
Abstract
Linear α-olefins or LAOs are produced by the catalytic oligomerisation of ethylene on a multimillion ton scale annually. A range of LAOs is typically obtained with varying chain lengths which follow a distribution. Depending on the catalyst, various types of distributions have been identified, such as Schulz–Flory, Poisson, alternating and selective oligomerisations such as ethylene trimerisation to 1-hexene and tetramerisation to 1-octene. A comprehensive mathematical analysis for all oligomer distributions is presented, showing the relations between the various distributions and with ethylene polymerisation, as well as providing mechanistic insight into the underlying chemical processes.
Similar content being viewed by others



Avoid common mistakes on your manuscript.
1 Introduction
Linear alpha olefins (LAOs, or 1-alkenes) find extensive use as co-monomers in olefin polymerisation and as intermediates to detergents, lubricants, and plasticisers [1,2,3]. Global consumption of LAOs grew at an average annual rate of 5.6% from 2012 to 2016, and global capacity for LAO production has been estimated at 6.2 million tonnes per year (mtpy) for 2018 [4, 5]. LAOs are generally prepared via the oligomerisation of ethylene, as is shown below in Scheme 1 [6].

Generic scheme for LAO synthesis via the oligomerisation of ethylene
LAO processes may be grouped into two types: wide-range and on-purpose [7]. Wide-range processes give a distribution of LAO products from C4 to C20+, whilst on-purpose processes predominantly give C4, C6 or C8 LAOs [7]. Examples of catalytic systems used in industry are given in Figs. 1 and 2 [6,7,8,9].

Wide-range industrial processes giving LAO distributions from C4 to C20+ (X = appropriate anion). Note: INEOS’s process gives a Poisson distribution of LAOs, whilst the SHOP and Gulfene processes give Schulz–Flory distributions of LAOs

On-purpose industrial processes predominantly giving C4, C6 or C8 LAOs (X = appropriate anion)
Three of the most common types of distributions from ethylene oligomerisation and polymerisation processes—namely Schulz–Flory, selective-LAO, and Poisson—are observed with the systems in Figs. 1 and 2. In addition, a number of alternating LAO distributions with chromium-based systems have been reported in the literature [11,12,13,14,15,16,17]. These four types of ethylene product distribution are depicted in Fig. 3.

Clockwise from top-left: illustrations of Schulz–Flory, alternating, selective-LAO, and Poisson distributions
Following on from the ground-breaking work 80 years ago by Schulz and Flory [18, 19], novel mathematical approaches for analysing product distributions from ethylene oligomerisation and polymerisation processes are presented in this paper. These approaches provide straightforward, applicable methods for characterising key mechanistic features of underlying catalytic processes, and demonstrate how Schulz–Flory, alternating, selective-LAO, and Poisson distributions may be considered as parts of a connected product distribution landscape.
2 Modelling Steady-State Oligomerisation Processes
2.1 First-Order Oligomerisation Processes
In 1935, Schulz characterised the mathematics behind first-order oligomerisation processes [18]. He noted the direct proportionality between mol(n)% (mol percentage for an oligomer of n ethylene units) and the product of a sequence featuring a constant propagation probability α:
Following a continuous function approximation, this implies (see SI.1.1 for derivation):
The relationship between wt(n)%, mol(n)% and n is:
where (RFM C2H4) is the Relative Formula Mass of ethylene.
Using the expression for mol(n)% from Eq. 1a and condensing:
Again making use of a continuous function approximation (see SI.1.2 for derivation), this implies:
The following year, in 1936, Flory presented precise derivations without making use of continuous function approximations [19]. Flory noted the dependence of mol(n), the amount of moles for each oligomer of n ethylene units, on the product of (n − 1) propagation steps and 1 elimination step along with R, the total mol of oligomers produced during the reaction time:
Consequently:
It follows that:
which gives (see SI.1.3 for derivation):
Comparing Eq. 1b with Eq. 3b, it is evident that both Schulz’s equation and Flory’s equation express:
The two Eqs. 1b and 3b are subtly different, as shown in Fig. 4, because \(- \left( {ln\alpha } \right)\) is the proportionality constant for Schulz and \((1 - \alpha)/\alpha\) is the proportionality constant for Flory. However, in practice, this makes little difference because \(\alpha\) is typically determined by taking natural logarithms to each side of Eq. 5a, plotting ln(mol(n) %) vs. n, performing a linear regression analysis to find the gradient lnα and then exponentiating the result as a power of e [20]. The value for \(\alpha\) determined in this way will be the same in both cases.

By comparing Eq. 2c with Eq. 4b, it may be seen that:
with \(\left( {ln\alpha } \right)^{2}\) as the proportionality constant for Schulz and \(\left( {1 - \alpha } \right)^{2} /\alpha\) as the proportionality constant for Flory (Table 1).
In the next section, a new derivation for Eq. 3a will be presented which utilises recurrence relations, followed by the presentation of a new back-solving method to determine α and R. The analysis lays the groundwork for an analogous consideration of second-order oligomerisation processes.
2.1.1 Derivation of the Schulz–Flory Equation via Recurrence Relations
Catalytic oligomerisation systems which give Schulz–Flory LAO distributions propagate either via a Cossee chain growth mechanism or via a metallacyclic mechanism (Schemes 2 and 3). Both mechanisms may be generically represented by Scheme 4. Deuterium labelling studies have shown that many chromium systems operate via the metallacyclic pathway [21,22,23].

