
Abstract
Lewis acids increase the catalytic activity of classical heterogeneous catalysts and molecular d0 tungsten oxo alkylidenes in a variety of olefin metathesis processes. The formation of labile adducts between the metal complex and the Lewis acid has been observed experimentally and suggested to be involved in the catalyst activity increase. In this contribution, DFT (M06) calculations have been performed to determine the role of Lewis acids on catalyst activity, Z-/E- selectivity and stability by comparing three W(E)(CHR)(2,5-dimethylpyrrolide)(O-2,6-dimesithylphenoxide) (E = oxo, imido or oxo-Lewis acid adduct) alkylidenes. Results show that the formation of the alkylidene—Lewis acid adducts influences the reactivity of tungsten oxo alkylidenes due to both steric and electronic effects. The addition of the Lewis acid on the E group increases its bulkiness and this decreases catalyst Z-selectivity. Moreover, the interaction between the oxo ligand and the Lewis acid decreases the donating ability of the former toward the metal. This is important when the oxo group has either a ligand in trans or in the same plane that is competing for the same metal d orbitals. Therefore, the weakening of oxo donating ability facilitates the cycloaddition and cycloreversion steps and it stabilizes the productive trigonal bipyramid metallacyclobutane isomer. The two factors increase the catalytic activity of the complex. The electron donating tuneability by the coordination of the Lewis acid also applies to catalyst deactivation and particularly the key β-hydride elimination step. In this process, the transition states show a ligand in pseudo trans to the oxo. Therefore, the presence of the Lewis acid decreases the Gibbs energy barrier significantly. Overall, the optimization of the E group donating ability in each step of the reaction makes tungsten oxo alkylidenes more reactive and this applies both for the catalytic activity and catalyst deactivation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Olefin metathesis is a key reaction in organic synthesis that implies the exchange of alkylidene substituents between alkenes. It has been applied to the synthesis of a large variety of molecules including raw materials, polymers, and drugs [1,2,3,4,5,6,7,8]. The reaction only takes place in presence of a catalyst. Silica and alumina supported molybdenum and tungsten oxides are within the first catalyst precursors and they have been applied in industrial applications util today [9]. The active species operating in these systems has not been characterized in detail, but they are thought to be metal oxo alkylidenes. Remarkably, addition of Lewis acids to these systems enhances their catalytic activity through a process that it is not well understood.
In 1971, Hérrison and Chauvin proposed the today’s accepted reaction mechanism (Scheme 1) [2, 10]. It states that metal alkylidenes are the catalytic active species and metallacyclobutanes the key intermediates. Moreover, the catalytic cycle implies two olefin metathesis processes, each one composed of a cycloaddition and a cycloreversion step. The reaction mechanism proposed by Chauvin paved the way for the synthesis of metal alkylidenes. Some of the first active well-defined molecular catalysts were tungsten oxo alkylidenes and oxo alkyl complexes that act as precursors of the metal alkylidene (1–4 in Scheme 2) [11,12,13,14]. These complexes are highly active towards olefin metathesis and their catalytic activity can usually be increased by adding Lewis acids in the media as in the case of classical heterogeneous catalysts. However, they deactivate fast and this, together with the development of d0 imido alkylidene complexes (5Mo-10Moand 5W-10W) [15,16,17,18,19,20,21,22,23,24,25] as well as ruthenium carbenes (i.e. 11 and 12) [26,27,28,29,30,31,32,33,34] prevented its further development since last decade.

Chauvin’s mechanistic proposal

Selected olefin metathesis catalysts and precursors. aOTPP = 2,3,5,6-Ph4(C6H); bHIMTO = 2,6-Mes2(C6H3); cHIPTO = 2,6-(2,4,6-iPr3(C6H2))2(C6H3);dH2IMes = 1,3-Mes2-2-imidazolidinylidene; edAdPO = 2,5-Ad2-4-CH3(C6H2)
In 2011, Schrock and co-workers reported the synthesis of a series of tungsten oxo alkylidenes bearing a pyrrolyl and a large alkoxy ancillary ligand (13 and 14 in Scheme 2) [35]. These complexes are efficient and highly Z-selective olefin metathesis catalysts particularly on the homocoupling of terminal olefin and the ring opening metathesis polymerization (ROMP) of substituted norbornenes. In the subsequent years, the Schrock group reported other tungsten oxo alkylidenes and the role of ancillary ligands on the catalytic activity and product selectivity were deeply discussed (i.e. 15 and 16) [36,37,38,39,40,41,42]. Moreover, Buchmeiser and co-workers synthetized a series of very active cationic tungsten oxo alkoxy alkylidene bearing an N-heterocyclic carbene (NHC) as fourth ligand (17–18) [43,44,45]. Both Schrock and Buchmeiser tungsten oxo alkylidenes were heterogenized by grafting on silica (19–20) [46,47,48]. Catalyst heterogenization prevented bimolecular deactivation and, as a consequence, the resulting complexes show very high catalytic activities and stabilities overperforming the imido analogues particularly when reacting with internal olefins. These high activities and stabilities are also reported for silica grafted tungsten oxo alkyl complexes that act as alkylidene precursors (21 in Scheme 2) [49,50,51].
The knowledge acquired on tungsten oxo alkylidenes allowed the synthesis of molybdenum and vanadium oxo analogues as well as other molybdenum precursors that are also active in metathesis (22, 23) [40, 52,53,54,55,56,57,58]. The number of existing molybdenum oxo alkylidenes is limited and their activity for olefin metathesis is usually lower than that of tungsten species.
With the aim of bridging the gap between the activity of classical heterogeneous catalyst and the most recently reported well-defined molecular tungsten and molybdenum alkylidene, the effect of Lewis acids on the well-defined molecular complexes has been recently analyzed [35, 59]. Addition of B(C6F5)3 as Lewis acid leads to the formation of a labile compound in which the Lewis acid is bonded to the oxo ligand (i.e. 13·B(C6F5)3). Moreover, an increase on the catalytic activity both towards terminal olefins and the ROMP of substituted norbornenes is usually observed. Remarkably, this activity increase is also commonly associated with a decrease of the Z-selectivity and more importantly catalyst stability. Analysis of the formed by-products shows the formation of propene and metallacyclopentene intermediates, suggesting that the deactivation occurs through β-hydride elimination and metal reduction without the reaction of additional ethene molecules as suggested for silica supported rhenium alkyl alkylidene alkylidyne complexes [60].
Theoretical studies on d0 olefin metathesis have contributed to the understanding of the electronic structure of metal-alkylidenes and metallacyclobutane intermediates as well as the factors controlling catalyst activity and deactivation [60,61,62,63,64,65,66,67,68,69,70,71]. Today, it is well accepted that olefin coordination occurs trans to the strongest σ-donating ancillary ligand and the metallacyclobutane involved in the reaction pathway presents a trigonal bipyramid structure (TBP) with the doubly bond ligand (E) and the weakest σ-electron donor ligand in apical positions (Scheme 3 for the homocoupling reaction of ethene) [66, 68, 69]. This implies the inversion of the configuration of the stereogenic metal center [72]. Moreover, the square based metallacyclobutane isomer (SBP) is a resting state of the catalytic process which is involved in unimolecular deactivation through β-hydride elimination, a process taking place trans to the weakest σ-donating ligand [60, 68,69,70]. Since X and Y ligands play a different role, complexes with different X and Y ligands can show higher catalytic activities than species with X = Y. Strong σ-donor X ligands favor alkene coordination and imido (in comparison with oxo and alkylidyne E ligands) and weak σ-donor Y ligands stabilize the metallacyclobutane intermediate [60, 68,69,70]. The overall reactivity is a balance of these two effects. Catalyst stability with respect β-hydride elimination also depends on the nature of E, X and Y. β-hydride elimination is hampered when replacing alkyl as X ligand by pyrrolyl and the imido ligand by the stronger donor oxo group [60, 69, 70].

