 105 cells/ml for epithelial and stromal fibroblasts respectively and 1 ml was were seeded into 75cm2 vented tissue culture flasks (Greiner Bio-One, Kremsmünster, Austria) with complete growth medium, which was stored in a 37 °C incubator in 5% CO2. Cells were allowed to reach 85% confluency before plating, with media changing every 48 h. Visual examination of cell morphology under the light microscope was used to check purity of cultures in addition to characterisation based on expression of differing cytoskeletal proteins.
Isolation and culture of PBMCs from whole blood
Bovine peripheral blood mononuclear cells (PBMCs) were isolated from whole blood samples collected in 9 ml vacutainers containing Heparin anticoagulant (Greiner Bio-One, Kremsmünster, Austria). Whole blood was diluted 1:1 with HBSS and mixed gently before 25 ml was layered onto 15 ml of sterile Ficoll-paque density gradient medium (Fisher Scientific, New Hampshire, US) in a 50 ml tube. The samples were then centrifuged at 800×g for 25 mins with the centrifuge break turned off. The mononuclear cells were carefully removed with a pastette into a new 50 ml tube and made up to 10 ml with wash buffer (HBSS and 5% FBS). Cells were pelleted by centrifugation at 300×g for 5 mins. Any red blood cells carried over during the separation process were lysed by re-suspending the cells in red blood cell lysis buffer (Sigma-Aldrich). The cell pellet was then re-suspended in complete RPMI media (containing 10% FBS and 1% Pen-Strep) and cells were counted using a haemocytometer and trypan blue staining. Cell were then seeded at appropriate densities in tissue culture plates containing media. The cells were incubated at 37 °C with 5% CO2.
Processing of endometrial samples for histological analysis
Endometrial biopsies were removed from 10% neutral buffered formalin (Sigma-Aldrich) and immersed in 70% IMS for 24 h before overnight paraffin wax embedding in Surgipath cassettes (Leica Biosystems, Newcastle, UK). Embedded biopsies were sectioned at 3 μm, dewaxed by immersion in histoclear (National Diagnostics, Atlanta, GA, USA) and rehydrated by immersion in 100%, 90%, 80% and 75% ethanol (Sigma-Aldrich). Haemotoxylin (Leica Biosystems) was applied to the tissue sections for 10 min before immersion in Scott’s tap water (30 g magnesium sulphate (Sigma-Aldrich) and 2 g sodium bicarbonate (Sigma-Aldrich) in 3 L tap water) for 5 mins. Tissue sections were progressively dehydrated by a reversal of the ethanol immersion steps used previously and mounted with Surgipath mounting medium (Leica Biosystems). Slides were examined using an Olympus BX51 upright microscope (Leica Biosystems).
Immunofluorescent staining of endometrial cell cytoskeletal proteins
Epithelial or stromal cells were seeded at a density of 1 × 105 cells/ml into 24-well tissue culture plates in complete growth medium, allowing one well each for cytokeratin staining (expressed by epithelial cells), vimentin staining (expressed by stromal cells), an IgG isotype control and an unstained well, and allowed to rest for 24 h. Growth medium was removed, the cell monolayer was rinsed three times with PBS and fixed with 500 μl 10% neutral-buffered formalin at room temperature for one hour. Formalin was removed and the cells washed three times with PBS before the primary antibody was added. Mouse anti-human cytokeratin antibody (Dako/Agilent Technologies, Santa Clara, CA, USA), mouse anti-human vimentin antibody (Sigma-Aldrich) or IgG1 isotype control (Sigma-Aldrich) were applied as appropriate using 400 μl per well of a 1:100 dilution in PBS. Cells were incubated at 4 °C with rocking overnight. An FITC-conjugated polyclonal goat anti-mouse secondary antibody (Abcam, Cambridge, UK)) was applied to all wells in a 1:250 dilution, incubating at 37 °C for 1 h. After rinsing three times in PBS, DAPI stain (Sigma-Aldrich) to detect nuclei was applied in a 1:250 dilution for 10 min at 37 °C. An Olympus IX81 inverted microscope was used to visualise immunofluorescent stained cells, with positive cytokeratin staining in the absence of vimentin staining taken to indicate a pure endometrial epithelial cell culture and the inverse being characteristic of stromal cells.