Cossee-type chain growth mechanism for Schulz–Flory LAO distributions. Active species and monomer shown in black, eliminated products shown in blue

Metallacyclic mechanism for Schulz–Flory LAO distributions. Active species and monomer shown in black, eliminated products shown in blue

Generic scheme for Schulz–Flory LAO distributions. Active species shown in black, rates and eliminated products shown in blue, probabilities of propagation and elimination pathways shown in green
In Scheme 4:
-
R is the total amount in mol of oligomers produced (from T1 onwards) during the reaction time i.e. the rate at which intermediate species arrive at
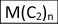 , in units of mol t−1.
, in units of mol t−1. -
α is the propagation probability from
 to
to
 etc.
etc.-
So (1 − α) is the elimination probability from
 etc.
etc.
-
-
\(T_{n}\) is the total mol of a particular LAO with chain length (C2)n produced during the reaction time (i.e. the rate at which that particular LAO is eliminated, in units of mol t−1), with T1 as the starting point in the sequence.
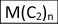 , in units of mol t−1.
, in units of mol t−1. to
to
 etc.
etc. etc.
etc.Expressions for each LAO fraction may be found by inspection of Scheme 4, or by using a recurrence relation methodology. These two techniques are demonstrated in the centre and right columns of Table 2 respectively.
The aim is to express Tn in terms of α and R. From inspection of the central column of Table 2, it may be inferred that the expression is:
Equation 6 is exactly the unnormalised Schulz–Flory equation, as per Eq. 3a.
Via the recurrence relation method, the general expression is:
Following a derivation (see SI.1.4), the general solution for this expression is:
i.e. the unnormalised Schulz–Flory equation, as per Eqs. 3a and 6.
With constant values for α and R, it is implicitly assumed that:
-
The sequence begins at a suitable T1 (e.g. a longer 1-alkene).
-
The oligomerisation proceeds under steady state conditions (i.e. negligible change in ethylene pressure, reaction temperature and quantity of catalytic species).
-
Under steady state conditions, the standing quantities of each catalytic species are constant. The sum of rates to any particular active species “box” in Scheme 4 are equal to the sum of rates from that “box”. This is equivalent to saying that the sum of probabilities for any particular active species “box” is 1 e.g. for
 \(R = \alpha R + \left( {1 - \alpha } \right)R \leftrightarrow 1 = \alpha + \left( {1 - \alpha } \right)\), for
\(R = \alpha R + \left( {1 - \alpha } \right)R \leftrightarrow 1 = \alpha + \left( {1 - \alpha } \right)\), for
 \(\alpha R = \alpha^{2} R + \alpha \left( {1 - \alpha } \right)R \leftrightarrow 1 = \alpha + \left( {1 - \alpha } \right)\), etc.
\(\alpha R = \alpha^{2} R + \alpha \left( {1 - \alpha } \right)R \leftrightarrow 1 = \alpha + \left( {1 - \alpha } \right)\), etc.
-


2.1.2 Back-Solving to Determine \(\alpha\) and R, and Checking for the Best Fit
In practice, the Tn terms are known (i.e. the amount in mol of each oligomer produced during the reaction time), and the aim is to find α and R. See SI.1.5 for a discussion on the method used to determine α and R, as well as a checking method to determine that the best fit has been found.
2.1.3 Application of the Fitting Procedure to Experiment
Bis(benzimidazole)pyridine chromium(III) chloride, in combination with MAO co-catalyst, gives a Schulz–Flory distribution of LAOs via a metallacyclic mechanism (Fig. 5) [24]. Fitting α either via a ln(mol(n)%) vs. n plot or via the recurrence relation method gives a resulting value of α = 0.84.

Schulz − Flory LAO distribution as mol(n) % vs n (units of ethylene) obtained with bis(benzimidazole)pyridine chromium(III) chloride (1 μmol) [16, 24]. Inset: \(ln\left( {mol\left( n \right)\% } \right)\) vs. \(n\) plot. Conditions: 4 bar of ethylene pressure, 30 °C, MAO (8 mmol), toluene (220 mL), 70 min. Activity = 5314 g mmol−1 h−1 bar−1
It is worth noting that the determination of α via the recurrence relation method offers a subtle advantage over ln(mol(n)%) vs. n plots, in that accurate fits can only be obtained for distributions with reasonably constant α values as per Scheme 4. Linear regression analysis via the ln(mol(n)%) vs. n method may permit adequate fits for data sets displaying non-constant α behaviour, leading to Schulz–Flory equations which deviate from experimental plots.
2.1.4 Derivations of Further Experimentally-Relevant Metrics
For polymer distributions, Mn, Mw, and PDI are defined as follows:
These definitions can be applied to Schulz–Flory distributions. Using Eq. 3a, 4a and 4b the following expressions are obtained (see SI.1.6 for derivations):
Note that α values are between 0 and 1. Consequently, PDI values are between 1 and 2 for Schulz–Flory distributions.
2.2 Second-Order Oligomerisation Processes
In 2006, Tomov et al. discovered a highly active chromium-based oligomerisation system, with activities in excess of 100,000 g mmol−1 h−1 bar−1, which gave an alternating distribution of 1-alkenes via a metallacyclic mechanism (Fig. 6) [15].