Olefin metathesis and unimolecular catalyst deactivation reaction mechanisms
In this contribution, we perform DFT calculations to get electronic insights on the role of Lewis acids on the catalytic activity and Z-/E- selectivity of tungsten oxo alkylidenes as well as their stability toward unimolecular decomposition through β-hydride elimination. For that, we compared the reactivity, selectivity, and stability of three existing complexes bearing the same ancillary ligands except the doubly bonded (E) (9W, 13 and 13·B(C6F5)3 Scheme 2).
2 Computational Details
2.1 Level of Theory
All calculations are performed with the Minnesota M06 hybrid density functional [73, 74]. This functional has been shown to reproduce the experimental trends for systems where weak interactions are important and this includes the study of the olefin metathesis reaction [75,76,77]. Geometry optimizations are performed representing main group elements with the 6-31G(d,p) basis sets [78, 79]. Tungsten is represented with the small core Stuttgart pseudopotential together with the associated basis set enlarged with a f polarization function (α = 0.823) [80, 81]. The nature of the stationary points (minima or transition states) is verified by vibrational analysis. Moreover, IRC calculations are performed to determine the minima connected through 13·B(C6F5)3-UU-TSII, 13·B(C6F5)3-UU-TSIII, 13·B(C6F5)3-TSAB, 13·B(C6F5)3-TSBC and 13·B(C6F5)3-TSBD. We assume that for all other systems and orientations the connected minima are equivalent based on the geometrical similarities found within all transition states. The final energetics are obtained by single point calculations with the larger 6–311 + G(d,p) basis sets [82, 83] for main group elements and the same representation of tungsten. In these single point calculations, solvent effects are included using the SMD continuum model [84] and toluene as solvent. Energies reported along the text are based on Gibbs energies (Gsolv = Ggp + ∆Gsolv, where Ggp is the gas phase free energy and ∆Gsolv stands for the solvation free energies) at 298.15 K and 1 atm. The thermal corrections are included at the smallest basis sets. All calculations are performed with the Gaussian09 package [85].
2.2 Model
The Z-/E- selectivity of tungsten oxo and imido olefin metathesis catalysts is analyzed by considering the 2-butene formation by reaction of propene with ethylidene of complexes 9W, 13 and 13·BX3 (9W-I, 13-I and 13·BX3-I, respectively). In this context, the Z-/E- selectivity arises from the relative feasibilities of the four potential productive pathways (Scheme 4). This approach was used before with great success when studying the Z-/E- selectivity of ruthenium complexes [86]. The tungsten complexes are represented with the full bulk of the ancillary ligands (oxo or N-2,6iPr-C6H3, 2,5-dimethylpyrrolyl and aryloxy ligands). The initial ethylidene species present two different isomers: syn with the methyl of ethylidene towards the oxo and anti with the methyl of the ethylidene away from the oxo and both are taken into account in the calculations. Moreover, two representations of the Lewis acid are considered: the experimentally used B(C6F5)3 and BF3 which mainly accounts for electronic effects.

Reaction pathways considered in this work
3 Results and Discussion
The aim of the present study is to give atomistic insights on the effect of Lewis acids on tungsten oxo alkylidenes’ catalytic activity for olefin metathesis, their Z-/E- selectivity and their stability. For that, we compare the results obtained for 13-I and 9W-I ethylidene complexes as well as the oxo complex interacting with two Lewis acids B(C6F5)3 (13·B(C6F5)3-I) and BF3 (13·BF3-I), see Scheme 2. First, we focus on the structure of the oxo complexes with the aim of validating the computational approach with existing experimental data [35]. After that, we study the productive pathways for the propene conversion to Z- or E-2-butene catalyzed by 13-I, 9W-I, 13·B(C6F5)3-I or 13·BF3-I. Finally, we discuss catalyst stability towards β-hydride elimination [59, 60]. The nomenclature is constructed by three parts: the arabic number that indicates the catalyst precursor according to Scheme 2, a roman number or a capital letter that indicates the reaction intermediate following Scheme 4 (roman number for the productive process and the capital letter for the catalyst deactivation) and a combination of two U or D capital letters that indicate the orientation of the methyl groups of the alkylidene and reacting olefin. U indicates that the methyl points towards the E group and D that the methyl points towards the aryloxy ligand. The first letter refers to the substituent of the initial alkylidene while the latter refers to the methyl group of propene (Scheme 4).
3.1 Methodology Validation
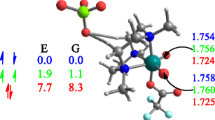
The interaction of a Lewis acid with tungsten oxo alkylidenes is labile according to experiments [35]. Thus, we decided to evaluate the ability of the methodology used to reproduce experimental data of complexes 13 and 14 (Scheme 2) and particularly the following three observations [35]: (i) the synthesis of complex 13 leads to a pentacoordinated with a coordinated phosphine as fifth ligand; (ii) the addition of two equivalents of B(C6F5)3 in a solution containing 13 leads to the formation of the 13·B(C6F5)3 adduct in which the Lewis acid is labile: and (iii) Substitution of 2,6-dimesithylphenoxide (HIMTO) ligand by the bulkier O-2,6-(2,4,6-iPrC6H2)2C6H3 (HIPTO) ligand allows the isolation of tetracoordinated syn-W(O)(CHtBu)(2,5-Me2Pyr)(HIPTO) complex (14). Table 1 compares the computed and X-Ray diffraction determined geometry parameters and Fig. 1 shows a detailed view of the optimized structures computed for 13, 14 and related adducts.

Detailed view of the optimized structures for syn- and anti- 13 and 14 alkylidenes and related adducts as well as the relative energies (in kcal mol−1) with respect to the ML4 species syn-13No-PMe2Ph or 14 and either PMe2Ph or B(C6F5)3
The syn-13No-PMe2Ph alkylidene presents a distorted tetrahedral coordination around tungsten with a short W=O (1.696 Å). The O=W=C angle is 105.0° and the W=C–H angle suggests the presence of an α–CH agostic interaction [65]. The anti isomer presents the same metal coordination without the α–CH agostic interaction and it is less stable by 6.8 kcal mol−1. Coordination of PMe2Ph to syn-13No-PMe2Ph is favorable by -1.5 kcal mol−1 and the resulting complex (syn-13) presents a trigonal bipyramid coordination around tungsten with apical phosphine and pyrrolyl ligands. The computed W-L distance are close to the X-Ray values (Table 1). Remarkably, the anti-13 is less stable by 10.6 kcal mol−1, thus suggesting that phosphine coordination is less favorable in anti-13No-PMe2Ph. Addition of B(C6F5)3 forms syn-13·B(C6F5)3 in which the Lewis acid interacts with the oxo group. The B···O distance is 1.532 Å and this lengthens the W = O bond to 1.759 Å. The syn-13·B(C6F5)3 is -1.1 kcal mol−1 lower in Gibbs energy than syn-13No-PMe2Ph, in agreement with the characterization of syn-13·B(C6F5)3 and the labile behavior of B(C6F5)3 in solution.
Regarding complex 14, calculations suggest that the substitution of the HIMTO ligand by the bulkier HIPTO stabilizes the syn- isomer with respect to the anti- one (Fig. 1). Moreover, they indicate that the largest bulkiness of the metal complex makes the coordination of PMe2Ph unfavorable by about + 4.3/ + 4.4 kcal mol−1 and this is consistent with the isolation of the tetracoordinated complex of syn-14.