Immunocytochemistry
Epithelial or stromal fibroblast cells were seeded at a density of 1 × 105 cells/ml or 0.5 × 105 cells/ml respectively into wells of the Millicell EZ SLIDE 8 well glass culture slides (Millipore), and allowed to rest for 24 h. Cells were washed three times in PBS before being fixed in ice-cold methanol for 10 mins at room temperature. Cells were then washed a further three times in PBS before incubation in 3% H2O2 (Sigma-Aldrich)) for 20 mins at room temperature, followed by blocking in 10% BSA (VWR) for 1 h at room temperature. Cells were then incubated overnight with mouse anti-human cytokeratin antibody, mouse anti-human vimentin antibody or IgG1 isotype control using 100 μl per well of a 1:100 dilution in PBS. Cells were then washed three times in PBS before incubation with the secondary antibody (Dako) for 30 mins at room temperature. Cells were washed a further three times before incubation with the chromogen diaminobenzidin (DAB) (Dako) for 10 mins. Cells were washed a further three times before being mounted with Surgipath mounting medium. Slides were examined using an Olympus BX51 upright microscope.
Western blotting
Epithelial or stromal fibroblasts cells (1 × 106 cells/ml) were harvested in RIPA buffer supplemented with the protease inhibitors leupeptin (2 μg/ml), aprotonin (2 μg/ml), sodium orthovanadate (4 μg/ml) and phenylmethylsulphonyl fluoride (100 μg/ml). Protein quantification was performed using BCA Protein Assay Reagents (Pierce Biotech) according to the manufacturer’s instructions. A standard curve was generated each time with BSA standards and the protein concentration of each sample was calculated from this. From these values, protein samples were normalised relative to the sample of lowest concentration in 20 μl dilutions. Loading buffer (4X Tris-glycine SDS β-mercapthenol) was added to each sample (5 μl) and boiled at 95 °C for 5 min.
Prior to SDS-PAGE a 15% running gel, topped with a 4% stacking gel, were prepared. The gels were loaded into the running tank and 1X Running buffer (0.025 M Tris, 0.192 M glycine, 0.1% SDS) was added and poured into the upper and lower reservoirs of the apparatus. The samples and 3 μl molecular marker was used per gel. The gel was run at 110 V for 45 mins. After the run, the stacking gel was removed and the running gel retained for transfer to PVDF membranes.
Transfer buffer (0.048 M Tris, 0.039 M glycine, 20% methanol, 0.00375% SDS), PVDF membrane (0.2 μm pore size) and blotting pads were prepared. The transfer membrane were cut to correct size and activated by immersion in methanol for 30 s and then in dH20 for 2 mins. The membrane and blotting pads were then soaked in transfer membrane for 5 mins. Once the gel had ran, it was removed from the gel apparatus and placed in transfer buffer for 5 mins. The gel was then transferred to the transfer machine where it was sandwiched between blotting pads and a transfer membrane. The transfer machine was connected to a voltmeter and run at 10 V (250 mA) for 20 mins.
Once the membrane was removed from the transfer machine, it was blocked to prevent non-specific binding of antibody with 5% milk in PBS tween (PBS-T) for 1 h. After blocking, the primary antibody was added and the blot was left rocking in the cold room overnight. Primary antibody was diluted to a concentration of 1:1500 in 30mls of PBS-T. Before adding the secondary antibody, the membrane was washed for 5 mins three times in PBS-T. Secondary antibodies were applied (1:5000 dilution in 5% milk) and the blot was left on the rocker for 1 h at room temperature. The blot was then washed again for 5 mins three times, before being incubated in ECL (Biorad) and visualised on the Biorad ChemiDoc MP System.
Cell treatments
For stimulations of epithelial and stromal fibroblast populations, cells were plated at a density of 1.5 × 105 cells/ml in a 24 well plate and left to rest for 24 h before stimulation. Following the 24 h rest period, medium was removed and replaced with control medium or medium containing 2 μg/ml LPS (Enzo Life Sciences, Exeter, UK) for 3, 6, 12 or 24 h. Following stimulation cells were lysed in TRIzol reagent (Thermo Fisher Scientific, Paisley, UK) for mRNA extraction and stored at −80 °C.