Distribution of 1-alkene oligomers as mol(n)% vs n (units of ethylene) produced via N,N-bis((1H-benzimidazol-2-yl)methyl)-N-methylamine chromium(III) chloride/MAO [16, 17]. Conditions: 32 nmol Cr, 4 bar continuous ethylene pressure, 50 °C, MAO (7 mmol), toluene (200 mL), 1 h. Activity = 102,300 g mmol−1 h−1 bar−1
The shape of this distribution cannot be described by first-order linear homogeneous recurrence relations with constant coefficients e.g. the Schulz–Flory equation. Variations on the first-order theme with orderly coefficients are likewise unable to accurately describe the experimentally-observed results. For instance, in a hypothetical alternating coefficients case, the quantities of the series of oligomers 1-hexene, 1-decene, 1-tetradecene etc. always decrease with n. Yet the distribution in Fig. 6 evidently displays some sort of periodicity.
All oligomerisations may be mathematically considered in terms of recurrence relations, so from an analytic point of view it is logical to proceed from a first-order treatment to a second-order treatment. Chemically speaking, this implies a mechanism involving both single and double coordination and insertion of ethylene. The double coordination and insertion of olefins has been proposed previously. Ystenes proposed a “trigger” mechanism whereby a coordinated olefin only inserts once a second olefin has coordinated to the metal [25]. Lemos and co-workers proposed a related mechanism based on kinetic studies of Ziegler–Natta catalysts [26], and a similar mechanism has been implicated in other oligomerisation processes [27, 28].
Building on the analysis from Sect. 2.1, a new “alternating equation” is derived here, which may be viewed as a direct analogue to the Schulz–Flory equation for second-order oligomerisation processes and which accurately describes alternating distributions as per Fig. 6. This is followed by the presentation of a new back-solving method for the determination of key mechanistic parameters.
2.2.1 Derivation of the Alternating Equation via Recurrence Relations
In Scheme 5:
-
R1 is the rate at which intermediate species arrive at
 , in units of mol t−1.
, in units of mol t−1. -
R2 is the rate at which intermediate species originating from before
 arrive at
arrive at
 , in units of mol t−1.
, in units of mol t−1.-
So the total amount in mol of oligomers, R, produced (from T1 onwards) during the reaction time is expressed by \(R = R_{1} + R_{2}\)
-
-
α is the propagation probability from
 to
to
 etc.
etc. -
β is the propagation probability from
 to
to
 etc.
etc.-
So (1 − α − β) is the probability of elimination from
 etc.
etc.
-
-
Tn is the total mol of a particular 1-alkene with chain length (C2)n produced during the reaction time (i.e. the rate at which that particular 1-alkene is eliminated, in units of mol t−1), with T1 as the starting point in the sequence.
 , in units of mol t−1.
, in units of mol t−1. arrive at
arrive at
 , in units of mol t−1.
, in units of mol t−1. to
to
 etc.
etc. to
to
 etc.
etc. etc.
etc.
Catalytic scheme for the production of alternating 1-alkene oligomer distributions. Active species shown in black, rates and eliminated products shown in blue, probabilities of propagation and elimination pathways shown in green
With reference to Scheme 5, Table 3 expresses each LAO via inspection (central column) or via recurrence relation (right column). The aim is to express Tn in terms of α, β, R1 and R2. In contrast to Sect. 2.1, obtaining a general expression via inspection is no longer feasible. The method of solving via a recurrence relation is much more straightforward, as will be demonstrated below.
The recurrence relation is:
Following a derivation (see SI.2.1), the general solution to this expression is: [16]
with:
i.e. the unnormalised alternating equation.
With constant values for α, β, R1 and R2, it is implicitly assumed that:
-
The sequence begins at a suitable T1 (e.g. a longer 1-alkene)
-
The oligomerisation proceeds under steady state conditions (i.e. negligible change in ethylene pressure, reaction temperature and quantity of catalytic species)
-
Under steady state conditions, the standing quantities of each catalytic species are constant. The sum of rates to any particular active species “box” are equal the sum of rates from that “box”. This is equivalent to saying that the sum of probabilities from any particular active species “box” is 1 e.g. for
 \(R_{1} = \alpha R_{1} + \beta R_{1} + \left( {1 - \alpha - \beta } \right)R_{1} \leftrightarrow 1 = \alpha + \beta + \left( {1 - \alpha - \beta } \right)\), for
\(R_{1} = \alpha R_{1} + \beta R_{1} + \left( {1 - \alpha - \beta } \right)R_{1} \leftrightarrow 1 = \alpha + \beta + \left( {1 - \alpha - \beta } \right)\), for
 \(\alpha R_{1} + R_{2} = \alpha \left( {\alpha R_{1} + R_{2} } \right) + \beta \left( {\alpha R_{1} + R_{2} } \right) + \left( {1 - \alpha - \beta } \right)\left( {\alpha R_{1} + R_{2} } \right) \leftrightarrow \alpha R_{1} = \alpha^{2} R_{1} + \beta \alpha R_{1} + \left( {1 - \alpha - \beta } \right)\alpha R_{1} \leftrightarrow 1 = \alpha + \beta + \left( {1 - \alpha - \beta } \right)\), etc.
\(\alpha R_{1} + R_{2} = \alpha \left( {\alpha R_{1} + R_{2} } \right) + \beta \left( {\alpha R_{1} + R_{2} } \right) + \left( {1 - \alpha - \beta } \right)\left( {\alpha R_{1} + R_{2} } \right) \leftrightarrow \alpha R_{1} = \alpha^{2} R_{1} + \beta \alpha R_{1} + \left( {1 - \alpha - \beta } \right)\alpha R_{1} \leftrightarrow 1 = \alpha + \beta + \left( {1 - \alpha - \beta } \right)\), etc.


