
Abstract
Ruthenium alkylidene complexes are commonly used as olefin metathesis catalysts. Initiation of the catalytic process requires formation of a 14-electron active ruthenium species via dissociation of a respective ligand. In the present work, this initiation step has been computationally studied for the Grubbs-type catalysts (H2IMes)(PCy3)(Cl)2Ru=CHPh, (H2IMes)(PCy3)(Cl)2Ru=CH-CH=CMe2 and (H2IMes)(3-Br-py)2(Cl)2Ru=CHPh, and the Hoveyda-Grubbs-type catalysts (H2IMes)(Cl)2Ru=CH(o-OiPrC6H4), (H2IMes)(Cl)2Ru=CH(5-NO2–2-OiPrC6H3), and (H2IMes)(Cl)2Ru=CH(2-OiPr-3-PhC6H3), using density functional theory (DFT). Additionally, the extended-transition-state combined with the natural orbitals for the chemical valence (ETS-NOCV) and the interacting quantum atoms (IQA) energy decomposition methods were applied. The computationally determined activity order within both families of the catalysts and the activation parameters are in agreement with reported experimental data. The significance of solvent simulation and the basis set superposition error (BSSE) correction is discussed. ETS-NOCV demonstrates that the bond between the dissociating ligand and the Ru-based fragment is largely ionic followed by the charge delocalizations: σ(Ru–P) and π(Ru–P) and the secondary CH…Cl, CH…π, and CH…HC interactions. In the case of transition state structures, the majority of stabilization stems from London dispersion forces exerted by the efficient CH…Cl, CH…π, and CH…HC interactions. Interestingly, the height of the electronic dissociation barriers is, however, directly connected with the prevalent (unfavourable) changes in the electrostatic and orbital interaction contributions despite the favourable relief in Pauli repulsion and geometry reorganization terms during the activation process. According to the IQA results, the isopropoxy group in the Hoveyda-Grubbs-type catalysts is an efficient donor of intra-molecular interactions which are important for the activity of these catalysts.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Well-defined ruthenium alkylidene complexes (Fig. 1) are commonly used as highly efficient catalysts for olefin metathesis. These systems show a high tolerance towards most of the functional groups in the substrates, air and moisture insensitivity, and thermal stability [1,2,3,4].

Example ruthenium alkylidene catalysts
The key step during the olefin metathesis process is initial formation of a 14-electron active complex [5,6,7,8,9,10,11,12,13,14]. In the case of the Grubbs-type catalysts (1–3), it occurs by dissociation of ligands L, whereas for the Hoveyda-Grubbs catalysts (4–6), it requires decoordination of the ether moiety (Fig. 2). Electron and steric properties of these ligands significantly affect the rate of the catalyst activation [6,7,8, 10,11,12, 14, 15]. In addition to this often postulated dissociative mechanism, where the dissociation/decoordination is the first step, associative and interchange mechanisms for the activation of the second and third generation ruthenium alkylidene catalysts were also proposed [8, 9, 12, 14, 16, 17]. In the case of the associative mechanism, the alkene molecule coordinates to the metal centre before dissociation of the appropriate ligand, whereas for the interchange mechanism, both steps take place simultaneously. Experimental kinetic studies for the Hoveyda-Grubbs catalysts indicated that two mechanisms, dissociative and interchange, are in work, depending on the steric and electronic properties of the ruthenium complex and the substrate employed [8]. Accordingly, the dissociative pathway was theoretically shown to be preferred over the interchange one in the case of the Hoveyda-Grubbs complexes bearing a large chelating alkoxy ligand, although the Hoveyda ligand decoordination during the cross-metathesis reaction is the rate-determining step [12]. For the Grubbs-type catalysts, the dissociative mechanism was rather computationally predicted as the kinetically favoured one, with the dissociation step being rate determining [12, 17]. Calculations also showed that the Gibbs energy barrier for phosphine dissociation from the Grubbs complex is solvent- and entropy-related [18].

Mechanism of olefin metathesis catalysed by ruthenium alkylidene complexes [7]
In this work, the formation of the propagating 14-electron alkylidene species from the Grubbs-type (1–3) and Hoveyda-Grubbs-type (4–6) initiators has been investigated using density functional theory (DFT). Our main objectives are to compare the activation parameters for catalysts with different ligands within a given family of compounds and to analyse in detail the nature of the interactions between the dissociating ligand L and the rest of the system. Bonding between the ligand L and the remaining Ru-based fragment is characterized for catalysts 1–3 and the corresponding transition state structures, using the extended-transition-state method combined with the natural orbitals of the chemical valence (ETS-NOCV) [19,20,21,22]. The Hoveyda-Grubbs catalysts have been additionally characterized by means of the interacting quantum atoms (IQA) [23] energy decomposition. This is the first time when these complementary theoretical approaches (ETS-NOCV and IQA) are used to get deeper insight into formation of active species from olefin metathesis catalysts.
Computational methods
Geometry optimization of all structures was carried out using the hybrid GGA PBE0 functional [24] combined with the split-valence def2-SVP basis set [25]. The 28 innermost electrons of Ru were replaced by the Stuttgart effective core potential [26]. Further single point energy calculations were performed with the hybrid meta-GGA M06 functional [27] and the triple-zeta valence def2-TZVPP basis set [25, 26]. The M06 method is recommended for main-group and transition metals thermochemistry, kinetics, and studies of noncovalent interactions [27]. A good performance of the M06 functional in predicting energies of reactions involving ruthenium alkylidene complexes [28,29,30,31,32,33], including the dissociation energy for the Grubbs catalysts [28,29,30,31], was proved and it was often used for investigations of real ruthenium systems [28,29,30,31,32,33,34,35,36]. On the other hand, the PBE0 method was shown to be accurate in reproducing experimental geometry of the second generation Grubbs catalyst (1) [28, 37], being even better than the M06 one [28].
Solvent effects were estimated by single point calculations on gas phase-optimized structures using the polarizable continuum model (PCM) [38]. In selected cases, the geometry optimization (PBE0/def2-SVP) has been also checked within the PCM framework.
Harmonic vibrational frequencies were calculated for each structure to confirm the potential energy minimum or the transition state structure and to obtain thermochemical quantities. The transition states were additionally verified by the IRC calculations [39, 40]. The PBE0/def2-SVP zero-point energy and thermal corrections were added to the gas phase single point energies to obtain a better estimate of gas phase enthalpies (Hg) and Gibbs energies (Gg). Enthalpies (Hs) and Gibbs energies (Gs) of the compounds in a solvent (toluene) were estimated analogously, involving the PCM single point energies. The counterpoise method [41] was used to estimate the basis set superposition error (BSSE). The reaction pathways are mainly discussed in terms of Gibbs energies calculated at the M06/def2-TZVPP//PBE0/def2-SVP level for simulated toluene solution.