Quantitative PCR (qPCR)
Total RNA was extracted using TRIzol reagent according to manufacturer’s instructions. RNA quantity was calculated using ND-1000 NanoDrop spectrophotometer (Thermo Fisher Scientific). One microgram of RNA was reverse transcribed into cDNA using the OmniScript kit (Qiagen, Crawley, UK) with Oligo (dT) primers (Qiagen) according to the manufacturer’s instructions. cDNA solutions were diluted 1:10 before qPCR analysis.
qPCR was performed using the following primers: IL-6F CAGGAACGAAAGAGAGCTCCA; IL-6R AATGGAGTGAAGGCGCTTGT; IL-8F ATTCCACACCTTTCCACCCC; IL-8R TTGCTTCTCAGCTCTCTTCACA; S100A8F ATTTTGGGGAGACCTGGTGG; S100A8R GCTTCCAGGCCCACCTTTAT; S100A9F GCTTCTCGGCTTGGTAGGAG; S100A9R CCTCCATTTTCCCGCCTTCT; H3F3AF CATGGCTCGTACAAAGCAGA; H3F3AR ACCAGGCCTGTAACGATGAG. Primers were designed using the Primer BLAST software to be intron spanning where possible. Optimal primer concentrations (IL8: 300 nM, IL6: 500 nM, S100A8: 300 nM, S100A9: 500 nM H3F3A: 100 nM) were assessed by titrating different final concentrations and dissociation curves were examined for the presence of a single product.
Quantitative PCR was run using the PowerUp SYBR Green Master Mix (Thermo Fisher Scientific) on a StepOnePlus Real-Time PCR System (Thermo Fisher Scientific) using the following parameters: 95 °C for 20 s, followed by 40 cycles of 95 °C for 3 s and 60 °C for 30 s and a final amplicon dissociation step at 95 °C for 15 s, 60 °C for 1 min and 95 °C for 15 s. H3F3A was found to be the most stably expressed reference gene from a panel of reference genes tested using GeNorm software and was subsequently used to generate normalized relative expression values (Vandesompele et al. 2002).
The HotStar Master Mix PCR kit (Qiagen) was used to carry out a PCR reaction to detect transcription of the protein tyrosine phosphatase, receptor type C (PTPRC) gene, encoding the pan-leukocyte marker CD45, within endometrial cell cultures. A 10 μl reaction volume contained 0.3 μl endometrial cell cDNA, 1X CoralLoad reaction buffer, 200 μM dNTP solution, 0.3 μl HotStarTaq polymerase enzyme and 300 nM PTPRC-specific primers (Forward: TGCAACCGCTCTCTCAACCATA, Reverse: CTTGCTTGGCTTTGCTGGATCT), with nuclease free water making up the remainder. cDNA prepared from bovine PBMCs was used as a positive control for PTPRC amplification. The constitutively expressed ribosomal protein S9 (RPS9F GCGTCTGTTCGAAGGTAATGC; RPS9R AAGTCGATGTGCTTCTGCGA) was amplified from all samples to ensure poor cDNA quality did not account for a lack of amplification. A non-template control without cDNA was run for both gene assays. The PCR reaction was carried out in a Techne Prime thermocycler (Bibby Scientific, Staffordshire, UK) using the following thermocycling conditions: 95 °C for 5 min and 40 cycles of 95 °C for 30 s, 60 °C for 1 min, 72 °C for 1 min. Results were assessed by presence or absence of a DNA product of expected size on a 2% agarose gel after electrophoresis.
Data analysis
For gene expression analysis, qPCR data was converted to gene expression fold changes using the 2-ΔΔCq method (where Cq represents the quantification cycle) (Schmittgen and Livak 2008). H3F3A was used as a reference gene following GeNorm analysis. Statistical analysis of qPCR data was performed using a non-parametric Kruskall-Wallis test with Dunns multiple comparison post-hoc test as implemented in Graphpad Prism 7 software.
Results
Optimisation of tissue isolation
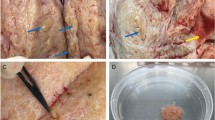
Dissection was performed on the uterine horn ipsilateral to the corpus luteum with the uterine horn dissected from the bifurcation of the uterine horns to the top of the uterine horn (Supplementary Figure 1A). The oestrous cycle stage of each tract was determined by examining the ovaries and identifying the presence of a stage I corpus luteum (Supplementary Figure 1B). Tracts in the early luteal phase of oestrous were chosen for basal levels of progesterone which would therefore not impact on inflammatory mediator production (Butts et al. 2007; Stites and Siiteri 1983). Tracts were collected from healthy cows who were on average 91.6 months old (7.6 years) and predominantly Holstein-Friesians (Supplementary Figure 1 C-D).