To convert the unnormalised alternating equation, Eq. 11, into the normalised form:
or equivalently (see SI.2.2):
Converting Eq. 11 into an unnormalised wt(n) equivalent:
which gives (see SI.2.3):
2.2.2 Back-Solving to Determine α, β, R1, and R2, and Checking for the Best Fit
In practice, the Tn terms are known (i.e. the amount in mol of each oligomer produced during the reaction time), and the aim is to find α, β, R1, and R2. See SI.2.4 for a discussion on the method used to determine α and R, as well as a checking method to determine that the best fit has been found.
2.2.3 Application of the Fitting Procedure to Experiment
See Sect. 2.4 for examples of fittings using the described recurrence relation method with N,N-bis((1H-benzimidazol-2-yl)methyl)-N-methylamine chromium(III) chloride/MMAO-12 systems.
2.2.4 Derivations of Further Experimentally-Relevant Metrics
As per Schulz–Flory distributions, the definitions of Mn, Mw and PDI in Eqs. 9a, 9b, 9c can be applied to alternating distributions. Using Eqs. 11, 14 and 15, and Eqs. 12ii and 14iii in SI, the following expressions are obtained (see SI.2.5 for derivations):
with:
Note that with \(q = 0, y = 1, \text{and}\,r_{1} = \alpha\), PDI matches the expression previously obtained with Schulz–Flory distributions (see Eq. 9f):
As with Schulz–Flory distributions, PDI values for alternating distributions lie between 1 and 2. This may be demonstrated by examining four limiting cases (see SI.2.6).
2.3 Connecting First-Order and Second-Order Oligomerisation Processes
By inspection of Eq. 11, two cases may be identified where the second-order general solution collapses to a first-order solution as per Eq. 10. In the first case, if β = 0, then from Eq. 10iv \(r_{1} = \alpha\) and from Eq. 10v \(r_{2} = 0\). With \(r_{2} = 0\), Eq. 10xiii gives \(R_{2} = 0\). Then, using Eqs. 10x and 10xi (in SI.2.1 and SI.2.4):
Since \(R_{2} = 0\), then \(R_{1} = R\). Using Eq. 11, the general solution is therefore:
i.e. the unnormalised Schulz–Flory equation.
In the second, more general case, if \(c_{2} = 0\) but \(\beta\) is not necessarily 0, then with Eqs. 10xii and 10xiii in Supplementary Information (SI.2.4):
Using Eq. 10v (Supplementary Information SI.2.1), Eq. 19 becomes:
Using Eq. 19 with Eqs. 10x and 10xi (in SI.2.1):
From Eq. 11, the general solution is therefore:
which gives the unnormalised Schulz–Flory equation if β = 0. The second case may be summarised by stating that given particular values for α and β, R2/R1 may be tuned to generate first-order behaviour.
In this sub-section, the mathematics of the second case is illustrated, followed by a method to describe the degree of similarity between second-order recurrence relations and first-order ones. This analysis has important implications for the fitting of more “gently” alternating oligomer distributions.
2.3.1 The First-Order Surface in R2/R1, α and β Space
Equation 20 features three variables, R2/R1, \(\alpha\) and β, which are physically interpretable as per Scheme 5. Conveniently, then, it is possible to plot in 3D space the combinations of \(R_{2} /R_{1}\), \(\alpha\) and β values which give rise to first-order behaviour. Furthermore, since r2 does not feature in Eq. 22 and since β is not necessarily 0, Eq. 10iv (Supplementary Information SI.2.1) now operates as a parametric equation where different combinations of α and β may give the same result for r1. A visualisation of the first-order surface in R2/R1, \(\alpha\) and \(\beta\) space, constructed by lines along which r1 is constant, is shown in Fig. 7 and 8. Note that first-order distributions following Eq. 18 are restricted to the alpha axis with β = 0 and R2/R1 = 0.

Visualisation of the first-order surface in \(R_{2} /R_{1}\), \(\alpha\) and \(\beta\) space along the \(\alpha\) axis (left) and along the \(\beta\) axis (right). Colours correspond to particular values of \(r_{1}\) (key centre)

Visualisation of the first-order surface in \(R_{2} /R_{1}\), \(\alpha\) and \(\beta\) space along the \(R_{2} /R_{1}\) axis. Optically identical first-order distributions with \(r_{1} = 0.50\) illustrated
2.3.2 Degree of Alternation
By sight, it is evident that second-order distributions can possess varying degrees of alternating character. To arrive at a definition for the degree of alternation in a second-order distribution, we need to consider the pictorial representation of Eq. 11.
Figure 9 illustrates how an alternating distribution is mathematically constructed by adding a constantly decreasing first-order recurrence relation to an alternating first-order recurrence relation (recall that with 0 ≤ α < 1, 0 ≤ β < 1, and 0 < (1 − α − β) < 1, then 0 < r1 < 1, \(- 1 < r_{2} < 0, {\text{and also}}\left| {r_{2} } \right| \le \left| {r_{1} } \right|\)). The degree of alternation is defined as:

Pictorial representation of Eq. 11
Absolute values are taken in the numerator to avoid cancellation of terms on summation (refer to Fig. 9). 0 < c1 (see Eq. 10viii in SuppIementary Information SI.2.1) so \(0 < \mathop \sum \nolimits_{n = 1}^{n = \infty } c_{1} r_{1}^{n}\). Given that \(- 1 < r_{2} < 0\), then \(\left| {r_{2} } \right|^{n} = \left( { - r_{2} } \right)^{n}\).
Evaluation of Eq. 23a (in SI.3.1) gives the following result:
Since \(0 < r_{1} < 1\), \(- 1 < r_{2} < 0, {\text{and }}\left| {r_{2} } \right| \le \left| {r_{1} } \right|\), then \(0 \le Deg.\;of\;Alt. < 1\) (i.e. \(Deg.\;of\;Alt.\) may be considered as a percentage).
As \(r_{1}\) and \(r_{2}\) may be expressed in terms of α and β, Eq. 23b gives the degree of alternation as a function of R2/R1,\(\alpha\) and β. In other words, every point in R2/R1,\(\alpha\) and β space has an associated degree of alternation. A visualisation of the degree of alternation map in R2/R1,\(\alpha\) and β space is shown in Fig. 10. Note that although \(0 \le R_{2} /R_{1}\), for convenience Fig. 10 runs only from \(0 \le \frac{{R_{2} }}{{R_{1} }} \le 1\).