It is well-established that the ground state for 16-electron and 14-electron ruthenium alkylidene complexes, studied in this work, is singlet [18, 42]. According to the PBE0/def2-SVP calculations, the triplet and quintet states of the 16-electron complex 1 are less stable than the singlet state, by 93 and 191 kJ mol−1, respectively. In the case of the 14-electron species 1_dis, formed after dissociation of 1, the corresponding differences are 75 and 171 kJ mol−1. Hence, the spin-restricted formalism is used for other computations. Furthermore, the DFT approach was proved sufficiently accurate in studying the reactivity of ruthenium alkylidene complexes [12, 13, 18, 28,29,30,31,32,33]. However, it shall be noticed, that for more difficult cases (e.g., open-shell transition metal complexes), single-determinant DFT shall be applied with special caution due to the approximated character of electron correlation, as well as a possible multireference character of wavefunction.
All the above calculations were done with the Gaussian 09 software [43].
For the Grubbs type catalysts, the charge and energy decomposition ETS-NOCV [19, 20] analysis was performed. DFT (BLYP [44, 45]) calculations were carried out with the Amsterdam Density Functional (ADF) program [21, 22], in which the ETS-NOCV scheme was implemented. The presented energies include DFT-D3(BJ) dispersion corrections [46, 47]. The standard triple-zeta STO basis set containing one set of polarization functions (TZP) was used for the ruthenium atom, whereas the double-zeta basis set (DZP) was employed for other elements as implemented in the ADF [21, 22]. The contours of deformation densities were plotted based on the ADF-GUI interface [21, 22].
ETS-NOCV charge and energy decomposition method
The natural orbitals for chemical valence (NOCV) ψi constitute the canonical representation for a differential density matrix ΔP (it is formed by subtracting the appropriate molecular fragments density matrices from a density matrix of a molecule under consideration) in which ΔP adopts a diagonal form. It gives rise to the corresponding eigenvalues vi and the related vectors ψi. NOCVs occur in pairs (ψ–k,ψk) related to |vk| and they decompose overall deformation density Δρ into bonding components with different symmetries (Δρk):
Usually, a few k allow to recover a major shape of Δρ. ETS-NOCV [19, 20] allows for obtaining the related energetics, ΔEorb(k), in addition to qualitative picture emerging from Δρk. ETS originally divides the total bonding energy between fragments, ΔEtotal, into five distinct components: ΔEtotal = ΔEelstat + ΔEPauli + ΔEorb + ΔEdispersion + ΔEdist. The ΔEelstat is an energy of quasi-classical electrostatic interaction between fragments. The next term, ΔEPauli, is responsible for repulsive Pauli interaction between occupied orbitals on the two fragments. The third component, ΔEorb, is stabilizing and shows formation of a chemical bond (including polarizations). In the ETS-NOCV scheme, ΔEorb is expressed in terms of the eigenvalues vk and diagonal Fock energy matrix elements \( {F}_{i,i}^{TS} \) (transformed into NOCV representation) as:
The ΔEdispersion denotes the semiempirical Grimme dispersion correction (D3), whereas the last term ΔEdist describes a change in the fragment’s energy when going from the optimized configurations to those present in the final complex. This method is applied for 1–3.
IQA energy decomposition method
IQA method [23] divides the total electronic energy E into atomic (\( {E}_{self}^A \)) and diatomic (\( {E}_{\mathrm{int}}^{AB} \)) contributions:
Diatomic part covers all interactions between particles of different atoms A and B: nucleus-nucleus (\( {V}_{nn}^{AB} \)), nucleus-electron(\( {V}_{ne}^{AB} \)), electron-nucleus (\( {V}_{en}^{AB} \)), and electron-electron (\( {V}_{ee}^{AB} \)):
where \( {V}_{eeC}^{AB} \) and \( {V}_{eeX}^{AB} \) are Coulomb and exchange contributions of \( {V}_{ee}^{AB} \). The former three terms together with \( {V}_{eeC}^{AB} \) are often combined into the overall Coulomb component ECoul, whereas the last, consisting of the exchange and correlation terms, describes the overall exchange-correlation contribution EXC. This complementary (to ETS-NOCV) method is applied for analyses of intramolecular contacts in the Hoveyda-Grubbs complexes. IQA analyses were performed on the wavefunction obtained in Gaussian 09 at the HF/def2-TZVP level of theory for the PBE0 geometries.
Results and discussion
Activation of the Grubbs-type catalysts
Assuming the commonly accepted dissociative mechanism [5, 6, 12, 13, 17, 18], the activation of the Grubbs-type complexes of general formula (H2IMes)(L)(Cl)2Ru=CHR requires dissociation of the ligand L (Fig. 2). For the phosphine-containing catalysts (1 and 2), rate determining transition states have been localized (Figs. 3 and 4), but the calculated enthalpy profiles (T = 298 K) are monotonic, indicating that these transition states do not affect the enthalpy barriers, which are determined by the reaction, not activation, enthalpies (Table 1). Hence, the Gibbs energy barriers for the reverse reaction (phosphine coordination to the 14-electron ruthenium alkylidene species) are mainly due to the entropy effect, as it was reported elsewhere [13, 18].

Gibbs energy profile (ΔGs, kJ mol−1) for the activation of the catalyst (H2IMes)(PCy3)(Cl)2Ru=CHPh (1). M06/def2-TZVPP//PBE0/def2-SVP calculations for simulated toluene solution

Gibbs energy profile (ΔGs, kJ mol−1) for the activation of the catalyst (H2IMes)(PCy3)(Cl)2Ru=CH-CH=CMe2 (2). M06/def2-TZVPP//PBE0/def2-SVP calculations for simulated toluene solution
The calculated activation parameters (ΔHs‡, ΔGs‡) for dissociation of the catalyst 1 are in a very good agreement with the corresponding experimental data, determined for the phosphine ligand exchange in toluene solution (Table 1). Other reported theoretical values of the activation Gibbs energy for phosphine dissociation from 1 are in the same range (90–99 kJ mol−1, toluene solution) [13, 17, 18] or significantly higher (123 kJ mol−1, dichloromethane solution) [12], compared to the present result (99 kJ mol−1).
There are two possible rotational isomers of the active ruthenium alkylidene species [18, 28,29,30, 35, 48]. Perpendicular orientation of the benzylidene group to the mesityl group (1_dis, Fig. 3) is energetically preferred, compared to the parallel orientation (1_dis′), which can be explained by electronic structure analysis [48]. The more stable isomer 1_dis is formed by dissociation of the 16-electron ruthenium complex 1. Rotation of the alkylidene ligand in the 14-electron species requires low activation barrier to overcome (ΔGs‡ = 37 kJ mol−1), which is accompanied by slight elongation of the carbene bond in the transition state 1_TS′, by about 0.025 Å. The less stable isomer 1_dis′ can be also generated directly from 1 by decoordination of the phosphine ligand according to a monotonic pathway.
In the case of the complex (H2IMes)(PCy3)(Cl)2Ru=CH-CH=CMe2 (2), the calculated activation Gibbs energy of phosphine dissociation (Fig. 4) is slightly higher, compared to 1 (Table 1). Hence, the latter can be more active than catalyst 2 in metathesis reactions. It was experimentally shown for ruthenium catalysts of general formula (L1)(L2)(Cl)2Ru=CHR that the substituent R significantly influences the rate of phosphine ligand exchange [6]. The activity of the ruthenium catalysts increases as R = H < < CH=CMe2 < Ph < Et, in accordance with the present results. Generally, bulky substituents R with electron-donor ability more effectively promote phosphine dissociation [6].