Dissection of the upper functional layer of the endometrium was optimised using a curved dissection scissors and forceps. The endometrial lining was dissected in thin strips using a curved scissors and forceps (Supplementary Figure 1G). The forceps was used to hold the edge of the endometrial lining while the curved scissors passed underneath the endometrial lining, cutting away the fibres that tie it to the lower functional layer composed of mainly stromal fibroblasts. Harvested tissue was immediately stored in transport media. We also investigated the use of a curette, an instrument with a sharp loop at the end of a long handle commonly used in human surgery to sample the endometrium. Tissue isolation using a curette involves running the loop over the endometrium to harvest the epithelial layer. While dissection with the curette was faster and required less skill, we found we recovered higher cell yields (particularly epithelial cells) by dissecting with the scissors, possibly due to less tissue recovered using the curette (Supplementary Figure 1H).
Histology images taken before and after dissection (Supplementary Figure 1 E, F) show that the uppermost functional layer containing the epithelium is removed during dissection. In the intact endometrium (Supplementary Figure 1E) the luminal epithelium forming a barrier overlying the stroma is observed. The stroma also contains glandular epithelium and spiral arterioles which maintain the blood supply.
Optimisation of tissue transport
Our initial protocol for cell isolation and transport involved tract collection at the abattoir and transportation on ice to the laboratory (transport time taking approximately 1–1.5 h). However cell viability using this approach was low. To improve cell viability, we harvested the endometrial tissue at the abattoir. Collected tracts were cleaned with 70% IMS and the exposed endometrium washed with PBS supplemented with 50 IU penicillin, 50 IU streptomycin and 2.5 μg/ml amphotericin B prior to dissection and transported the dissected tissue back in culture media (RPMI supplemented with 10% FBS). This resulted in problems with bacterial contamination of our isolated cultures, probably due to the lack of a proper aseptic environment while performing the dissection in the abattoir. To address this issue we investigated several different transport conditions. Tissue was transported in media with or without FBS and with or without antibiotics. The tissue was also transported on ice or at room temperature. Use of media lacking FBS but containing antibiotics and transported at room temperature was found to result in high yields of viable cells with no contamination.
Cell isolation by differential size filtration
Disruption of the harvested tissue was performed using both mechanical dissociation and by enzymatic dissociation. Mechanical dissociation was performed by using a sharp scissors to chop the tissue finely (1-3 mm in size) before proceeding to enzymatic dissociation. An enzymatic digestion solution had previously been optimised containing trypsin (375 BAEE units), collagenase (50 mg), BSA (100 mg) and DNase (10 mg) made up to 100 ml HBSS (Fortier et al. 1988; Herath et al. 2006). Low concentrations of trypsin (375 BAEE units) were used to avoid damage to the cells. Trypsin concentration was determined using activity units rather than volume due to large batch-batch variation in trypsin activity (MacKintosh et al. 2013). We found collagenase was able to digest the extracellular matrix, separating the epithelial and stromal cells but did not dissociate epithelial cells, allowing them to remain in viable clusters or islands. DNase was used to remove DNA released from lysed cells which can promote re-aggregation of dissociated cells. Incubation times ranging from 1 to 2 h have been reported (Cheng et al. 2003; Fortier et al. 1988; Herath et al. 2006). We found incubation at 37 °C for 1 h in a shaking incubator was sufficient for cell dissociation.
The initial protocol we used for isolation of pure populations of epithelial and stromal fibroblast cells involved adding the total cell isolate to a tissue culture flask and allowing 24 h for stromal fibroblast cells to adhere before the epithelial cells still in suspension were harvested and re-plated in a new flask (Herath et al. 2006). However, this approach did not yield pure populations of either cell type and so we separated by size using filters to address this problem. Stromal cells easily passed through 40 μm filters while epithelial islands were retained. Flipping the filters and backwashing the epithelial cells allows for the easy isolation of pure epithelial populations. Any residual stromal cell contamination of these epithelial populations could be removed in the flasks by accutase, as stromal cells lift in the first 5 min of accutase treatment while epithelial cells take a minimum of 15 min to lift. Removal of red blood cells was performed by the addition of 1 ml sterile water to the cell pellet followed by gentle pipetting. Initial attempts at removing red blood cells included the use of red blood cell lysis buffer and vortexing for 30 s. However, this resulted in a higher proportion of epithelial or stromal cell death.