Front (left) and back (right) view of the degree of alternation map in \(R_{2} /R_{1}\), \(\alpha\) and \(\beta\) space. Colours correspond to particular values for degree of alternation (key centre)
The red areas in Fig. 10 represent those areas with the largest degree of alternation, i.e. where \(R_{2} /R_{1}\) or \(\beta\) are highest. The light blue layer corresponds to the first-order surface shown in Figs. 7 and 8, where distributions display no degree of alternation. This layer acts as a crossing-point, above which distributions display “normal” alternating behaviour, and below which distributions display “inverse” alternation. Figure 11 illustrates these results. Note that in Fig. 11, \({\alpha}\) and \({\beta}\) remain constant while \(R_{2} /R_{1}\) varies. While experimentally so far only “normal” alternating distributions have been found, the discovery of “inverse” alternating behaviour is theoretically possible.

Examples of distributions obtained above the first-order surface (top) showing “normal” alternating behaviour, on the first-order surface (middle), and below the first-order surface (bottom) showing “inverse” alternating behaviour
2.3.3 Implications for Second-Order Fitting with “Gently” Alternating Distributions
“Gently” alternating distributions display significantly less \(c_{2} r_{2}^{n}\) character than \(c_{1} r_{1}^{n}\) character. Such distributions lie close to the first-order surface in \(R_{2} /R_{1}\), \(\alpha\) and \(\beta\) space as illustrated in Figs. 7 and 8. In back-solving to determine \(\alpha\), \(\beta\), \(R_{1}\), and \(R_{2}\), “gently” alternating distributions present a unique difficulty in the fitting process when experimental error is taken into account.
The first-order surface in Figs. 7 and 8 is constructed by lines along which \(r_{1}\) is constant and along which distributions appear identical. Second-order distributions which are similar in appearance, but which lie close to the first-order surface, may accurately be described using quite different \(R_{2} /R_{1}\), \(\alpha\) and \(\beta\) values.
By way of illustration, consider the hypothetical second-order distribution shown in Fig. 12, as may be detected via GC from 1-hexene onward. If, due to experimental error, a quantity of 1-hexene evaporates prior to GC measurement, then the back-solving method produces a different fit for the data set slightly perturbed at C6.

Hypothetical second-order “gently” alternating distribution
Comparing the fitted parameters in Fig. 12 with those in Fig. 13, there are significant differences between values for \(\alpha\), \(\beta\), \(R_{2} /R_{1}\), \(c_{2}\) and \(r_{2}\). The only reliable metric for “gently” alternating distributions is the first-order \(c_{1} r_{1}^{n}\) term. In borderline cases where a second-order distribution lies too close to the first-order surface to obtain reliable values for \(\alpha\), \(\beta\), \(R_{1}\), and \(R_{2}\), pressure studies (see Sect. 2.4) and/or repeat experiments will be necessary.

Hypothetical second-order “gently” alternating distribution with some 1-hexene missing
2.4 The Effect of Pressure in Second-Order Steady-State Oligomerisation
In Sect. 2.2, a catalytic scheme (Scheme 5) for the production of alternating 1-alkene oligomer distributions was presented. The terms \(M\left( {C_{2} } \right)_{n}\), \(M\left( {C_{2} } \right)_{n + 1}\) etc. group together all intermediates involving \(n\) ethylene units. In order to investigate the effect of changing ethylene pressure on \(\alpha\) and \(\beta\), it is necessary to propose a more detailed scheme with the individual elementary reaction steps that occur within each active species “box” (i.e. between each type of \(M\left( {C_{2} } \right)_{n}\) species). This so-called candidate scheme is kept as general as possible, for application to any processes proceeding via a metallacyclic mechanism (Scheme 6):

Candidate scheme proposed for individual reaction steps occurring between each type of metallacyclic \(M\left( {C_{2} } \right)_{n}\) species, with selected rate constants labelled
Following a derivation (see SI.4.1), it can be shown that the ratio between the probabilities for double ethylene insertion β and single insertion α is defined as:
So long as \(k_{2}, k_{7} \ne 0\), the relationship between the probabilities \(\alpha\) and \(\beta\) and ethylene pressure \(P_{et}\) can be expressed as:
where:
2.4.1 Comparison of Experimentally Determined Results vs. Fitted Data
A series of oligomerisation experiments have been performed at different ethylene pressures, using N,N-bis((1H-benzimidazol-2-yl)methyl)-N-methylamine chromium(III) chloride/MMAO-12 as the catalyst (experimental details as per ref. 17). The \(\alpha\) and \(\beta\) values for each run were obtained using the back-solving method described in Sect. 2.2, and the values of \({\beta }/{\alpha }\) were plotted vs. ethylene pressure. A curve following Eq. 24b was fitted to these calculated values, using a minimisation of the sum of squared differences to determine the most suitable values for \(g_{1}\) and \(g_{2}\). The results are given in Table 4 and Fig. 14.