Overall, calculations on the precursors show that the methodology used in this work reproduces the structure of these complexes as well as the associated energetics. Particularly, calculations predict: i) the largest stability for the syn- isomer; ii) the marginally favorable phosphine coordination to 13No-PMe2Ph but the unfavorable phosphine coordination to 14; and iii) the weak interaction between B(C6F5)3 and the oxo group.
3.2 Effect of Lewis Acids on Catalyst Activity
The role of Lewis acids on the catalytic activity for olefin metathesis of tungsten oxo alkylidene complexes is discussed by comparing the propene metathesis with the ethylidene complexes 13-I, 9W-I, 13·B(C6F5)3-I and 13·(BF3)3-I. Table 2 summarizes the relative Gibbs energies with respect to separated reactants N-I-U + propene following the labelling of Schemes 2 and 4 and the relative reaction rates with respect to the UU route. The optimized structures can be found in the Supplementary Information.
The propene metathesis occurs through today’s accepted reaction mechanism that involves the formation of an olefin complex, a cycloaddition step that leads to a metallacyclobutane intermediate with a trigonal pyramid structure and the reverse cycloreversion and olefin decoordination steps [60, 68,69,70]. Moreover, the square based metallacyclobutane isomer is a resting state of the reaction. Remarkably, we could not locate the transition state associated to olefin coordination and olefin decoordination. This is indicative that the barrier likely arises from the entropic term and thus can not be located by exploring the potential energy surface. This was highlighted in previous contributions [87, 88] and is particularly relevant when dispersion forces are taken into account at least partially.
The computed energy barriers with the tungsten oxo ethylidene 13-I show that all steps are easy (within 1.5 and 10.1 kcal mol−1) and thus all intermediates and transition states lie at energies that are accessible at reaction conditions. The highest in Gibbs energy structure is 13-TSIII-DD and it lies 24.4 kcal mol−1 over 13-I-U and propene. As found in model calculations [70], the reactive TBP metallacycle is significantly higher in energy than the SBP isomer (between 7.5 and 12.4 kcal mol−1) and this is rationalized by the strong trans effect of the oxo ligand, which presents a very short W = O distance (1.71 Å). Overall, the lowest intermediate is separated reactants except for the UD pathway, where the SBP metallacycle (13-A-UD) is slightly lower in energy (-2.2 kcal mol−1). The highest in Gibbs energy transition state is in all cases the cycloreversion step (13-TSIII), thus the energy span of the reaction is defined as the energy difference between 13-A-UD and the cycloreversion transition state (13-TSIII) and it varies between 17.1 and 26.7 kcal mol−1.
The energetics for the same processes catalyzed with tungsten imido alkylidene (9W-I) present several similarities with the reactivity of 13-I as well as remarkable differences. As for 13-I, the highest in energy transition state is that associated to the cycloreversion (9W-TSIII), while the global minimum is 9W-III-UU. Moreover, the computed energy spans are similar to those computed for 13-I albeit marginally higher (between 18.1 and 25.6 kcal mol−1). In contrast, the relative stability between TBP and SBP metallacyclobutane (III vs. A) isomers is inversed. That is, the reactive TBP isomer is more stable than the SBP resting state for the imido complex by 0.8–10.3 kcal mol−1. This indicates that most of the initial catalyst remains on the productive pathway, and it does not accumulate on an isomer not involved in the olefin metathesis process. The stabilization of III can be rationalized by the lower trans effect of the imido ligand with respect to the oxo group, while the destabilization of A is likely due to steric repulsion between the large aryloxy and imido ligands. Overall, the small difference in the energy spans of 9W-I and 13-I and the different relative stabilities of the metallacyclobutane isomers suggest that the two complexes should present similar reactivities in absence of catalyst deactivation.
The presence of B(C6F5)3 as Lewis acid leads to the formation of 13·B(C6F5)3-I in which B(C6F5)3 is weakly interacting with the oxo group through a O-B bond of 1.571 Å. The formation of the syn-13·B(C6F5)3-I adduct is exergonic by -1.9 kcal mol−1 (-3.7 kcal mol−1 for the anti- isomer) and the O-B(C6F5)3 produces a weakening of the W = O bond (W = O distance elongation of about 0.05 Å), as in the case of the catalyst precursor. The reaction of 13·B(C6F5)3-I with propene shows that the formation of 13·B(C6F5)3 adduct is in general marginally exergonic for all intermediates and transition states with respect to the analogous structures of 13 and B(C6F5)3. The main exception is the SBP metallacyclobutane intermediate. In this case, although we find the adduct as a minimum of the potential energy surfaces, calculations predict that the Lewis acid–metal complex formation is unfavorable by 2.7–6.4 kcal mol−1. The larger stabilization due to the presence of the Lewis acid is observed for the cycloreversion transition state. Consequently, the energy span defined by the difference between separated reactants (the global minimum of the reaction) and the transition state for either cycloaddition (TSII) or cycloreversion (TSIII) decreases significantly and becomes between 13.3 and 19.7 kcal mol−1 and, this agrees with the higher catalytic activity of tungsten oxo alkylidenes in presence of Lewis acids.
With the aim of isolating the steric and electronic effects we considered BF3 as a smaller model of Lewis acid. Results for 13·BF3-I are equivalent to those obtained with 13·B(C6F5)3: (i) the formation of the adduct for all intermediates and transition states involved in the productive process is exergonic with respect to the analogues structures and separated BF3; (ii) The BF3-oxo interaction weakens the W=O bond; (iii) the BF3-oxo group interaction is unfavorable for the SBP isomer; and (iv) the energy span decreases significantly, thus indicating that the presence of the Lewis acid increases the catalyst activity. Consequently, the effect of the Lewis acid has mainly an electronic contribution that is stronger at the cycloaddition and cycloreversion transition states as well as the at the TBP metallacyclobutane isomer but not on the SBP one. Remarkably, these are the species in which the oxo ligand has either a ligand in trans or the oxo group is in the same plane of the alkylidene and aryloxy ligands. This suggest that the higher catalytic activity of tungsten oxo alkylidenes in presence of a Lewis acid can be mainly attributed to two factors: i) the decrease of the electron donating ability of the oxo group, thus the trans influence of the oxo ligand and ii) and increase of the electrophilicity of the metal center when the N·BX3 adduct is formed as evidenced by the Natural Population Charges of tungsten in the initial ethylidene species (q13 = 1.39, q13·B(C6F5)3 = 1.63 and q13·BF3 = 1.51). The higher metal electrophilicity favors the olefin coordination, while the decrease of the trans effect of the oxo ligand stabilizes the TBP metallacyclobutane intermediate and the cycloaddition and cycloreversion transition states. Overall, the Lewis acid allows adapting the electron donating ability of the oxo ligand in each step of the reaction and this leads to a smoother Gibbs energy profile.
3.3 Effect of Lewis Acids on Catalyst Z-/E-Selectivity
The analysis of the Z-/E- selectivity of each catalyst implies comparing the relative Gibbs energies and rates of the most favorable pathway leading to Z-2-butene and that of the most favorable pathway leading to E-2-butene (Table 2). For that, we first focus on how the Gibbs energies of each intermediate and transition state varies as function of the position of the methyl groups and then we discuss the energetics, in terms of energy span, of the different pathways.