Cell culture, characterisation and cryopreservation
During initial isolations, flasks were coated with 0.1% gelatin to promote cell adherence. We also examined the tissue culture growth media and its components. Using different amounts of FBS and trying supplements such as Insulin-Transferrin-Selenium (ITS-X) we found that the optimal growth media contained 10% FBS and 5% ITS-X. Purified cell populations were identified by morphology. Epithelial cells grow in small islands that ultimately combine to form a confluent monolayer (Fig. 1a, c) while stromal fibroblast cells were found scattered throughout the flask (Fig. 1b, d). Using the above process, stromal fibroblast cells (1.88 × 106 cells per uterine horn) and epithelial cells (3.05 × 105 cells per uterine horn) of >95% viability were isolated following this isolation process (Fig. 1e). The ability of both cell populations to proliferate in culture is shown by the cell culture images and growth curve in Fig. 1, with both cell populations maintaining a steady increase in cell numbers over the first 12 days of culture (Fig. 1f-g).

Endometrial epithelial and stromal fibroblast cells exhibit distinct morphological features. Representative images of endometrial epithelial and stromal cells showing distinct morphological differences between the two cell populations. a Small cluster of epithelial cells visible after 2 days of culture. b Stromal cells appear scattered throughout the flask following 2 days of culture. c Pure population of endometrial cells following 14 days of culture. d Pure population of endometrial stromal fibroblast cells following 6 days of culture. Scale bar indicates 100 μm. Images were taken using an Olympus IX81 inverted microscope with camera attached. e Average epithelial cell yield isolated from tissue was 3.08 × 105 ± 0.66 × 105, average stromal cell yield was 1.88 × 106 ± 5.44 × 105 (n = 15). Following isolation cell pellets were counted under a light microscope using a haemocytometer and trypan blue staining. Values presented are mean cell counts per uterine horn ± SEM. f Growth curve showing the increase in cell populations over 12 days of culture. Endometrial epithelial (▪) and stromal (•) cells were plated at a density of 9 × 103 cells into six-well tissue culture plates and counted over the course of 12 days using a haemocytometer and trypan blue staining. Values presented are mean cell count ± SEM. g Growth curve showing the increase in cell populations over a period of 96 h as measured by the xCelligence real time cell analyser. h Growth curve showing increase in cell populations following cryopreservation. i Doubling time across consecutive passages of both endometrial epithelial (▪) and stromal cells (•). Cells were plated at a density of 9 × 103 cells into six-well tissue culture plates and over 16 days of culture, cells were detached using trypsin, diluted in trypan blue and counted using a haemocytometer before being reseeded into the 6 well plate at the same density
We next investigated whether endometrial cells could be cryopreserved or passaged using standard protocols. The growth curve in Fig. 1(h-i) shows that both cell populations recover well from cryopreservation and continue to proliferate in culture. No morphological differences were observed in the cryopreserved cells after reconstitution. Beyond two passages morphological differences begin to appear in both cell populations and both populations become slower to proliferate, taking approximately 5–6 days to double their population as opposed to 2 days after just one passage.
Purity of the epithelial and stromal cells populations was demonstrated by the expression of cell specific cytoskeletal proteins (Fig. 2). Epithelial cells express cytokeratin while stromal cells express vimentin (Franquemont et al. 1991; Wonodirekso et al. 1993) (Fig. 2(a & b). Immunofluorescent, immunocytochemical and western blotting staining for the cytoskeletal proteins cytokeratin and vimentin confirmed qPCR data (Fig. 2c-h).