Experimentally determined \(\beta /\alpha\) values vs. pressure, together with fitted curve according to Eq. 24b
The fitting of the data in Table 4 with Eq. 24b gives values for \(g_{1} = 0.177\) and \(g_{2} = 0.176\). Note that \(g_{2}\) is equivalent to the ratio of rate constants for single insertion vs. double insertion from a double-ethylene associated intermediate. There is a good degree of correlation between experimentally determined and the fitted values of \({\beta}/{\alpha }\) in Fig. 14 (goodness of fit R2 = 0.974).
A final note should be added, that for ethylene oligomerisations at lower pressures with these catalysts, it is possible that the most abundant oligomeric product, 1-butene in this case, is incorporated during the oligomerisation reaction. We have observed this with some of the most active catalysts, resulting in a minor distribution of 2-ethyl-1-alkenes with the opposite alternating pattern, alongside the major distribution of 1-alkenes with the normal alternating behaviour [17].
2.5 Steady-State Oligomerisation with Nonconstant Propagation Probabilities
In assuming constant values for the propagation probabilities \(\alpha\) and \(\beta\) in Schemes 4 and 5 (Sects. 2.1 and 2.2 respectively), recurrence relations with constant coefficients are generated. The solution of these may be expressed in equation format (see Eqs. 8 and 11). If, however, the propagation probabilities adopt nonconstant values in the catalytic cycle, then recurrence relations with variable coefficients are generated. The aim in such cases is to determine a unique value for each unknown parameter, and/or to elucidate the key mechanistic features of the system.
In this sub-section, a method for handling first-order recurrence relations with variable coefficients is presented. The method finds application in oligomerisation systems where \(\alpha\) changes at some point in the catalytic cycle. Second-order systems with nonconstant propagation probabilities will be discussed later, with a particular focus on selective-tetramerisation.
2.5.1 First-Order Recurrence Relations with Nonconstant Propagation Probabilities
Experimental LAO distributions which deviate from Schulz–Flory have been observed, for example chromium(III) bis(carbene)pyridine systems reported by McGuinness and co-workers (Fig. 15) [29]. The observation has been rationalised by less favourable elimination at the early stages of metallacyclic growth [29]. In Fig. 15, \(\alpha\) is variable before C8, then adopts a constant value from C8 onwards.

Distribution of 1-alkene oligomers as relative mol(n) vs n (units of ethylene) produced with complex 2,6-bis(1-(2,6-diisopropylphenyl)imidazol-2-ylidene)pyridine chromium(III) chloride. Conditions: 10 μmol Cr, 5 bar continuous ethylene pressure, 25 °C, MAO (5 mmol), toluene (50 mL), 30 min. Activity (C4–C20 LAOs) = 159 g mmol−1 h−1 bar−1. Variable \(\alpha\) and constant \(\alpha\) regions indicated
Data from C8 (n = 4) onward may be treated with the back-solving method described in Sect. 2.1 to find the constant \({\alpha}\) value and R, the total mol of C8+ oligomers produced during the reaction time (i.e. the rate at which intermediate species arrive at
 . Adapting Scheme 4 to show rates, the first nonconstant term \(T_{1}^{\prime}\) (i.e. C6) may be appended, along with \(R^{\prime}\), the rate at which intermediate species arrive at
. Adapting Scheme 4 to show rates, the first nonconstant term \(T_{1}^{\prime}\) (i.e. C6) may be appended, along with \(R^{\prime}\), the rate at which intermediate species arrive at
 (Scheme 7).
(Scheme 7).
To find \(\alpha^{\prime}\) (i.e. the \(\alpha\) value at C6) and \(R^{\prime}\), two distinct equations involving the two unknown variables may be identified (note that \(T_{1}^{\prime}\) is the experimental data point for 1-hexene):
These equations sum to give:
Given that there are two distinct equations for two unknowns (\(\alpha^{\prime}\) and \(R^{\prime}\)), a unique solution does exist. \(R^{\prime}\) may be found with Eq. 25c, then substituted into Eqs. 25a or 25b to find \(\alpha^{\prime}\). The second non-constant term \(T_{2}^{\prime}\) (i.e. C4) may now be appended to Scheme 7, and the \(\alpha\) value at C4 found using the same methodology. The \(\alpha\) values calculated for Fig. 15 via this methodology are 0.86, 0.65 and 0.54 from C4, C6 and C8+ respectively. In other words, for this particular chromium catalyst, the termination probability is less favourable at the early stages and increases as the metallacycle grows from a metallacyclopentane to—heptane and—nonane. Once the metallacycle has reached a certain size, the termination probability becomes constant.

Catalytic scheme for Fig. 15 at the boundary of the variable \(\alpha\) and constant \(\alpha\) regions. Active species shown in black, rates and eliminated products shown in blue
2.5.2 Second-Order Recurrence Relations with Nonconstant Propagation Probabilities
McGuinness, Britovsek and co-workers produced a detailed experimental and theoretical study of single- and double-coordination mechanisms in ethylene tri- and tetramerisation with Cr/PNP catalysts [30]. With PNP = Ph2PN(iPr)PPh2, high selectivity for 1-octene is observed (~ 70 mol%), together with approximately 16 mol% 1-hexene, 3 mol% C10+ α-olefins, and various other coproducts (Fig. 16).

Computational studies employing the M06L functional on simplified model compounds have indicated that a single- and double-coordination mechanism operates (see Fig. 17) [30,31,32,33]. Elimination from a chromacyclopentane intermediate to give 1-butene is calculated to be energetically unfavourable [30]. Competing processes to 1-hexene and 1-octene formation diverge at a singly-coordinated chromacyclopentane intermediate 7, with 1-hexene formed predominately via single insertion from intermediate 7, and 1-octene formed predominantly via a bis(ethylene) metallacyclic intermediate 17, with additional involvement of a mono(ethylene) route through intermediate 18 [30].