The relative stability of the olefin metathesis intermediates involved in the productive process (I, II, III and IV) and the transition states connecting them (TSII and TSIII) for all considered complexes 13-I, 9W-I, 13·B(C6F5)3 and 13·BF3 follow the same general trend with very few exceptions. In general, UU is the most stable isomer and DD the highest in energy species in each stationary point. The detailed order is UU < UD < DU < DD. The few exceptions are mainly associated with those species presenting the bulkier E ligands (imido or the O–B(C6F5)3). In the UU pathway the two substituents point toward the E group and in the case of the less favorable DD route, the substituents points toward the aryloxy ligand. Therefore, the relative stability of the different routes is mainly controlled by the ligand bulkiness. That is, since the E group is generally smaller than the aryloxy ligand, the preferred relative orientation shows the two methyl groups pointing to E, the two pathways presenting one methyl towards E and the other towards the bulky aryloxy ligand have intermediate energies and the route with the two methyls pointing toward the aryloxy ligand is the less favorable one.
The relative stabilities of the SBP metallacyclobutane resting state do not follow the same order. The SBP metallacyclebutane stability trend follows: UD > UU ~ DD > DU. The puckered form of the metallacyclebutane fragment leads to the presence of equatorial and axial sites. Thus, the structure with the two methyl groups in equatorial (A-UD) is lower in Gibbs energy than the structures with one methyl in axial position (A-DD and A-UU), and A-DU, with the two substituents in axial posions, is usually the less stable one. Overall, the relative stabilities of the SBP isomer are controlled by the local structure of the metallacyclobutane fragment and not by the bulkiness of the ligands. Consequently, the relative stabilities of the different reactant-alkylidene orientation in the SBP metallacyclobutane isomer are not sensitive to the alkylidene ancillary ligands. However, this relative stabilities of the SBP metallacyclobutane species does not have any effect on defining the Z-/E- selectivity, since according to the energy span model this essentially originates from the relative stabilities of the transition state for cycloreversion.
Overall, the Z-/E- selectivity is defined by the relative Gibbs energies of N-TSIII-UU and N-TSIII-UD, except for 13·B(C6F5)3, which is defined by the relative Gibbs energies between 13·B(C6F5)3-TSII-UU and 13·B(C6F5)3-TSII-UD. For the case, of 13-I with a very small oxo ligand as E, the energy difference is large (3.1 kcal mol−1), thus suggesting a > 99% selectivity for the Z-product at least at the initial stages of the reaction (the rate of the UD pathway is 4.5·10–3 times slower than that of the UU route). The Gibbs energy difference between the same transition states of the tungsten imido ethylidene analogue (9W-I) is smaller (1.7 kcal mol−1) suggesting a smaller selectivity that is related to the bulkiness increase of the E group (the ratio between kUD and kUU is 6.7·10–2). Interestingly, the preference for the UU pathway is also small for 13·BF3-I (∆δG = 0.8 kcal mol−1 and kUD/kUU = 0.26) and 13·B(C6F5)3-I, thus explaining the loss in Z-selectivity when adding a Lewis acid in the reaction mixture.
Analysis of TSIII transition state geometries (Fig. 2) give further support to the importance of the ligand bulkiness. When going from 13 to 13·BF3 and 13·B(C6F5)3, there is a general increase of E group size in all directions due to tetrahedral environment around boron. This implies an opening of the W···Cole-CCH3 angle in the UU-TSIII pathway only (Fig. 2) and a significant variation of the dihedral angle defined by the reacting carbon atoms. These two facts indicates that the released olefin orientation in the UU route is influenced by the bulk of E in the larger systems, preventing the achievement of the optimal structure in 9W, 13·BF3 and 13·B(C6F5)3. Moreover, comparison between the UU and DD pathways as limit cases, indicates that the W–O-Caryloxy angle associated to the Y ligand increases when substituents point toward Y. This suggests that the Y ligand also adapts to avoid repulsive interactions with the methyl groups of the releasing olefin. Consequently, TSIII is destabilized in the UD, DU and DD routes. In summary, despite the reported values could be sensitive to the conformational exploration, the computed data indicate that the interaction between the Lewis acid and the oxo group makes the resulting adduct to have a large E ligand that decreases the preference for the Z- product. In this way, the use of smaller Lewis acids or alternatively small Y ligands that could favor the DD route may eventually increase the catalyst activity and retain a larger amount of Z- selectivity.

Detailed view of the metal coordination of the optimized structures for the UU and DD cycloreversion transition states (TSIII) for 13, 9W and 13·B(C6F5)3. Distances are in Å and angles in degrees. The reported dihedral corresponds to that defined by the Cole–Cole–W–Cene atoms
3.4 Effect of Lewis Acids on Catalyst Deactivation
Based on the species formed after olefin metathesis, two main catalyst deactivation pathways for d0 alkylidenes have been reported in the literature: (i) bimolecular coupling of two alkylidenes [89] and (ii) β-hydride elimination from the unsubstituted SBP metallacyclobutane (A) [59, 60, 90]. While the former does not seem to be favored by the presence of Lewis acids (it may even disfavor it due to the ligand bulkiness increase), the addition of Lewis acid leads to the formation of a W(IV) olefin complex compatible with the β-hydride elimination at A.[59] Here, we studied deactivation through β-hydride elimination of the methylidene complexes of 13, 9W and 13·B(C6F5)3 catalysts. We considered two different final products: the W(IV) olefin complex (D) and the alkyl allyl complex (C) resulting from ethene addition to the allyl hydride intermediate (B), which has been proposed to be the first step for side product formation and other catalyst deactivation processes [60]. Results for catalyst deactivation are summarized in Fig. 3 (the nomenclature is based on that used in Schemes 2 and 3) and Table 3 reports the energetics for the degenerate metathesis of ethene for comparison.

Gibbs energy profile (in kcal mol−1) for the unimolecular deactivation through β-hydride elimination at the SBP metallacycle. Values in parenthesis correspond to the interaction energy between the metal complex and the Lewis acid
The Gibbs energies associated to the ethene metathesis with methylidene follows the same trends described before and particularly, the addition of B(C6F5)3 favors the cycloaddition step and stabilizes the TBP metallacyclobutane (III) intermediate with respect to the SBP (A) one. When comparing with the propene metathesis with the ethylidene complex, one observes that most of the intermediates and transition states are stabilized with respect to separated reactants as a consequence of reducing the reactant bulkiness. Therefore, the nature of the olefin and alkylidene substituents tunes the energetics but does not modify the previously described trends.
The deactivation of 13 starts with the β-hydride elimination. This step takes place with the hydride being transferred trans to the weakest σ-donating aryloxy ligand. The computed Gibbs energy barrier is high (35.0 kcal mol−1), thus suggesting a large stability of 13 if bimolecular coupling does not occur. The resulting allyl hydride intermediate (B) is 15.3 kcal mol−1 higher in energy than the SBP metallacycle (A) thus, it must further evolve to achieve catalyst deactivation. The ethene insertion to the metal hydride presents an even higher in Gibbs energy transition state (42.7 kcal mol−1 with respect to methylidene + ethene). This indicates that despite the process is thermodynamically favorable (∆G°298 = − 12.0 kcal mol−1) ethene insertion is a rare event. In contrast, the reductive coupling between the hydride and the allyl ligands that leads to the formation a W(IV) olefin complex is largely favored thermodynamically (∆G°298 = − 27.4 kcal mol−1) and the associated energy barrier is low (∆G‡°298 = 4.7 kcal mol−1). Indeed, the transition state for the reductive coupling is located 29.4 kcal mol−1 lower in Gibbs energy than the transition state for ethene insertion, indicating that the most likely deactivation process for 13 is the formation of a W(IV) olefin complex and the highest in Gibbs energy transition state is that associated with the β-hydride process. This is in contradiction with the DFT results reported for silica grafted (OSi)Re(CtBu)(CHtBu)(CH2tBu) [60], but this could be either related to the modeling strategy or to the different reactivity of the two complex as suggested by the experimentally observed side products in each case [59, 60].