Endometrial cell populations can be identified based on their differential expression of cytoskeletal proteins. Relative keratin-18 and vimentin mRNA gene expression in (a) endometrial epithelial and (b) stromal cell populations. Immunofluorescence staining of (c, e) endometrial epithelial and (d, f) stromal fibroblast cells with antibodies against cytoskeletal proteins cytokeratin and vimentin. Scale bar indicates 100 μm. Western blotting staining of (g) endometrial epithelial and (h) stromal fibroblast cell lysates with antibodies against cytokeratin and vimentin. Gel electrophoresis image of PCR products for the PTPRC gene (which encodes CD45 protein) and RPS9 (reference gene) from (i) endometrial epithelial and (j) stromal fibroblast cells. cDNA from PBMCs was included as a positive control
As we aim to use our primary culture model to investigate the innate immune responses of endometrial cells we needed to be certain that there was no immune cell contamination of our cultures. A PCR assay for PTPRC, the gene that encodes CD45, a marker of immune cells was optimised. PBMC cDNA was used as a positive control. The housekeeping gene RPS9 was included to ensure cDNA quality. Cultures were negative for CD45 (Fig. 2i-j) indicating absence of immune cell contamination, again confirming the purity of our endometrial cell populations.
Expression of inflammatory mediators
Having generated pure endometrial cell populations, a time course of the response of these cells to the microbial PAMP, LPS, was performed to investigate their ability to detect and respond to inflammatory stimuli compared to the more classically defined immune cells present in PBMCs. Results indicate that epithelial and stromal fibroblast populations display significantly divergent response profiles. Constitutive expression of IL8 and IL6 mRNA, as quantified using qPCR, was higher in epithelial cells than stromal fibroblasts or unmatched PBMCs (in which levels were comparable) (Fig. 3a and e). IL-8 mRNA was significantly induced at earlier time-points (3 h post-stimulation) in epithelial cells while stromal cells show a higher induction of IL8 at 24 h post-stimulation, similar to PBMCs which demonstrate peak IL8 mRNA production at 12 h post stimulation (Fig. 3b-d). Induction of IL6 mRNA demonstrated a similar pattern, with peak production of IL6 occurring in epithelial cells 3 h post stimulation, and lower induction of IL6 by stromal fibroblast cells and PBMCs at the same time-points (Fig. 3f-h). In contrast, constitutive expression of the antimicrobial genes S100A8 and S100A9 was highest in PBMCs compared to the low levels detected in endometrial cells (Fig. 3i and m). However, upon stimulation with LPS endometrial cells were more responsive compared to PBMCs, with epithelial cells demonstrating stronger induction of both S100A8 (Fig. 3j-l) and S100A9 (Fig. 3n-p) compared to stromal fibroblasts.

Endometrial epithelial and stromal fibroblast cells express IL6, IL8, S100A8 and S100A9 mRNA in response to LPS stimulation. Following an LPS time-course stimulation, endometrial cells were harvested in TRIzol reagent, mRNA was extracted and reverse transcribed to cDNA before IL8, IL6, S100A8 and S100A9 gene expression was examined by qPCR. (a-d) IL8 mRNA levels in endometrial cells and PBMCs following stimulation (n = 3). (e-h) IL6 mRNA levels in endometrial cells and PBMCs following stimulation (n = 3). (i-l) S100A8 mRNA levels in endometrial cells and PBMCs following stimulation (n = 3). (m-p) S100A9 mRNA levels in endometrial cells and PBMCs following stimulation (n = 3)
Discussion
Infection of the endometrium by microbial and viral pathogens can result in pathological inflammation that is harmful to cow health and agricultural sustainability (Bromfield et al. 2015; Sheldon et al. 2008). Epithelial and stromal fibroblast cell populations play important but distinct roles in innate defence of the endometrium. Understanding the inflammatory pathways and mechanisms of response in endometrial cells is crucial to developing therapeutics targeted towards the specific inflammatory insult, thereby improving cow health and fertility and ultimately increasing profitability. Elucidating these inflammatory pathways requires a reliable and well characterised model in order to elucidate which cell type is contributing to the inflammation.
Bovine endometrial epithelial cells and stromal fibroblasts have been grown with varying degrees of success since the first reports describing their isolation (Fortier et al. 1988; Herath et al. 2006). Our optimised protocol gives consistently pure and viable populations of epithelial and stromal cells, confirmed by the positive staining for their respective cytoskeletal proteins cytokeratin and vimentin and by the easily observed differences in cell morphology. We have also confirmed the absence of immune cell contamination by PCR for PTPRC which encodes CD45, the immune cell surface marker.