It is known that type of ligand L has a great impact on the activity of the Grubbs-type catalysts [6, 7]. As 3-bromopyridine is a much more labile ligand than PCy3, the phosphine free catalyst 3 is highly active in olefin metathesis, with the initiation rate constant higher by several orders of magnitude, compared to 1 [7]. It can also effectively catalyse polymerization [35, 49] and other transformations of terminal alkynes [36, 50, 51].
The computational results clearly indicate that (H2IMes)(3-Br-py)2(Cl)2Ru=CHPh (3) most easily forms the active species, among all the tested catalysts (Table 1). Due to the presence of two 3-bromopyridine ligands, the two-step dissociation pathway is expected (Fig. 5). Considering decoordination of the first ligand, 3-bromopyridine in cis position to the NHC group (pathway a) should dissociate faster than the 3-Br-py ligand located in the trans position (pathway b), based on the calculated Gibbs energy barriers. As it has been suggested in other theoretical studies [17], where the pathway a was also calculated, the higher lability of 3-bromopyridine in cis position to the NHC moiety can be also predicted on the basis of the Ru-N distance which is significantly longer, compared to the ligand in trans position (2.398 and 2.221 Å, respectively).

Gibbs energy profiles (ΔGs, kJ mol−1) for two possible pathways of the activation of the catalyst (H2IMes)(3-Br-py)2(Cl)2Ru=CHPh (3): a dissociation of the first 3-Br-py ligand in cis position to the NHC ligand, b dissociation of the first 3-Br-py ligand in trans position to the NHC ligand. M06/def2-TZVPP//PBE0/def2-SVP calculations for simulated toluene solution
In the partially dissociated complex (H2IMes)(3-Br-py)(Cl)2Ru=CHPh (3_dis1a and 3_dis1b), the 3-bromopyridine ligand is located in trans position to the NHC ligand, with the Ru-N distance in the range of 2.160–2.169 Å. Not surprisingly, further dissociation to form 14-electron ruthenium alkylidene species 1_dis requires a higher activation barrier to overcome (ΔGs‡ = 56 kJ mol−1), compared to the first step, in agreement with other authors [17]. However, this process is still much more kinetically accessible than dissociation of the phosphine-containing catalysts 1 and 2 (ΔGs‡ = 99 and 100 kJ mol−1, respectively). Moreover, the latter reactions are also highly endergonic (ΔGs = 73 and 83 kJ mol−1, respectively), in contrast to only slightly endergonic dissociation of both 3-bromopyridine ligands (ΔGs = 23 kJ mol−1). Consequently, the concentration of the active 14-electron ruthenium species should be larger for catalyst 3 than for 1 and 2, in agreement with the experimentally observed higher activity of initiator 3, compared to 1 [7]. On the other hand, the thermodynamic equilibrium between the initiator 3 and the mono(3-bromopyridine) ruthenium alkylidene species (3_dis1a, 3_dis1b) can be expected (Fig. 5).
The estimated BSSE correction slightly reduces the calculated dissociation energies for the catalysts 1–3 by about 10 kJ mol−1, and the activation barriers by approximately 3–7 kJ mol−1 (Table 1). The dissociation and activation energies computed for the simulated toluene solution are lower by 10–17 and 5–14 kJ mol−1, respectively, compared to the gas phase values. The solvent effects influence most significantly the dissociation energy of two 3-bromopyridine ligands. Even if relatively small, the solvent effects can change the conclusions concerning the relative activities of the catalysts (compare the ΔGg‡ and ΔGs‡ values for the catalysts 1 and 2, Table 1). As both BSSE and simulation of the solvent cause decrease of the dissociation energies and activation barriers, the summary correction to these quantities may be quite significant, in the range of 9–27 kJ mol−1.
In the case of the dissociation pathways for 1 and 2, we have also examined how including the solvent effects already at the geometry optimization stage influences the final Gibbs energy profiles (Fig. S1, Supplementary Material). It can be seen that the differences are quite small, up to 5 kJ mol−1, compared to the single point PCM calculations based on the gas phase optimized geometries. Importantly, the predicted trends in the activation barriers and dissociation energies have been preserved. Indeed, the structures calculated using the continuum solvent model are slightly changed, compared to the gas phase geometry optimization, particularly in the case of the minima. For instance, the differences in Ru=C and Ru-P bond lengths are up to 0.002 and 0.009 Å, respectively. On the other hand, the Ru-P distance in the transition states is more significantly affected by the inclusion of the solvent effects, being longer up to about 0.1 Å, compared to the gas phase geometry.
Activation of the Hoveyda-Grubbs-type catalysts
The absence of released phosphine, which might re-coordinate to the 14-electron ruthenium alkylidene species, is the key feature of the Hoveyda-Grubbs-type catalysts. The activation of the Hoveyda-Grubbs-type complexes occurs by breaking the Ru–O bond and simultaneous rotation of the Ccarbene–Caromatic bond in the benzylidene moiety (Fig. 2) if the dissociative mechanism [8, 12, 52,53,54] is assumed. It is known that the activity of this type of catalysts can be significantly modified by changing the steric and/or electronic properties of the Ru-chelating isopropoxy fragment. Experimental works [55, 56] showed that introduction of the strong electron-withdrawing nitro group into the aromatic ring of 4, resulting in complex 5, known as the Grela catalyst, causes a significant increase in the catalytic activity. This might be explained by the fact that electron-withdrawing substituents located in position para to the alkoxy group mostly reduce the electron density at the oxygen atom and, consequently, weaken the Ru–O interaction, just facilitating the activation process [57]. However, the sterically activated ruthenium complex 6 [58] is even more active than 5 [56]. The calculated activation barriers (ΔGs‡) for the decoordination of the isopropoxy moiety in the catalysts 4–6, being of 77, 73 and 54 kJ mol−1, respectively (Fig. 6, Table 1), are in agreement with these experimental observations.

Gibbs energy profiles (ΔGs, kJ/mol) for the activation of the Hoveyda-Grubbs-type catalysts: a (H2IMes)(Cl)2Ru=CH(o-OiPrC6H4) (4), b (H2IMes)(Cl)2Ru=CH(5-NO2–2-OiPrC6H3) (5), c (H2IMes)(Cl)2Ru=CH(2-OiPr-3-PhC6H3) (6). M06/def2-TZVPP//PBE0/def2-SVP calculations for simulated toluene solution
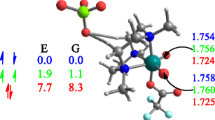
We have further performed IQA energy decomposition study in order to better understand the most facile breaking of the Ru–O bond in 6. Since the Ru–O bond distances are quite similar for 5 and 6 and they are different than in 4 (2.28 Å for 4 and 2.30 Å for 5 and 6), we have focused our attention on comparison between 4 and 6. We have determined, in line with the trend in bond distances, that the overall diatomic Ru–O interaction energy \( {E}_{int}^{AB} \) in 6 is higher (less negative), by 16 kJ mol−1 than in 4 (Fig. 7). IQA further indicates, in line with the previous suggestion [57], that Ru–O is electrostatically dominated (ECoul covers ~86% of the overall stabilization ECoul + EXC), though, the non-classic quantum exchange-correlation term (EXC) is also non-negligible (~14%) (Fig. 7).