Substitution of the oxo group by an imido ligand makes the β-hydride elimination and the alkene insertion easier. The Gibbs energies with respect to separated reactants of the associated transition states are 25.4 and 40.2 kcal mol−1 for the β-hydride and the ethene insertion respectively. Again, ethene insertion appears as an unlikely process, thus suggesting that another process should take place. In this context, reductive coupling leading to D is thermodynamically favorable (∆G°298 = − 15.4 kcal mol−1) and the associated Gibbs energy barrier lower than that of the ethene insertion (30.5 vs. 40.2 kcal mol−1). Noteworthily, the transition state for the reductive coupling is higher in Gibbs energy than that of the β-hydride elimination. Therefore, 9W decomposition through β-hydride elimination leads to also to a W(IV) olefin complex, but, in contrast to 13, the highest in Gibbs energy transition state is that associated with the formation of the olefin complex. Overall, deactivation of 9W is easier than the deactivation of 13.
The 13·B(C6F5)3 adducts associated to the deactivation of 13 in presence of a Lewis acid are minima of the potential energy surfaces. The species are especially favorable for the allyl hydride intermediate (B) and the transition states associated to the β-hydride elimination (TSAB) and ethene insertion (TSBC) (values in parenthesis in Fig. 3). Consequently, both the β-hydride elimination from 13·B(C6F5)3-A and the ethene insertion to 13·B(C6F5)3-B are easier than the analogous processes without the presence of the Lewis acid. The relative Gibbs energies of 13·B(C6F5)3-TSAB and 13·B(C6F5)3-TSBC with respect to separated 13·B(C6F5)-V and ethene are 31.3 and 36.6 kcal mol−1, thus ethene insertion is more challenging than the β-hydride. The Gibbs energy barrier for the reductive coupling leading to D is 20.2 kcal mol−1 and, although the process is more challenging than in absence of the Lewis acid, it is still largely more accessible than the ethene insertion. Therefore, similarly to 9W and 13, the 13·B(C6F5)3 intramolecular deactivation leads to a W(IV) olefin complex, and it involves two steps, β-hydride elimination and the reductive coupling between the allyl and hydride ligands. The transition state for the β-hydride is the highest transition state in the catalyst deactivation process, indicating a mechanistic difference between the oxo complexes (rate determining step is the β-hydride elimination) and the imido alkylidene (the highest in energy step is the reductive coupling). The comparison of the relative Gibbs energies of the highest transition states with respect to separated species indicate that 13·B(C6F5)3 is more prone to deactivate through a unimolecular process than 13.
Trends between the three complexes for the relative Gibbs energies of TSAB, TSBC and TSBD can be rationalized in terms of the donating ability of the E ligand. As shown in Fig. 4, both the β-hydride elimination and the alkene insertion imply the presence of a ligand in (pseudo)-trans to the E group. In the case of the β-hydride elimination transition state, one of the Cα carbons of the metallacycle is in pseudo trans to E (Fig. 4). On the other hand, ethene insertion takes place in the vacant site trans to the E and the transition state presents a short W···Cene (between 2.44 and 2.51 Å (Fig. 4). Consequently, in both cases the transition state stability is penalized when the E group is a strong donating group (i.e. oxo). Moreover, the two processes become easier when the donating ability of E becomes weaker, which is achieved either by replacing the oxo for an imido ligand or by adding a Lewis that tunes the electron density of the oxo group.

Detailed view of the optimized structures for TSAB, TSBC and TSBD for 13, 9W and 13·B(C6F5)3 complexes. Distances in Å angles in degrees
The relative stabilities of the transition states for the reductive coupling of 13, 9W and 13·B(C6F5)3 are inversed with respect to TSAB and TSBC and a strong donating E ligand favors the process. In this case, part of the metal-hydride bond is loss in the TSBD transition state when compared with the allyl hydride and thus the presence of strong donating ligands better compensate the loss in electron density on the metal center. This is particularly the case of the tungsten oxo complex 13, which presents a low energy barrier, while the opposite extreme is represented by the imido complex 9W.
4 Conclusions
DFT (M06) calculations have been performed to rationalize the effect of Lewis acids on the catalytic activity for olefin metathesis, Z-/E- selectivity and catalyst stability of tungsten oxo alkylidene complexes. For that, we compared three W(E)(CHR)(2,5-dimethylpyrrolide)(2,6-mesithylphenoxide) complexes differing only on the nature of the doubly bonded E group: oxo, imido and an oxo group interacting with a Lewis acid. Results show that the formation of a stable oxo-Lewis acid adduct occurs mainly when the metal is tetracoordinated and more significantly when the E group has another ligand in trans or in the same plane competing for the same empty metal d orbitals. This is particularly the case of the cycloaddition and cycloreversion transition states, the productive TBP metallacycle isomer and the transition state for β-hydride elimination, which is the highest in energy transition state associated to the unimolecular deactivation of tungsten oxo alkylidene complexes. In these adducts, the resulting E group is bulky due to the presence of the phenyl rings of the B(C6F5)3 Lewis acid and the ligand steric hindrance is comparable to that of the large aryloxy ligand. Therefore, the Z-/E- selectivity, defined by the Gibbs energy difference between the lowest transition state leading to the Z- and E- product, decreases significantly. Moreover, the Lewis acid has also an electronic influence. The oxo-Lewis acid interaction decreases the oxo electron donating ability, which decreases its trans influence and makes the metal slightly more electrophilic. Consequently, the cycloaddition and cycloreversion Gibbs energy barriers with respect to separated reactants are lowered and the TBP metallacyclobutane isomer, which is much higher in Gibbs energy than the SBP metallacycle resting state for the oxo complex, is stabilized. The two factors lead to an increase of catalyst activity in presence of the Lewis acid, thus rationalizing the experimental observations. Unfortunately, the tuneability of the oxo electron donating ability also affects the transition state for the β-hydride elimination, the rate determining transition state in the catalyst deactivation. Since the H-transfer implies the presence of the Cα carbon of the original metallacycle fragment pseudo trans to the E group, the decrease of the trans influence of the oxo group by the interaction with a Lewis acid stabilizes the transition state and favors catalyst deactivation when the Lewis acid—oxo adduct is formed.
Overall, the presence of Lewis acid in the reacting media allows adapting the electron donating ability of the oxo ligand in each step of the reaction. This takes place by forming a Lews acid—oxo alkylidene adduct when a weak electron donor E group is needed and by the decoordination of the Lewis acid when a strong electron donating E group stabilizes one intermediate or transition state. This makes the oxo alkylidene more reactive both for catalyzing the olefin metathesis reaction and in those processes involving catalyst deactivation. Therefore, the optimal use of Lewis acids requires a subtle balance between the catalytic activity increase and the alkylidene stability loss. Despite present conclusions are obtained for a reduced series of molecular tungsten oxo alkylidenes, similar effects of the Lewis acid are expected for other metal oxo complexes, and they could also be envisaged in classical heterogeneous catalysts.
Data Availability
The Cartesian Coordinates of all optimized structures can be found as an xyz and pdf files in the Supplementary Information.