Here we describe several key modifications to previously published protocols. Dissection and harvesting of the endometrium occurs immediately after recovering the tract in the abattoir as opposed to transporting the tract back to the laboratory on ice. We found this greatly improved cell viability and numbers of cells recovered. Separating cells based on size using differential filtering also greatly improved the purity of the distinct cell populations. We found the brand of cell strainer used impacted on cell recovery. In addition, any residual stromal cell contamination of our epithelial cultures was removed with accutase treatment based on differential lifting time. Finally, supplementing the growth media with insulin, transferrin, selenium solution (ITS-X) also had a marked impact on the growth of our cell populations in culture.
The induction of inflammatory cytokine mRNA in response to LPS stimulation confirms that isolated and cultured endometrial cells retain the ability to respond to bacterial stimuli in-vitro. While the induction of the cytokines IL8 and IL6 by endometrial cells has been previously reported (Cronin et al. 2012; Healy et al. 2015), their induction here, in addition to the antimicrobial S100A8 and S100A9, confirms that our improved protocol for isolating and culturing endometrial cells provides a practical option for the study of bovine inflammatory responses in-vitro.
This model will facilitate investigations into the production of immune molecules such as cytokines, chemokines, HDPs and acute phase proteins under defined inflammatory conditions. It is our aim that the protocol detailed here will provide a useful basis for others wishing to establish a similar model within their laboratory.
Data availability
The datasets analysed during the current study are available from the corresponding author on reasonable request.
References
Borges AM, Healey GD, Sheldon IM (2012) Explants of intact endometrium to model bovine innate immunity and inflammation ex vivo. Am J Reprod Immunol (New York, NY : 1989) 67:526–539. https://doi.org/10.1111/j.1600-0897.2012.01106.x
Bromfield JJ, Santos JE, Block J, Williams RS, Sheldon IM (2015) PHYSIOLOGY AND ENDOCRINOLOGY SYMPOSIUM: Uterine infection: linking infection and innate immunity with infertility in the high-producing dairy cow. J Anim Sci 93:2021–2033. https://doi.org/10.2527/jas.2014-8496
Butts CL et al (2007) Progesterone inhibits mature rat dendritic cells in a receptor-mediated fashion. Int Immunol 19:287–296. https://doi.org/10.1093/intimm/dxl145
Cheng Z, Elmes M, Abayasekara DRE, Wathes DC (2003) Effects of conjugated linoleic acid on prostaglandins produced by cells isolated from maternal intercotyledonary endometrium, fetal allantochorion and amnion in late pregnant ewes. Biochim Biophys Acta 1633:170–178. https://doi.org/10.1016/S1388-1981(03)00123-9
Cronin JG, Turner ML, Goetze L, Bryant CE, Sheldon IM (2012) Toll-like receptor 4 and MYD88-dependent signaling mechanisms of the innate immune system are essential for the response to lipopolysaccharide by epithelial and stromal cells of the bovine endometrium. Biol Reprod 86:51. https://doi.org/10.1095/biolreprod.111.092718
Davies D, Meade KG, Herath S, Eckersall PD, Gonzalez D, White JO, Conlan RS, O'Farrelly C, Sheldon IM (2008) Toll-like receptor and antimicrobial peptide expression in the bovine endometrium. Reprod Biol Endocrinol 6:53. https://doi.org/10.1186/1477-7827-6-53
Donofrio G, Herath S, Sartori C, Cavirani S, Flammini CF, Sheldon IM (2007) Bovine herpesvirus 4 is tropic for bovine endometrial cells and modulates endocrine function. Reproduction (Cambridge, England) 134:183–197. https://doi.org/10.1530/rep-07-0065
Fortier MA, Guilbault LA, Grasso F (1988) Specific properties of epithelial and stromal cells from the endometrium of cows. J Reprod Fertil 83:239–248
Frandson RD, Wilke WL, Fails AD (2009) Anatomy and physiology of farm animals. Wiley, Hoboken
Franquemont DW, Frierson HF Jr, Mills SE (1991) An immunohistochemical study of normal endometrial stroma and endometrial stromal neoplasms. Evidence for smooth muscle differentiation. Am J Surg Pathol 15:861–870
Healy LL, Cronin JG, Sheldon IM (2015) Polarized epithelial cells secrete interleukin 6 apically in the bovine endometrium. Biol Reprod 92:151. https://doi.org/10.1095/biolreprod.115.127936
Herath S et al (2006) Expression and function of toll-like receptor 4 in the endometrial cells of the uterus. Endocrinology 147:562–570. https://doi.org/10.1210/en.2005-1113