Selected IQA diatomic interaction energies Eint (kJ mol−1) and their constituents for catalysts 4 and 6. Eint = EXC + ECoul
Interestingly, we have further revealed by the IQA calculations that bulky OiPr fragment is able to form the ancillary non-covalent interactions CH…Cl and agostic CH…Ru with the neighbouring metal-fragment (Fig. 7). We have determined that the overall chlorine–hydrogen interaction energy \( {E}_{int}^{AB} \) (CH43…Cl) + \( {E}_{int}^{AB} \) (CH47…Cl) = −36 kJ mol−1 in 4, whereas the corresponding value for 6 is significantly more pronounced, i.e. –69 kJ mol−1 (Fig. 7). Furthermore, solely in the case of 6 additional CH…π interactions are recovered between the C–H43 bond of OiPr and the phenyl substituent, \( {E}_{int}^{AB} \)(CH43…C75) = −21 kJ mol−1 (Fig. 7). Such efficient and more pronounced non–covalent interactions in 6 with respect to 4 and 5 might be responsible for both the Ru–O interaction energies and accordingly for the observed activation energies. These are very intriguing outcomes in the light of the intuitive claim on the solely steric-hindrance power of the isopropoxy group. It is to be added, consistently with the above discussion, that the removal of the mentioned non-covalent interactions due to the simple substitution of bulky OiPr by the significantly smaller in size metoxy unit (OMe) results in the significant elongation of Ru–O distances by ~ 0.02 Å (Fig. S2, Supplementary Material).
Previous computations suggested that the initiator 4 should more easily decoordinate than the initiator 5, only the computational analysis of the full metathesis pathway, including the addition of the first olefin molecule, indicated a higher activity of the catalyst 5 [12]. However, in that work, the isopropoxy group was replaced by ethoxy group—the latter, due to smaller size, can exert different non-covalent interactions with the metal-fragment and accordingly diverge the Ru–O distances (and activation barriers). The present study (which considers real bulky OiPr unit) shows that both the catalysts 5 and 6 are better metathesis initiators than 4, already at the step of the ether moiety decoordination (Fig. 6, Table 1).
Finally, the solvent effects estimated for the initiation step of the Hoveyda-Grubbs-type catalysts 4–6 are less significant than in the case of the Grubbs complexes 1–3 (Table 1). The energies of the decoordination reactions obtained for the simulated toluene solution are decreased by 5–9 kJ mol−1, compared to the gas phase calculations, whereas the predicted activation barriers are lower by approximately 5 kJ mol−1.
ETS-NOCV analysis
In order to understand factors which determine bond energies in the Grubbs-type catalysts, we have performed the ETS-NOCV analysis for the substrates 1–3. Subsequently, we have done similar analysis for the corresponding transition states in order to shed some light on the calculated energy barriers. The ETS energy decomposition results are gathered in Table 2 (for more details on the method, see Section 2 and Refs. [19, 20]).
Among the systems under study, the lowest total bonding energy ΔEtotal (the strongest bond) is predicted for the catalysts 1 and 2 (Table 2). The small difference between the ΔEtotal values and between the corresponding constituents obtained for 1 and 2 is consistent with the similar electronic energy barriers ΔE0‡ (137 and 131 kJ mol−1, respectively). The highest total interaction energy (the weakest bond) is predicted for the complex 3, which consequently most easily dissociates. The dissociation energies for both 3-bromopyridine ligands are very close to each other (ΔEtotal for 3cis and 3trans; see also Fig. 5). On the other hand, the 3-bromopyridine ligand located in cis position to the NHC moiety dissociates with lower activation barrier, compared to that in trans position (27 and 53 kJ mol−1, respectively; see also Fig. 5). This difference is mainly due to stronger stabilization of the latter, arising from the electrostatic and orbital interaction terms, although the Pauli repulsion term is less favourable for 3trans than for 3cis (Table 2). In accordance with the calculated Gibbs energy profiles (Fig. 5), the total interaction energy for the partially dissociated complex 3_dis1a is lower, compared to 3cis and 3trans.
It shall be emphasized that the electrostatic stabilization ΔEelstat dominates in each case, followed by the orbital interaction and dispersion terms (Table 2). The dispersion contributions ΔEdispersion appeared to cover from 9.4% (for 3_dis1a) of the overall stabilization (ΔEdispersion + ΔEorb + ΔEelstat) up to 18.2% (for 3cis). Particularly, ΔEdispersion is extremely significant at absolute level; it is −141 kJ mol−1 for 2 and − 144 kJ mol−1 for 1 (Table 2). It is in accord with the recent findings which highlight that bulky substituents might lead to enhancement of the molecular stability since their London dispersion donating properties might easily outweigh the corresponding steric (Pauli/kinetic) repulsion [59,60,61,62,63,64]. In the forthcoming paragraphs, electron density reorganization channels due to formation of various types of non-covalent interactions are identified and discussed in detail.
It is seen from Fig. 8 that the most relevant deformation density channel, Δρ1, that characterizes the Ru–P bond in 1 depicts donation from the lone electron pair of phosphorus to the metal centre (ΔEorb(1) = −164 kJ mol−1). Quantitatively less important are the π-contributions Δρ2 and Δρ3 related to ΔEorb(2 + 3) = −42 kJ mol−1 which stem from back-donation from 4d orbitals of Ru to the empty σ*(P–C). In addition, we can see secondary contributions from non-covalent interactions of type CH…Cl, CH…π. The former involves charge transfer from the lone pair of chlorine to the empty σ*(C–H), whereas the latter consist of donation from σ(C–H) to the empty π* of phenyl ring. Additionally, homopolar dihydrogen close contacts CH…HC lead to the two ways charge delocalization channels σ(C–H)➔ σ*(C–H) (and vice-versa) [61,62,63,64]. They all amount to ΔEorb(4 + 5 + 6) = −14 kJ mol−1 (Fig. 8). Apart from these charge delocalization components within CH…Cl and CH…π and CH…HC interactions, one can observe, in addition, the crucial stabilization stemming from dispersion as already discussed above.

Primary deformation density contributions Δρ1, Δρ2, and Δρ3, with the corresponding orbital interaction energies ΔEorb(1–3) describing Ru–P bond in complex 1. In addition, formation of secondary non-covalent interactions of the types CH…Cl, CH…π, and CH…HC are presented (Δρ4, Δρ5, Δρ6). Blue/red contours correspond to accumulation/depletion of electron density
An interesting question emerges at this point, namely, how the relative importance of all bonding contributions will change when going from substrates to the corresponding transition states. As an example, the most relevant deformation density contributions for the transition state 1_TS are presented in Fig. 9.