References
Calderon N, Chen HY, Scott KW (1967) Olefin metathesis—a novel reaction for skeletal transformations of unsaturated hydrocarbons. Tetrahedron Lett 34:3327–3329
Chauvin Y (2006) Olefin metathesis: the early days (Nobel lecture). Angew Chem Int Ed 45:3740–3747. https://doi.org/10.1002/anie.200601234
Grubbs RH (2006) Olefin-metathesis catalysts for the preparation of molecules and materials (Nobel lecture). Angew Chem Int Ed 45:3760–3765. https://doi.org/10.1002/anie.200600680
Schrock RR (2006) Multiple metal-carbon bonds for catalytic metathesis reactions (Nobel lecture). Angew Chem Int Ed 45:3748–3759. https://doi.org/10.1002/anie.200600085
Samojłowicz C, Bieniek M, Grela K (2009) Ruthenium-based olefin metathesis catalysts bearing N-heterocyclic carbene ligands. Chem Rev 109:3708–3742. https://doi.org/10.1021/cr800524f
Ogba OM, Warner NC, O’Leary DJ, Grubbs RH (2018) Recent advances in ruthenium-based olefin metathesis. Chem Soc Rev 47:4510–4544. https://doi.org/10.1039/c8cs00027a
Copéret C, Berkson ZJ, Chan KW et al (2021) Olefin metathesis: what have we learned about homogeneous and heterogeneous catalysts from surface organometallic chemistry? Chem Sci 12:3092–3115. https://doi.org/10.1039/d0sc06880b
Hoveyda AH, Liu Z, Qin C et al (2020) Impact of ethylene on efficiency and stereocontrol in olefin metathesis: when to add it, when to remove it, and when to avoid it. Angew Chem Int Ed 59:22324–22348. https://doi.org/10.1002/anie.202010205
Mol J (2004) Industrial applications of olefin metathesis. J Mol Catal A 213:39–45. https://doi.org/10.1016/j.molcata.2003.10.049
Hérisson J-L, Chauvin Y (1970) Catalyse de transformation des oléines par les complexes du tungstene. Die Makromol Chem 141:161–176
Wengrovius JH, Schrock RR, Churchill MR et al (1980) Tungsten-oxo alkylidene complexes as olefin metathesis catalysts and the crystal structure of W(O)(CHCMe3)PEt3)Cl2. J Am Chem Soc 309:4515–4516
Kress JRM, Russell MJM, Wesolek MG, Osborn JA (1980) Tungsten(VI) and molybdenum(VI) oxo-alkyl species. Their role in the metathesis of olefins. J Chem Soc Chem Commun 2:431–432. https://doi.org/10.1039/C39800000431
Kress J, Wesolek M, le Ny JP, Osborn JA (1981) Molecular complexes for efficient metathesis of olefins. The oxo-ligand as a catalyst-cocatalyst bridge and the nature of the active species. J Chem Soc Chem Commun. https://doi.org/10.1039/C39810001039
Wengrovius JH, Schrock RR (1982) Synthesis and characterization of tungsten oxo neopentylidene complexes. Organometallics 1:148–155. https://doi.org/10.1021/om00061a026
Murdzek JS, Schrock RR, Complexes A, Ar M (1987) Well-characterized olefin metathesis catalysts that contain molybdenum. Organometallics 6:1373–1374
Schrock RR, DePue RT, Feldman J et al (1988) Preparation and reactivity of several alkylidene complexes of the type W(CHR’)(N-2, 6–C6H3-i-Pr2)(OR)2 and related tungstacyclobutane complexes. controlling metathesis activity through the choice of alkoxide ligand. J Am Chem Soc 110:1423–1435. https://doi.org/10.1021/ja00213a014
Schrock RR, Murdzek JS, Bazan GC et al (1990) Synthesis of molybdenum imido alkylidene complexes and some reactions involving acyclic olefins. J Am Chem Soc 112:3875–3886. https://doi.org/10.1021/ja00166a023
Bazan GC, Oskam JH, Cho H et al (1991) Living ring-opening metathesis polymerization of. 2,3-difunctionalized 7-oxanorbornene and 7-oxanorbornadienes by Mo(CHCMe2R)(N-2,6–C6H3-i-Pr2)(O-t-Bu)2 and Mo(CHCMe2R)(N-2,6–C6H3-i-Pr2)(OCMe2CF3)2. J Am Chem Soc 113:6899–6907
Schrock RR (2009) Recent advances in high oxidation state Mo and W imido alkylidene chemistry. Chem Rev 109:3211–3226. https://doi.org/10.1021/cr800502p
Townsend EM, Schrock RR, Hoveyda AH (2012) Z-selective metathesis homocoupling of 1,3-dienes by molybdenum and tungsten monoaryloxide pyrrolide (MAP) complexes. J Am Chem Soc 134:11334–11337. https://doi.org/10.1021/ja303220j
Jiang AJ, Zhao Y, Schrock RR, Hoveyda AH (2009) Highly Z-selective metathesis homocoupling of terminal olefins. J Am Chem Soc 131:16630–16631. https://doi.org/10.1021/ja908098t
Wang C, Yu M, Kyle AF et al (2013) Efficient and selective formation of macrocyclic disubstituted Z alkenes by ring-closing metathesis (RCM) reactions catalyzed by Mo- or W-based monoaryloxide pyrrolide (MAP) complexes: applications to total syntheses of epilachnene, yuzu lactone, ambrettolide, Epothilone C, and Nakadomarin A Chem. Eur J 19:2726–2740. https://doi.org/10.1002/chem.201204045
Meek SJ, Obrien, Llaveria J RV et al (2011) Catalytic Z-selective olefin cross-metathesis for natural product synthesis. Nature 471:461–466. https://doi.org/10.1038/nature09957
Flook MM, Jiang AJ, Schrock RR et al (2009) Z-selective olefin metathesis processes catalyzed by a molybdenum hexaisopropylterphenoxide monopyrrolide complex. J Am Chem Soc 131:7962–7963. https://doi.org/10.1021/ja902738u
Jiang AJ, Simpson JH, Müller P, Schrock RR (2009) Fundamental studies of tungsten alkylidene imido monoalkoxidepyrrolide complexes. J Am Chem Soc 131:7770–7780. https://doi.org/10.1021/ja9012694
Schwab P, Grubbs RH, Ziller JW (1996) Synthesis and applications of RuCl2(=CHR′)(PR3)2: the influence of the alkylidene moiety on metathesis activity. J Am Chem Soc 118:100–108. https://doi.org/10.1021/ja952676d
Nguyen ST, Johnson LK, Grubbs RH, Ziller JW (1992) Ring-opening metathesis polymerization (ROMP) of norbornene by a Group VIII carbene complex in protic media. J Am Chem Soc 114:3974–3975. https://doi.org/10.1021/ja00036a053
Scholl M, Ding S, Lee CW, Grubbs RH (1999) Synthesis and activity of a new generation of ruthenium-based olefin metathesis catalysts coordinated with 1,3-dimesityl-4,5-dihydroimidazol-2-ylidene ligands. Org Lett 1:953–956. https://doi.org/10.1021/ol990909q
Kingsbury JS, Harrity JPA, Bonitatebus PJ, Hoveyda AH (1999) A recyclable Ru-based metathesis catalyst. J Am Chem Soc 121:791–799. https://doi.org/10.1021/ja983222u
Garber SB, Kingsbury JS, Gray BL, Hoveyda AH (2000) Efficient and recyclable monomeric and dendritic Ru-based metathesis catalysts. J Am Chem Soc 122:8168–8179. https://doi.org/10.1021/ja001179g
Michrowska A, Bujok R, Harutyunyan S et al (2004) Nitro-substituted Hoveyda-Grubbs ruthenium carbenes: enhancement of catalyst activity through electronic activation. J Am Chem Soc 126:9318–9325. https://doi.org/10.1021/ja048794v
Keitz BK, Endo K, Herbert MB, Grubbs RH (2011) Z -selective homodimerization of terminal olefins with a ruthenium metathesis catalyst. J Am Chem Soc 133:9686–9688. https://doi.org/10.1021/ja203488e