Condensed energy profiles for 1-hexene and 1-octene formation from the singly-coordinated chromacyclopentane intermediate 7 [30]. Gaussian09, M06L/quadruple-ζ valence def2-QZVP basis set and SDD ECP on Cr, 6-311 + G(2d,p) basis set on all other atoms
Given the varying differences in energy between the early intermediates in Fig. 17, the selective-tetramerisation process in Fig. 16 may be mathematically considered as a second-order recurrence relation with variable coefficients. Data from C16 onwards corresponds to the tail end of an alternating distribution, where \(\alpha\) and \(\beta\) values are constant by virtue of negligible geometry and energy differences between larger chromacyclic species. For the purpose of the present discussion, setting aside the difficulties in fitting gently alternating distributions as outlined in Sect. 2.3, the region from C16 onwards may conceivably be treated with the back-solving method described in Sect. 2.2 to find the constant values for \(\alpha\) and \(\beta\) as well as \(R_{1}\), the rate at which intermediate species arrive at
 , and \(R_{2}\), the rate at which intermediate species originating from before
, and \(R_{2}\), the rate at which intermediate species originating from before
 arrive at
arrive at
 . Adapting Scheme 5 to show rates, the first non-constant term \(T_{1}^{\prime}\) (i.e. C14) may be appended, along with \(R_{1}^{\prime}\), the rate at which intermediate species arrive at
. Adapting Scheme 5 to show rates, the first non-constant term \(T_{1}^{\prime}\) (i.e. C14) may be appended, along with \(R_{1}^{\prime}\), the rate at which intermediate species arrive at
 , and \(R_{2}^{\prime}\), the rate at which intermediate species originating from before
, and \(R_{2}^{\prime}\), the rate at which intermediate species originating from before
 arrive at
arrive at
 (Scheme 8).
(Scheme 8).

Catalytic scheme for Fig. 16 at the boundary of the variable \(\alpha, \beta\) and constant \(\alpha , \beta\) regions
In an effort to find \(\alpha^{\prime}\) and \(\beta^{\prime}\) (i.e. the \(\alpha\) and \(\beta\) values at C14) and \(R_{1}^{\prime}\) and \(R_{2}^{\prime}\), three distinct equations involving the four unknown variables may be identified (note that \(T_{1}^{\prime}\) is the experimental data point for 1-tetradecene):
These equations sum to give:
Given that there are three distinct equations for four unknowns (\(\alpha^{\prime}, \beta^{\prime}, R_{1}^{\prime} and\,R_{2}^{\prime}\)), a unique solution does not exist in this case. If no further information is available/applied from e.g. theoretical studies, an assumption (such as \(\frac{{\alpha^{\prime}}}{{\beta^{\prime}}} = \frac{\alpha }{\beta }\)) is required as a fourth equation to determine unique solutions for the four unknown variables.
3 Modelling Non-Steady-State Polymerisation Processes
3.1 Connecting Schulz–Flory Distributions and Poisson Distributions
The mathematics behind Schulz–Flory ethylene oligomer distributions was first accurately described in 1936 [19]. In 1940, Flory went on to give a kinetic derivation of Poisson’s equation for polymer distributions obtained from a monofunctional monomer (ethylene oxide) [34]. Yet the mathematical connection between Schulz–Flory distributions and Poisson distributions has not been fully elucidated. In this section, the relationship between the two is made explicit from first principles.
It should be noted that the treatment presented below is described with ethylene as the monomer, but is generally applicable to oligomerisation and polymerisation processes involving the sequential incorporation of monomers in forming linear product chains.
3.1.1 First Principles
The mechanics of a first-order ethylene oligomerisation or polymerisation process whereby propagation proceeds via metal alkyl or metallacyclic species is summarised in Scheme 9. In this model and the following mathematical treatment, we assume that the rate constants kp and ke are constant over time and independent of chain length. Furthermore, all active species must start the polymerisation instantaneously and there is no catalyst deactivation.

Catalytic scheme for first-order propagation
\(M\left( n \right)\) represents catalyst intermediates involving \(n\) ethylene units (e.g. \(M\left( 0 \right)\) = metal hydride species, \(M\left( 1 \right)\) = metal ethyl or metallacyclopropane species, \(M\left( 2 \right)\) = metal butyl or metallacyclopentane species etc.), \(k_{p}\) is the rate constant of propagation, \(k_{e}\) is the rate constant of elimination, and \(L\) is the total precatalyst loading.
A general expression for \(M\left( n \right)\), as a function of time, may be derived (see SI.5.1):