Deformation density contributions Δρ1, Δρ2, Δρ3, and Δρrest with the corresponding orbital interaction energies ΔEorb(1–3) and ΔEorb(rest) describing Ru–P bond in the transition state 1_TS. Blue/red contours correspond to accumulation/depletion of electron density
It is clearly seen that predominantly non-covalent close contacts of the types CH…Cl and CH…π described by Δρ1–3 stabilize the transition state; it corresponds to the electronic stabilization ΔEorb(1 + 2 + 3) = −12 kJ mol−1. Interestingly, the remaining part Δρrest appears to be dominant (ΔEorb(rest) = −16 kJ mol−1) and it is also associated with the formation of CH…Cl, CH…π and CH…HC augmented further by the intra-fragment density reorganization (Fig. 9). Importantly, it is seen from Table 3 that dispersion is decisively the most important stabilizing factor (ΔEdispersion = −74 kJ mol−1) in 1_TS. Similar conclusions on the crucial role of London dispersion forces are valid when considering the remaining transition state structures (Table 3). Similarly, critical role of van der Waals forces has been recently recognized in Diels-Alder cycloadditions as nicely reviewed in Ref. [60]—the authors have also stated correctly, that, although a rational tuning of a reaction barrier through various types of non-covalent interactions (inducing pure London dispersion) is still in “….its infancy” [60], it constitutes a promising new way toward better understanding and designing of chemical reactions.
The changes in bonding constituents when going from the considered substrates (1–3) to the corresponding transition states are collected in Table 4. Such analysis allows for decomposition of the electronic barrier ΔE0≠ into changes in bonding contributions. It can immediately be seen that the lowest barrier noted for dissociation of the 3-bromopyridine ligand located in cis position to the NHC moiety (3cis) is related to the least unfavourable changes in both the electrostatic and orbital interactions contributions; such changes are only + 213 and + 90 kJ mol−1, respectively. Considering decoordination of 3-bromopyridine in trans position (3trans), the corresponding changes in ΔEelstat and ΔEorb values are more repulsive, + 292 and + 135 kJ mol−1, respectively, what results in the higher barrier ΔE0‡, by 25 kJ mol−1, as compared to 3cis. In both cases, a drop in Pauli term is noted; however, it is the least negative for activation of 3cis (Table 4). Variations in dispersion and distortion terms are less pronounced. As opposed, the highest barrier noted for 1 can be traced back to the most significant unfavourable changes in both the electrostatic and orbital interactions contributions which are + 501 and + 238 kJ mol−1, respectively, despite the most favourable relief in Pauli repulsion (− 629 kJ mol−1) and distortion energy term (− 43 kJ mol−1).
Summarizing the above ETS-NOCV based results, one can state that in the complexes 1–3, the bond between the dissociating ligand and the Ru-based fragment is dominated by electrostatic contribution, followed by charge transfer components of σ and π symmetry. In addition, we have identified and quantified the strength of secondary interactions stemming from non-covalent contacts of type CH…Cl, CH…π, and CH…HC. Charge transfer stabilization due to such interactions is between 10 and 15 kJ mol−1, depending on the type of complex. Dispersion contribution appeared to be relatively the least important, though, it can, at absolute level, contribute as much as − 144 kJ mol−1 for complex 1. Exactly opposite situation is valid when considering the transition state structures—namely, the majority of overall stabilization originates from dispersion forces within CH…Cl, CH…π, and CH…HC contacts. It was experimentally shown that initiation rates for olefin metathesis catalysed by Grubbs complexes increase with the change of the halide ligand from Cl, through Br, to I, which was explained mainly by increase in steric bulk promoting phosphine dissociation [6]. According to the present results, stronger London dispersion interactions that stabilize the transition states is another reason for this effect.
Despite crucial role of dispersion in absolute stabilization of the transition states, we have found that the height of the electronic barriers is predominantly controlled by dominating (unfavourable) changes in electrostatic and orbital interaction contributions despite favourable relief in Pauli repulsion and geometry reorganization terms (when going from substrates to the corresponding transition states). In the case of orbital interaction constituent, electron donation from the ligand to the ruthenium centre is the most important, which is in accordance with the observed fast activation of the Grubbs-type complexes bearing ligands that exhibit weak electron donor ability, like pyridine, 3-bromopyridine, or 4-phenylpyridine [7]. Replacing the PCy3 ligand by less electron donating PPh3 also results in increasing the initiation rate [6]. Smaller ligand size might also favour dissociation because of lower dispersion component in total stabilization of a complex. These results demonstrate that any barrier of chemical transformation must originate from variations of all chemical bonding constituents and it is impossible to envision a priori which term will be of prime importance—to this end, quantum chemical analyses of bonding contributions in both substrates and transition states or even better along the entire reaction pathways (e.g. the powerful Activation Strain Model [65], the related approaches [66,67,68,69,70,71], as well as Electron Localization Function and Bonding Evolution Theory [72,73,74]) are in our view crucial for detailed understanding and more rational design of chemical reactions.
Conclusions
Activation of the Grubbs-type catalysts (H2IMes)(PCy3)(Cl)2Ru=CHPh (1), (H2IMes)(PCy3)(Cl)2Ru=CH-CH=CMe2 (2), and (H2IMes)(3-Br-py)2(Cl)2Ru=CHPh (3) has been computationally studied by exploring the reaction pathways for the phosphine and 3-bromopyridine ligands dissociation, combined with the ETS-NOCV analysis of bond energies. Formation of 14-electron propagating species from the Hoveyda-Grubbs-type initiators (H2IMes)(Cl)2Ru=CH(o-OiPrC6H4) (4), (H2IMes)(Cl)2Ru=CH(5-NO2–2-OiPrC6H3) (5), and (H2IMes)(Cl)2Ru=CH(2-OiPr-3-PhC6H3) (6) has also been considered, assuming the dissociative mechanism. Additionally, the IQA energy decomposition has been performed to describe intramolecular interactions in the Hoveyda-Grubbs complexes.
The theoretically determined activity order for the Grubbs-type catalysts, 2 ≤ 1 < < 3, is consistent with the respective initiation rates reported for these systems. Whereas the concentration of the 14-electron ruthenium alkylidene species should be much higher for the initiator 3, compared to the catalysts 1 and 2, a thermodynamic equilibrium between the complex 3 and mono(3-bromopyridine) ruthenium alkylidene species is predicted. The calculated barriers for decoordination of the isopropoxy fragment of the Hoveyda ligand are also in accordance with observed relative activities of the catalysts 4–6.
Although the solvent effects estimated for dissociation of the Grubbs-type complexes are not very significant, they may sometimes be decisive for the predictions concerning the relative activities of these catalysts. In particular, a summary correction to the dissociation energies and barriers, including both solvent simulation and BSSE, is often not negligible. In the case of the initiation step for the Hoveyda-Grubbs-type catalysts, the solvent effects are less important.