Keitz BK, Endo K, Patel PR et al (2012) Improved ruthenium catalysts for Z-selective olefin metathesis. J Am Chem Soc 134:693–699. https://doi.org/10.1021/ja210225e
Khan RKM, Torker S, Hoveyda AH (2013) Readily accessible and easily modifiable Ru-based catalysts for efficient and Z-selective ring-opening metathesis polymerization and ring-opening/cross- metathesis. J Am Chem Soc 135:10258–10261. https://doi.org/10.1021/ja404208a
Peryshkov DV, Schrock RR, Takase MK et al (2011) Z-selective olefin metathesis reactions promoted by tungsten Oxo alkylidene complexes. J Am Chem Soc 133:20754–20757. https://doi.org/10.1021/ja210349m
Peryshkov DV, Schrock RR (2012) Synthesis of tungsten oxo alkylidene complexes. Organometallics 31:7278–7286. https://doi.org/10.1021/om3008579
Forrest WP, Axtell JC, Schrock RR (2014) Tungsten oxo alkylidene complexes as initiators for the stereoregular polymerization of 2,3-dicarbomethoxynorbornadiene. Organometallics 33:2313–2325. https://doi.org/10.1021/om5002364
Forrest WP, Weis JG, John JM et al (2014) Stereospecific ring-opening metathesis polymerization of norbornadienes employing tungsten oxo alkylidene initiators. J Am Chem Soc 136:10910–10913. https://doi.org/10.1021/ja506446n
Townsend EM, Hyvl J, Forrest WP et al (2014) Synthesis of molybdenum and tungsten alkylidene complexes that contain sterically demanding arenethiolate ligands. Organometallics 33:5334–5341. https://doi.org/10.1021/om500655n
Boudjelel M, Zhai F, Schrock RR et al (2021) Oxo 2-adamantylidene complexes of Mo(VI) and W(VI). Organometallics 40:838–842. https://doi.org/10.1021/acs.organomet.1c00086
Yan T, Venkatramani S, Schrock RR, Müller P (2019) Synthesis of tungsten oxo alkylidene biphenolate complexes and ring-opening metathesis polymerization of norbornenes and norbornadienes. Organometallics 38:3144–3150. https://doi.org/10.1021/acs.organomet.9b00377
Kruger AG, Brucks SD, Yan T et al (2021) Stereochemical control yields mucin mimetic polymers. ACS Central Sci 7:624–630. https://doi.org/10.1021/acscentsci.0c01569
Buchmeiser MR (2018) Molybdenum imido, tungsten imido and tungsten oxo alkylidene N-heterocyclic carbene olefin metathesis catalysts. Chem Eur J 24:14295–14301. https://doi.org/10.1002/chem.201802497
Schowner R, Frey W, Buchmeiser MR (2015) Cationic tungsten-oxo-alkylidene-N-heterocyclic carbene complexes: highly active olefin metathesis catalysts. J Am Chem Soc 137:6188–6191. https://doi.org/10.1021/jacs.5b03788
Hauser PM, Musso, Frey W, Buchmeiser MR JV (2021) Cationic tungsten oxo alkylidene N-heterocyclic carbene complexes via hydrolysis of cationic alkylidyne progenitors. Organometallics 40:927–937. https://doi.org/10.1021/acs.organomet.1c00035
Conley MP, Mougel V, Peryshkov DV et al (2013) A well-defined silica-supported tungsten oxo alkylidene is a highly active alkene metathesis catalyst. J Am Chem Soc 135:19068–19070. https://doi.org/10.1021/ja410052u
Pucino M, Mougel V, Schowner R et al (2016) Cationic silica-supported N-heterocyclic carbene tungsten oxo alkylidene sites: highly active and stable catalysts for olefin metathesis. Angew Chem Int Ed 55:4300–4302. https://doi.org/10.1002/anie.201510678
Conley MP, Forrest WP, Mougel V et al (2014) Bulky aryloxide ligand stabilizes a heterogeneous metathesis catalyst. Angew Chem Int Ed 53:14221–14224. https://doi.org/10.1002/anie.201408880
Mazoyer E, Merle N, de Mallmann A et al (2010) Development of the first well-defined tungsten oxo alkyl derivatives supported on silica by SOMC: towards a model of WO3/SiO2 olefin metathesis catalyst. Chem Commun 46:8944–8946. https://doi.org/10.1039/c0cc02507k
Bouhoute Y, Garron A, Grekov D et al (2014) Well-defined supported mononuclear tungsten oxo species as olefin metathesis pre-catalysts. ACS Catal 4:4232–4241. https://doi.org/10.1021/cs501294j
Qureshi ZS, Hamieh A, Barman S et al (2017) SOMC-designed silica supported tungsten Oxo imidazolin-2-iminato methyl precatalyst for olefin metathesis reactions. Inorg Chem 56:861–871. https://doi.org/10.1021/acs.inorgchem.6b02424
Bukhryakov KV, Schrock RR, Hoveyda AH et al (2018) Syntheses of molybdenum oxo alkylidene complexes through addition of water to an alkylidyne complex. J Am Chem Soc 140:2797–2800. https://doi.org/10.1021/jacs.8b00499
Zhai F, Bukhryakov KV, Schrock RR et al (2018) Syntheses of molybdenum oxo benzylidene complexes. J Am Chem Soc 140:13609–13613. https://doi.org/10.1021/jacs.8b09616
de Jesus SJ, Pucino M, Zhai F et al (2021) Boosting the metathesis activity of molybdenum oxo alkylidenes by tuning the anionic ligand σ donation. Inorg Chem 60:6875–6880. https://doi.org/10.1021/acs.inorgchem.0c03173
Pucino M, Zhai F, Gordon CP et al (2019) Silica-supported molybdenum oxo alkylidenes: bridging the gap between internal and terminal olefin metathesis. Angew Chem Int Ed 58:11816–11819. https://doi.org/10.1002/anie.201903325
Merle N, le Quéméner F, Barman S et al (2017) Well-defined silica supported bipodal molybdenum oxo alkyl complexes: a model of the active sites of industrial olefin metathesis catalysts. Chem Commun 53:11338–11341. https://doi.org/10.1039/c7cc06041f
Merle N, le Quéméner F, Bouhoute Y et al (2017) Well-defined molybdenum oxo alkyl complex supported on silica by surface organometallic chemistry: a highly active olefin metathesis precatalyst. J Am Chem Soc 139:2144–2147. https://doi.org/10.1021/jacs.6b11220
Belov DS, Fenoll DA, Chakraborty I, et al (2021) Synthesis of vanadium oxo alkylidene complex and its reactivity in ring-closing olefin metathesis reactions. Organometallics acs.organomet.1c00425. https://doi.org/10.1021/acs.organomet.1c00425
Peryshkov DV, Forrest WP, Schrock RR et al (2013) B(C6F5)3 activation of oxo tungsten complexes that are relevant to olefin metathesis. Organometallics 32:5256–5259. https://doi.org/10.1021/om4007906
Leduc A-M, Salameh A, Soulivong D et al (2008) beta-h transfer from the metallacyclobutane: a key step in the deactivation and byproduct formation for the well-defined silica-supported rhenium alkylidene alkene metathesis catalyst. J Am Chem Soc 130:6288–6297. https://doi.org/10.1021/ja800189a
Rappé AK, Goddard WA (1982) Olefin metathesis. a mechanistic study of high-valent group 6 catalysts. J Am Chem Soc 162:448–456
Folga E, Ziegler T (1993) Density functional study on molybdacyclobutane and its role in olefin metathesis. Organometallics 12:325–337. https://doi.org/10.1021/om00026a018