Kaur G, Dufour JM (2012) Cell lines: Valuable tools or useless artifacts. Spermatogenesis 2:1–5. https://doi.org/10.4161/spmg.19885
MacKintosh SB, Schuberth HJ, Healy LL, Sheldon IM (2013) Polarised bovine endometrial epithelial cells vectorially secrete prostaglandins and chemotactic factors under physiological and pathological conditions. Reproduction (Cambridge, England) 145:57–72. https://doi.org/10.1530/rep-12-0253
Narciandi F, Lloyd AT, Chapwanya A, O'Farrelly C, Meade KG (2011) Reproductive tissue-specific expression profiling and genetic variation across a 19 gene bovine beta-defensin cluster. Immunogenetics 63:641–651. https://doi.org/10.1007/s00251-011-0551-7
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108
Sheldon IM, Williams EJ, Miller AN, Nash DM, Herath S (2008) Uterine diseases in cattle after parturition. Vet J 176:115–121. https://doi.org/10.1016/j.tvjl.2007.12.031
Staggs KL, Austin KJ, Johnson GA, Teixeira MG, Talbott CT, Dooley VA, Hansen TR (1998) Complex induction of bovine uterine proteins by interferon-tau. Biol Reprod 59:293–297
Stites DP, Siiteri PK (1983) Steroids as immunosuppressants in pregnancy. Immunol Rev 75:117–138
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:research0034.0031. https://doi.org/10.1186/gb-2002-3-7-research0034
Whelehan CJ, Barry-Reidy A, Meade KG, Eckersall PD, Chapwanya A, Narciandi F, Lloyd AT, O'Farrelly C (2014) Characterisation and expression profile of the bovine cathelicidin gene repertoire in mammary tissue. BMC Genomics 15:128. https://doi.org/10.1186/1471-2164-15-128
Wonodirekso S, Au CL, Hadisaputra W, Affandi B, Rogers PA (1993) Cytokeratins 8, 18 and 19 in endometrial epithelial cells during the normal menstrual cycle and in women receiving Norplant. Contraception 48:481–493
Acknowledgements
The authors would like to acknowledge Robert Walsh, Paul Delaney and all staff at Kildare Chilling Company for their assistance with sample collection. The authors would also like to acknowledge Dr. Aspinas Chapwanya for assistance with sample collection and staging of the ovaries.
Funding
This work was funded by a Research Grant to KM and COF from the Irish Department of Agriculture (DAFM grant 13/S/472).
Author information
Authors and Affiliations
Contributions
Conceived the study: KGM and COF. Developed primary culture model: PK, ABR and AB. Performed experiments and interpreted data: PK. Wrote the manuscript: PK, KGM and COF. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Declarations
All authors declare that they have no competing interests.
Ethics statement
All procedures described were conducted under ethical approval and experimental license from the Irish Health Products Regulatory Authority in accordance with the Cruelty to Animals Act 1876 and in agreement with the European Union (Protection of Animals Used for Scientific Purposes) regulations 2012 (S.I. No. 543 of 2012).
Consent for publication
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Fig. 1
Optimisation of dissection technique. Collection of female reproductive tracts and isolation of endometrial tissue was initially optimised using a curved scissors dissection or dissection with a curette. (A). Image of a uterus following dissection of the endometrial lining of the uterine horn ipsilateral to the corpus luteum. (B) Example of ovary staging for tract collection. A stage 1 corpus luteum (or corpus hemorrhagicum) is clearly visible. (C) Histogram detailing the age categories of cows from which endometrial samples were obtained. (D) Details of the breed of cows from which endometrial samples were obtained. (E & F) Representative histological image of the endometrium before and after dissection of the epithelial layer respectively. Lum=endometrial lumen, LE=luminal epithelium, Str=stroma, GE=glandular epithelium. Scale bar indicates 200μm. (G) Image of tissue isolated from the functional layer of the endometrium. (H) Comparison of tissue weight isolated using either a scissors or curette for dissection (PDF 499 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kelly, P., Barry-Reidy, A., Brewer, A. et al. Improved filtration method to isolate pure populations of primary bovine endometrial epithelial and stromal cells for immunological studies. Vet Res Commun 44, 29–39 (2020). https://doi.org/10.1007/s11259-020-09770-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11259-020-09770-3