According to the ETS-NOCV results, in the Grubbs-type complexes, the bond between the dissociating ligand and the Ru-based fragment is dominated by electrostatic contribution, followed by charge delocalization components; the latter contains primary σ(Ru–P) and π(Ru–P) components together with the secondary non-covalent CH…Cl, CH…π, and CH…HC interactions. Additionally, London dispersion forces appeared to be (at absolute level) also very efficient (between ~50 and 150 kJ mol−1, depending on the catalyst). In contrary, in the case of the corresponding transition state structures, the majority of overall stabilization originates predominantly from London dispersion forces exerted by the pronounced CH…Cl, CH…π, and CH…HC interactions. The height of the electronic barriers is associated with the prevailing (unfavourable) variations in the electrostatic and orbital interaction contributions despite the favourable relief in Pauli repulsion and geometry reorganization terms (when going from substrates to the corresponding transition states). From the IQA analysis, it is found that the isopropoxy group in the Hoveyda-Grubbs-type catalysts forms numerous intra-molecular CH…Cl, CH…π, and CH…Ru interactions which can be crucial for the activity of these catalysts.
References
Samojłowicz C, Bieniek M, Grela K (2009) Ruthenium-based olefin metathesis catalysts bearing N -heterocyclic carbene ligands. Chem. Rev. 109:3708–3742. https://doi.org/10.1021/cr800524f
Vougioukalakis GC, Grubbs RH (2010) Ruthenium-based heterocyclic carbene-coordinated olefin metathesis catalysts. Chem. Rev. 110:1746–1787. https://doi.org/10.1021/cr9002424
Chikkali S, Mecking S (2012) Refining of plant oils to chemicals by olefin metathesis. Angew. Chem. Int. Ed. 51:5802–5808. https://doi.org/10.1002/anie.201107645
Ogba OM, Warner NC, O’Leary DJ, Grubbs RH (2018) Recent advances in ruthenium-based olefin metathesis. Chem. Soc. Rev. 47:4510–4544. https://doi.org/10.1039/c8cs00027a
Sanford MS, Ulman M, Grubbs RH (2001) New insights into the mechanism of ruthenium-catalyzed olefin metathesis reactions. J. Am. Chem. Soc. 123:749–750. https://doi.org/10.1021/ja003582t
Sanford MS, Love JA, Grubbs RH (2001) Mechanism and activity of ruthenium olefin metathesis catalysts. J. Am. Chem. Soc. 123:6543–6554. https://doi.org/10.1021/ja010624k
Love JA, Morgan JP, Trnka TM, Grubbs RH (2002) A practical and highly active ruthenium-based catalyst that effects the cross metathesis of acrylonitrile. Angew. Chem. Int. Ed. 41:4035–4037. https://doi.org/10.1002/1521-3773(20021104)41:21<4035::AID-ANIE4035>3.0.CO;2-I
Thiel V, Hendann M, Wannowius K-J, Plenio H (2012) On the mechanism of the initiation reaction in Grubbs-Hoveyda complexes. J. Am. Chem. Soc. 134:1104–1114. https://doi.org/10.1021/ja208967h
Ashworth IW, Hillier IH, Nelson DJ, Percy JM, Vincent MA (2013) Olefin metathesis by Grubbs-Hoveyda complexes: computational and experimental studies of the mechanism and substrate-dependent kinetics. ACS Catal. 3:1929–1939. https://doi.org/10.1021/cs400164w
Engle KM, Lu G, Luo S-X, Henling LM, Takase MK, Liu P, Houk KN, Grubbs RH (2015) Origins of initiation rate differences in ruthenium olefin metathesis catalysts containing chelating benzylidenes. J. Am. Chem. Soc. 137:5782–5792. https://doi.org/10.1021/jacs.5b01144
Chu CK, Lin T-P, Shao H, Liberman-Martin AL, Liu P, Grubbs RH (2018) Disentangling ligand effects on metathesis catalyst activity: experimental and computational studies of ruthenium-aminophosphine complexes. J. Am. Chem. Soc. 140:5634–5643. https://doi.org/10.1021/jacs.8b02324
Nuñez-Zarur F, Solans-Monfort X, Rodriguez-Santiago L, Sodupe M (2012) Differences in the activation processes of phosphine-containing and Grubbs-Hoveyda-type alkene metathesis catalysts. Organometallics 31:4203–4215
Minenkov Y, Occhipinti G, Jensen VR (2013) Complete reaction pathway of ruthenium-catalyzed olefin metathesis of ethyl vinyl ether: kinetics and mechanistic insight from DFT. Organometallics 32:2099–2111. https://doi.org/10.1021/om301192a
Urbina-Blanco CA, Poater A, Lebl T, Manzini S, Slawin AMZ, Cavallo L, Nolan SP (2013) The activation mechanism of Ru-indenylidene complexes in olefin metathesis. J. Am. Chem. Soc. 135:7073–7079. https://doi.org/10.1021/ja402700p
Solans-Monfort X, Pleixats R, Sodupe M (2010) DFT mechanistic study on diene metathesis catalyzed by Ru-based Grubbs-Hoveyda-type carbenes: the key role of π-electron density delocalization in the Hoveyda ligand. Chem - A Eur J 16:7331–7343. https://doi.org/10.1002/chem.200903525
Ashworth IW, Hillier IH, Nelson DJ, Percy JM, Vincent MA (2011) What is the initiation step of the Grubbs-Hoveyda olefin metathesis catalyst? Chem. Commun. 47:5428–5430. https://doi.org/10.1039/c1cc11230a
Trzaskowski B, Grela K (2013) Structural and mechanistic basis of the fast metathesis initiation by a six-coordinated ruthenium catalyst. Organometallics 32:3625–3630. https://doi.org/10.1021/om400233s
Minenkov Y, Occhipinti G, Heyndrickx W, Jensen VR (2012) The nature of the barrier to phosphane dissociation from Grubbs olefin metathesis catalysts. Eur. J. Inorg. Chem.:1507–1516. https://doi.org/10.1002/ejic.201100932
Mitoraj MP, Michalak A, Ziegler T (2009) A combined charge and energy decomposition scheme for bond analysis. J. Chem. Theory Comput. 5:962–975
Sagan F, Mitoraj MP (2019) Kinetic and potential energy contributions to a chemical bond from the charge and energy decomposition scheme of extended transition state natural orbitals for chemical valence. J. Phys. Chem. A 123:4616–4622. https://doi.org/10.1021/acs.jpca.9b01420
Te Velde G, Bickelhaupt FM, Baerends EJ, Fonseca Guerra C, van Gisbergen SJA, Snijders JG, Ziegler T (2001) Chemsitry with ADF. J. Comput. Chem. 22:931–967