Wu YD, Peng ZH (1997) Theoretical studies on alkene addition to molybdenum alkylidenes. J Am Chem Soc 119:8043–8049. https://doi.org/10.1021/ja970644f
Goumans TPM, Ehlers AW, Lammertsma K (2005) The asymmetric Schrock olefin metathesis catalyst. A Comput Study Organometal 24:3200–3206. https://doi.org/10.1021/om050109l
Solans-Monfort X, Clot E, Copéret C, Eisenstein O (2005) Understanding structural and dynamic properties of well-defined rhenium-based olefin metathesis catalysts, Re(=CR)(=CHR)(X)(Y), from DFT and QM/MM calculations. Organometallics 24:1586–1597. https://doi.org/10.1021/om048997s
Solans-Monfort X, Clot E, Copéret C, Eisenstein O (2005) d(0) Re-based olefin metathesis catalysts, Re(=CR)(=CHR)(X)(Y): The key role of X and Y ligands for efficient active sites. J Am Chem Soc 127:14015–14025. https://doi.org/10.1021/ja053528i
Herz K, Podewitz M, Stöhr L et al (2019) Mechanism of olefin metathesis with neutral and cationic molybdenum imido alkylidene N-hetercyclic carbene complexes. J Am Chem Soc 141:8264–8276. https://doi.org/10.1021/jacs.9b02092
Poater A, Solans-Monfort X, Clot E et al (2007) Understanding d(0)-olefin metathesis catalysts: which metal, which ligands? J Am Chem Soc 129(8207):8216. https://doi.org/10.1021/ja070625y
Solans-Monfort X, Copéret C, Eisenstein O (2010) Shutting down secondary reaction pathways: the essential role of the pyrrolyl ligand in improving silica supported d(0)-ML4 alkene metathesis catalysts from DFT calculations. J Am Chem Soc 132:7750–7757. https://doi.org/10.1021/ja101597s
Solans-Monfort X, Copéret C, Eisenstein O (2012) Oxo vs imido alkylidene d(0)-metal species: how and why do they differ in structure, activity, and efficiency in alkene metathesis? Organometallics 31:6812–6822. https://doi.org/10.1021/om300576r
Solans-Monfort X, Copéret C, Eisenstein O (2015) Metallacyclobutanes from Schrock-type d(0) metal alkylidene catalysts: structural preferences and consequences in alkene metathesis. Organometallics 34(1668):1680. https://doi.org/10.1021/acs.organomet5b00147
Meek SJ, Malcolmson SJ, Li B et al (2009) The significance of degenerate processes to enantioselective olefin metathesis reactions promoted by stereogenic-at-Mo complexes. J Am Chem Soc 131:16407–16409. https://doi.org/10.1021/ja907805f
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals Theor. Chem Acc 120:215–241. https://doi.org/10.1007/s00214-007-0310-x
Zhao Y, Truhlar DG (2008) Density functionals with broad applicability in chemistry. Acc Chem Res 41:157–167. https://doi.org/10.1021/ar700111a
Zhao Y, Truhlar DG (2007) Attractive noncovalent interactions in the mechanism of Grubbs second-generation Ru catalysts for olefin metathesis. Org Lett 9:1967–1970. https://doi.org/10.1021/ol0705548
Minenkov Y, Occhipinti G, Jensen VR (2009) Metal−phosphine bond strengths of the transition metals: a challenge for DFT. J Phys Chem A 113:11833–11844. https://doi.org/10.1021/jp902940c
Paredes-Gil K, Solans-Monfort X, Rodriguez-Santiago L et al (2014) DFT study on the relative stabilities of substituted Ruthenacyclobutane intermediates involved in olefin cross-metathesis reactions and their interconversion pathways. Organometallics 33:6065–6075. https://doi.org/10.1021/om500718a
Hehre WJ, Ditchfield R, Pople JA (1972) Self—consistent molecular orbital methods. XII. Further extensions of Gaussian—type basis sets for use in molecular orbital studies of organic molecules. J Chem Phys 56:2257–2261. https://doi.org/10.1063/1.1677527
Francl MM, Pietro WJ, Hehre WJ et al (1982) Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J Chem Phys 77:3654–3665. https://doi.org/10.1063/1.444267
Andrae D, Häussermann U, Dolg M et al (1990) Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor Chimica Acta 77:123–141. https://doi.org/10.1007/BF01114537
Ehlers AW, Böhme M, Dapprich S et al (1993) A set of f-polarization functions for pseudo-potential basis sets of the transition metals Sc-Cu, Y-Ag and La-Au. Chem Phys Lett 208:111–114. https://doi.org/10.1016/0009-2614(93)80086
Hariharan PC, Pople JA (1973) The influence of polarization functions on molecular orbital hydrogenation energies. Theor Chimica Acta 28:213–222. https://doi.org/10.1007/BF00533485
Frisch MJ, Pople JA, Binkley JS (1984) Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J Chem Phys 80:3265–3269. https://doi.org/10.1063/1.447079
Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113:6378–6396. https://doi.org/10.1021/jp810292n
Frisch MJ, Trucks GW, Schlegel HB, et al (2009) Gaussian09, Revision D.01
Liu P, Xu X, Dong X et al (2012) Z -selectivity in olefin metathesis with chelated Ru catalysts: computational studies of mechanism and selectivity. J Am Chem Soc 134:1464–1467. https://doi.org/10.1021/ja2108728
Solans-Monfort X (2014) DFT study on the reaction mechanism of the ring closing enyne metathesis (RCEYM) catalyzed by molybdenum alkylidene complexes. Dalton Trans 43:4573–4586. https://doi.org/10.1039/c3dt53242a
Kesharwani MK, Elser I, Musso JV et al (2020) Reaction mechanism of ring-closing metathesis with a cationic molybdenum imido alkylidene N-heterocyclic carbene catalyst. Organometallics 39:3146–3159. https://doi.org/10.1021/acs.organomet.0c00311
Lopez LPH, Schrock RR (2004) Formation of dimers that contain unbridged W(IV)/W(IV) double bonds. J Am Chem Soc 126:9526–9527. https://doi.org/10.1021/ja0400988
Tsang WCP, Hultzsch KC, Alexander JB et al (2003) Alkylidene and metalacyclic complexes of tungsten that contain a chiral biphenoxide ligand. synthesis, asymmetric ring-closing metathesis, and mechanistic investigations. J Am Chem Soc 125:2652–2666. https://doi.org/10.1021/ja0210603
Acknowledgements
Financial support from the Ministerio de Ciencia e Innovación (PID2020-112715GB-I00) and the Generalitat de Catalunya (2017SGR1323) is acknowledged. XSM is grateful for the Professor Agregat Serra Húnter position.
Funding
Open Access Funding provided by Universitat Autonoma de Barcelona. Ministerio de Ciencia e Innovación—Grant: PID2020-112715 GB-I00 and Generalitat de Catalunya—Grant: 2017SGR1323.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Merino, J.H., Bernad, J. & Solans-Monfort, X. Effect of Lewis Acids on the Catalyst Activity for Alkene Metathesis, Z-/E- Selectivity and Stability of Tungsten Oxo Alkylidenes. Top Catal 65, 433–447 (2022). https://doi.org/10.1007/s11244-021-01534-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-021-01534-w