Baerends EJ, Ziegler T, Autschbach J, Bashford D, Bérces A, Bickelhaupt FM, Bo C, Boerrigter PM, Cavallo L, Chong DP, Deng L, Dickson RM, Ellis DE, van Faassen M, Fan L, Fischer TH, Fonseca Guerra C, Franchini M, Ghysels A, Giam-mona A, van Gisbergen SJA, Götz AW, Groeneveld JA, Gritsenko O V, Grüning M, Gusarov S, Harris FE, van den Hoek P, Jacob CR, Jacobsen H, Jensen L, Kaminski JW, van Kessel G, Kootstra F, Kovalenko A, Krykunov M V, van Lenthe E, McCormack DA, Michalak A, Mitoraj M, Morton SM, Neugebauer J, Nicu VP, Noo-dleman L, Osinga VP, Patchkovskii S, Pavanello M, Philipsen PHT, Post D, Pye CC, Ravenek W, Rodríguez JI, Ros P, Schipper, PRT, Schreckenbach G, Seldenthuis JS, Seth M, Snijders JG, Solà M, Swart M, Swerhone D, Te Velde G, Vernooijs P, Versluis L, Visscher L, Visser O, Wang F, Wesolowski TA, van Wezenbeek EM, Wiesenekker G, Wolff SK, Woo TK, Yakovlev AL ADF2012, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com
Blanco MA, Pendás AM, Francisco E (2005) Interacting quantum atoms: a correlated energy decomposition scheme based on the quantum theory of atoms in molecules. J. Chem. Theory Comput. 1:1096–1109. https://doi.org/10.1021/ct0501093
Adamo C, Barone V (1999) Simple reliable density functional method without adjustable parameters: the PBE0 model. J. Chem. Phys. 110:6158–6170
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7:3297–3305. https://doi.org/10.1039/b508541a
Andrae D, Häußermann U, Dolg M, Stoll H, Preuß H (1990) Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 77:123–141. https://doi.org/10.1007/BF01114537
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Accounts 120:215–241. https://doi.org/10.1007/s00214-007-0310-x
Śliwa P, Handzlik J (2010) Assessment of density functional methods for the study of olefin metathesis catalysed by ruthenium alkylidene complexes. Chem. Phys. Lett. 493:273–278. https://doi.org/10.1016/j.cplett.2010.05.066
Benitez D, Tkatchouk E, Goddard III WA (2009) Conformational analysis of olefin-carbene ruthenium metathesis catalysts. Organometallics 28:2643–2645. https://doi.org/10.1021/om900041j
Śliwa P, Kurleto K, Handzlik J, Rogalski S, Żak P, Wyrzykiewicz B, Pietraszuk C (2016) Regioselectivity of stoichiometric metathesis of vinylsilanes with second-generation Grubbs catalyst: a combined DFT and experimental study. Organometallics 35:621–628. https://doi.org/10.1021/acs.organomet.5b00878
Zhao Y, Truhlar DG (2007) Attractive noncovalent interactions in the mechanism of Grubbs second-generation Ru catalysts for olefin metathesis. Org. Lett. 9:1967–1970. https://doi.org/10.1021/ol0705548
Pandian S, Hillier IH, Vincent MA, Burton NA, Ashworth IW, Nelson DJ, Percy JM, Rinaudo G (2009) Prediction of ring formation efficiency via diene ring closing metathesis (RCM) reactions using the M06 density functional. Chem. Phys. Lett. 476:37–40. https://doi.org/10.1016/j.cplett.2009.06.021
Poater A, Pump E, Vummaleti SVC, Cavallo L (2014) The right computational recipe for olefin metathesis with Ru-based catalysts: the whole mechanism of ring-closing olefin metathesis. J. Chem. Theory Comput. 10:4442–4448. https://doi.org/10.1021/ct5003863
Liu P, Xu X, Dong X, Keitz BK, Herbert MB, Grubbs RH, Houk KN (2012) Z-selectivity in olefin metathesis with chelated Ru catalysts: computational studies of mechanism and selectivity. J. Am. Chem. Soc. 134:1464–1467. https://doi.org/10.1021/ja2108728
Śliwa P, Handzlik J, Czeluśniak I (2014) Alkynol polymerization catalysed by Grubbs-type and Hoveyda-Grubbs ruthenium alkylidene complexes: a computational study. J. Organomet. Chem. 767:6–15. https://doi.org/10.1016/j.jorganchem.2014.05.019
Gierada M, Czeluśniak I, Handzlik J (2017) Terminal-alkyne-induced decomposition of a phosphine-free ruthenium alkylidene catalyst. ChemCatChem 9:2284–2291. https://doi.org/10.1002/cctc.201601647
Paredes-Gil K, Jaque P (2016) Theoretical characterization of first and second generation Grubbs catalysts in styrene cross-metathesis reactions: insights from conceptual DFT. Catal Sci Technol 6:755–766. https://doi.org/10.1039/c5cy00826c
Tomasi J, Mennucci B, Cammi R (2005) Quantum mechanical continuum solvation models. Chem. Rev. 105:2999–3093. https://doi.org/10.1021/CR9904009
Gonzalez C, Schlegel HB (1989) An improved algorithm for reaction path following. J. Chem. Phys. 90:2154–2161. https://doi.org/10.1063/1.456010
Gonzalez C, Schlegel HB (1990) Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 94:5523–5527. https://doi.org/10.1021/j100377a021
Boys SF, Bernardi F (1970) The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 19:553–566. https://doi.org/10.1080/00268977000101561
Engel J, Smit W, Foscato M, Occhipinti G, Törnroos KW, Jensen VR (2017) Loss and reformation of ruthenium alkylidene: connecting olefin metathesis, catalyst deactivation, regeneration, and isomerization. J. Am. Chem. Soc. 139:16609–16619. https://doi.org/10.1021/jacs.7b07694
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2013) Gaussian 09, revision D.01. Gaussian, Inc., Wallingford CT
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38:3098–3100. https://doi.org/10.1103/PhysRevA.38.3098
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37:785–789. https://doi.org/10.1103/PhysRevB.37.785
Grimme S, Antony J, Ehrlich S, Krieg H (2010) A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 132:154104-1-154104–19. https://doi.org/10.1063/1.3382344
Grimme S, Ehrlich S, Goerigk L (2011) Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32:1456–1465. https://doi.org/10.1002/jcc.21759
Straub BF (2007) Ligand influence on metathesis activity of ruthenium carbene catalysts: a DFT study. Adv. Synth. Catal. 349:204–214. https://doi.org/10.1002/adsc.200600465
Czeluśniak I, Handzlik J (2009) Polymerization of hydroxyacetylenes by ruthenium alkylidene complexes. J. Organomet. Chem. 694:1427–1434. https://doi.org/10.1016/j.jorganchem.2008.12.037
Czeluśniak I, Handzlik J, Gierada M, Szymańska-Buzar T (2015) Catalytic transformation of phenylacetylene mediated by phosphine-free ruthenium alkylidene complexes. J. Organomet. Chem. 786:31–39. https://doi.org/10.1016/j.jorganchem.2015.03.025
Gierada M, Czeluśniak I, Handzlik J (2019) Dimerization and cyclotrimerization of terminal arylalkynes initiated by a phosphine-free ruthenium alkylidene complex. Mol Catal 469:18–26. https://doi.org/10.1016/j.mcat.2019.02.026
de Brito SÉ, de Matos JME (2015) Ring closing metathesis by Hoveyda-Grubbs catalysts: a theoretical approach of some aspects of the initiation mechanism and the influence of solvent. Inorganica Chim Acta 426:20–28. https://doi.org/10.1016/j.ica.2014.11.007
Trzaskowski B, Goddard WA, Grela K (2017) Faster initiating olefin metathesis catalysts from introducing double bonds into cyclopropyl, cyclobutyl and cyclopentyl derivatives of Hoveyda-Grubbs precatalysts. Mol Catal 433:313–320. https://doi.org/10.1016/j.mcat.2016.12.018
Trzaskowski B, Ostrowska K (2017) A computational study of structures and catalytic activities of Hoveyda-Grubbs analogues bearing coumarin or isopropoxycoumarin moiety. Catal. Commun. 91:43–47. https://doi.org/10.1016/j.catcom.2016.12.014
Grela K, Harutyunyan S, Michrowska A (2002) A highly efficient ruthenium catalyst for metathesis reactions. Angew. Chem. Int. Ed. 41:4038–4040. https://doi.org/10.1002/1521-3773(20021104)41:21<4038::AID-ANIE4038>3.0.CO;2-0
Michrowska A, Bujok R, Harutyunyan S, Sashuk V, Dolgonos G, Grela K (2004) Nitro-substituted Hoveyda-Grubbs ruthenium carbenes: enhancement of catalyst activity through electronic activation. J. Am. Chem. Soc. 126:9318–9325. https://doi.org/10.1021/ja048794v
Nuñez-Zarur F, Poater J, Rodríguez-Santiago L, Solans-Monfort X, Solà M, Sodupe M (2012) On the electronic structure of second generation Hoveyda-Grubbs alkene metathesis precursors. Comput Theor Chem 996:57–67. https://doi.org/10.1016/j.comptc.2012.07.015
Wakamatsu H, Blechert S (2002) A new highly efficient ruthenium metathesis catalyst. Angew. Chem. Int. Ed. 41:2403–2405. https://doi.org/10.1002/1521-3773(20020703)41:13<2403::AID-ANIE2403>3.0.CO;2-F
Wagner JP, Schreiner PR (2015) London dispersion in molecular chemistry - reconsidering steric effects. Angew Chemie - Int Ed 54:12274–12296. https://doi.org/10.1002/anie.201503476
Liptrot DJ, Power PP (2017) London dispersion forces in sterically crowded inorganic and organometallic molecules. Nat Rev Chem 1:0004
Mitoraj MP, Sagan F, Babashkina MG, Isaev AY, Chichigina YM, Safin DA (2019) N-Thiophosphorylthioureas RNHC(S)NHP(S)(OiPr)2 as an excellent platform for studying the synergy between hydrogen-hydrogen bonding and other families of non-covalent interactions. European J Org Chem 2019:493–503. https://doi.org/10.1002/ejoc.201801041
Mitoraj MP, Babashkina MG, Robeyns K, Sagan F, Szczepanik DW, Seredina YV, Garcia Y, Safin DA (2019) Chameleon-like nature of anagostic interactions and its impact on metalloaromaticity in square-planar nickel complexes. Organometallics 38:1973–1981. https://doi.org/10.1021/acs.organomet.9b00062
Mitoraj MP, Babashkina MG, Isaev AY, Chichigina YM, Robeyns K, Garcia Y, Safin DA (2018) London dispersion forces in crystal packing of thiourea derivatives. Cryst. Growth Des. 18:5385–5397. https://doi.org/10.1021/acs.cgd.8b00783
Wang C, Mo Y, Wagner JP, Schreiner PR, Jemmis ED, Danovich D, Shaik S (2015) The self-association of graphane is driven by London dispersion and enhanced orbital interactions. J. Chem. Theory Comput. 11:1621–1630. https://doi.org/10.1021/acs.jctc.5b00075
Fernández I, Bickelhaupt FM (2014) The activation strain model and molecular orbital theory: understanding and designing chemical reactions. Chem. Soc. Rev. 43:4953–4967. https://doi.org/10.1039/C4CS00055B
Alkorta I, Thacker JCR, Popelier PLA (2018) An interacting quantum atom study of model SN2 reactions (X–···CH3X, X = F, Cl, Br, and I). J. Comput. Chem. 39:546–556. https://doi.org/10.1002/jcc.25098
Díaz S, Brela MZ, Gutiérrez-Oliva S, Toro-Labbé A, Michalak A (2017) ETS-NOCV decomposition of the reaction force: the HCN/CNH isomerization reaction assisted by water. J. Comput. Chem. 38:2076–2087. https://doi.org/10.1002/jcc.24856
Toro-Labbé A (1999) Characterization of chemical reactions from the profiles of energy, chemical potential, and hardness. J. Phys. Chem. A 103:4398–4403. https://doi.org/10.1021/jp984187g
Jaque P, Toro-Labbé A, Politzer P, Geerlings P (2008) Reaction force constant and projected force constants of vibrational modes along the path of an intramolecular proton transfer reaction. Chem. Phys. Lett. 456:135–140. https://doi.org/10.1016/j.cplett.2008.03.054
Morokuma K (1971) Molecular orbital studies of hydrogen bonds. III. C=O···H–O hydrogen bond in H2CO···H2O and H2CO···2H2O. J. Chem. Phys. 55:1236–1244. https://doi.org/10.1063/1.1676210
Ess DH, Houk KN (2007) Distortion/interaction energy control of 1,3-dipolar cycloaddition reactivity. J. Am. Chem. Soc. 129:10646–10647. https://doi.org/10.1021/ja0734086
Berski S, Latajka Z (2011) A mechanism of the 1,3-dipolar cycloaddition between the hydrogen nitryl HNO2 and acetylene HCCH: the electron localization function study on evolution of the chemical bonds. Int. J. Quantum Chem. 111:2378–2389. https://doi.org/10.1002/qua.22532
Kalinowski J, Berski S, Latajka Z (2011) AIM and BET approach for ionic and covalent bond evolution in reaction of hydrogen elimination from ammonia and lithium hydride. Chem. Phys. Lett. 501:587–593. https://doi.org/10.1016/j.cplett.2010.11.053
Berski S, Latajka Z, Gordon AJ (2010) Ab initio and quantum chemical topology studies on the isomerization of HONO to HNO2. Effect of the basis set in QCT. J Comput Chem 31:2555–2567. https://doi.org/10.1002/jcc.21547
Funding
This research was supported in part by the PL-Grid Infrastructure. M.P.M. acknowledges the financial support of the Polish National Science Center within the Sonata Bis Project 2017/26/E/ST4/00104.
Author information
Authors and Affiliations
Corresponding authors
Additional information
This paper is dedicated to Professor Zdzisław Latajka, honouring his significant contribution to quantum chemistry.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This paper belongs to the Topical Collection Zdzisław Latajka 70th Birthday Festschrift
Electronic supplementary material
ESM 1
(PDF 991 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Śliwa, P., Mitoraj, M.P., Sagan, F. et al. Formation of active species from ruthenium alkylidene catalysts—an insight from computational perspective. J Mol Model 25, 331 (2019). https://doi.org/10.1007/s00894-019-4202-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-4202-5