The label “non-steady-state” refers to time dependent terms, whilst the label “steady-state” refers to time independent terms. It is worth noting again that Scheme 9 assumes \(k_{p}\) and \(k_{e}\) are constant over time and independent of \(M\left( n \right)\), i.e. no distinction in propagation or elimination kinetics is made between the various intermediate groups in the catalytic scheme. In addition, by combining \(M\left( n \right)\) species in forming the kinetics equations, it is implicitly assumed that a single species within each \(M\left( n \right)\) group dominates. For instance, the \(M\left( 0 \right)\) group includes both metal hydrides with a vacant site and ethylene-associated metal hydrides. This assumption is no longer required for the Schulz–Flory case, given that steady-state kinetics operate.
3.1.2 Derivation of the Schulz–Flory Equation for First-Order Steady-State Oligomerisation
Referring to Scheme 9, the scenario where steady state is achieved from \(t_{0}\) (i.e. instantaneously from the beginning of reaction) with \(k_{e} \ne 0\) corresponds to the limiting case of first-order steady-state oligomerisation. In such a case, by ignoring non-steady-state terms, Eq. 27simplifies to:
Given that elimination occurs at a constant rate, the interest lies only in the sum of linear alpha olefins of chain length \(n\) ethylene units continuously eliminated from \(t_{0}\) to \(t_{end}\):
Equation 29a is the unnormalised Schulz–Flory equation, and Eq. 29b is the normalised Schulz–Flory equation. Note in Eq. 29a that the total mol of oligomers produced during the reaction time \(R = L\alpha k_{e} t_{end}\). Given that both \(\frac{{Lk_{e}^{2} t_{end} }}{{k_{p} }}\) (where \(L\) and \(t_{end}\) are known) and \(\alpha\) may be fitted, \(k_{p}\) and \(k_{e}\) may be inferred.
3.1.3 Derivation of Poisson’s Equation for First-Order Living Polymerisation
Referring again to Scheme 9, setting \(k_{e} = 0\), i.e. there is no chain termination process, corresponds to the limiting case of first-order non-steady-state living polymerisation. In such a case, Eq. 27 simplifies to:
Given that no termination occurs throughout, the interest lies only in the distribution of intermediate species at \(t_{end}\) (i.e. the end of reaction), which would give n-alkene products on thermal termination (as per the INEOS (Ethyl) process [8]) or n-alkane products upon hydrolysis. At this point:
Equation 31a is Poisson’s equation scaled by the precatalyst loading \(L\), and Eq. 31b is Poisson’s equation scaled by the factor \(\left( {1 - e^{ - \lambda } } \right)^{ - 1}\) (which quickly approaches 1 as \(\lambda\) increases). Note that \(\lambda\) is the mean number of ethylene units per chain. Given that \(\lambda\) may be fitted (where \(t_{end}\) is known), \(k_{p}\) may be inferred.
3.2 Non-Steady-State Living Polymerisation with Catalysed Chain Growth
In 1952, Ziegler et al. reported the Aufbaureaktion involving the formation of long chain trialkylaluminium species via stepwise insertion of ethylene monomers into Al–C bonds (starting with triethylaluminium, for instance) [35]. The effect of transition metals, such as colloidal Ni, in suppressing chain growth in the Aufbaureaktion to selectively form 1-butene was subsequently discovered [36]. This prompted a screening of the d-block transitional metals for other metal effects and, in the case of titanium, this led to the first reported transition metal catalysed ethylene polymerisation [37, 38].
In their early work, Ziegler et al. described the process as a transition metal catalysed Aufbaureaktion, with the alkylaluminium functioning as the polymerisation catalyst and the transition metal compound functioning as the co-catalyst [37]. Later it was found that the polymer chain grows on the transition metal centre, rather than on aluminium. More recently however, several processes have been reported where the polymer chain is transferred during the course of the reaction from a transition metal to a main group metal such as aluminium, magnesium or zinc [39,40,41,42,43,44]. If this chain transfer is very fast and reversible, then it appears as if the polymer chains grow on the main group metal centres instead. Such a reaction is generally described as a catalysed chain growth (CCG) process or a coordinative chain-transfer polymerisation (CCT), or, to employ the original terminology, a transition metal catalysed Aufbaureaktion [44,45,46].
Tremendous advances have been made in CCG during the past 2 decades, but the mathematics of generating product distributions via CCG has not yet been reported. Such a treatment from first principles is presented here and then applied next.
3.2.1 First Principles
Scheme 10 summarises the mechanics of a first-order ethylene oligomerisation or polymerisation process whereby propagation proceeds via metal alkyl or metallacyclic species, and chain transfer to and amongst a population of non-propagating agents is possible. A similar caveat as in Sect. 3.1 applies in that in this model and the following mathematical treatment, we assume that the rate constants kp, ke and kct are constant over time and independent of chain length. Furthermore, all active species must start the polymerisation instantaneously and there is no catalyst deactivation.

Catalytic scheme for first-order propagation with chain transfer
\(M\left( n \right)\) represents transition metal intermediates involving \(n\) ethylene units, \(CT\left( n \right)\) represents the population of chain transfer species bearing an alkyl chain of length \(n\) ethylene units, \(k\left( {M\left( n \right)} \right)_{p}\) is the rate constant of propagation for \(M\left( n \right)\), \(k\left( {M\left( n \right)} \right)_{e}\) is the rate constant of elimination for \(M\left( n \right)\), and \(k_{ct}\)’s are rate constants of chain transfer between species. Scheme 10 is not labelled with individual \(k_{ct}\)’s given the general nature of the present treatment.
It is possible to show (see SI.5.2) that:
This expression for change does not show dependence on chain transfer terms. In other words, the underlying \(M\left( n \right)\) system drives towards the distribution that it would adopt in the absence of chain transfer agent. However, the nature of instantaneous steady state for CT species slows down the overall rate at which the \(M\left( n \right)\) system evolves vs. the case with no chain transfer.
3.2.2 Numerical Simulations
Two illustrative examples are provided to demonstrate the preceding mathematics. Numerical simulations of evolving oligomerisation or polymerisation systems which involve chain transfer, as per Scheme 10, are possible with Microsoft Excel (see SI.5.3 for details).
3.2.3 Illustrative Example 1: Living Polymerisation with Catalysed Chain Growth
See Scheme 11, Figs. 18, 19, 20.

Scheme for a living polymerisation system with CCG

System evolution and product distribution with CT agent loading = 0 μmol dm−3 (0 eqv. vs. transition metal catalyst). Note: M(n) and CT(n) up to n = 5 illustrated left and centre, all n captured right

System evolution and product distribution with CT agent loading = 10 μmol dm−3 (1 eqv. vs. transition metal catalyst). Note: M(n) and CT(n) up to n = 5 illustrated left and centre, all n captured right

System evolution and product distribution with CT agent loading = 100 μmol dm−3 (10 eqv. vs. transition metal catalyst). Note: M(n) and CT(n) up to n = 5 illustrated left and centre, all n captured right
3.2.4 Illustrative Example 2: First-Order Oligomerisation with Catalysed Chain Growth
See Scheme 12, Figs. 21, 22, 23.

Scheme for a first-order oligomerisation system with CCG

System evolution and product distribution with CT agent loading = 0 μmol dm−3 (0 eqv. vs. transition metal catalyst). Note: M(n) and CT(n) up to n = 5 illustrated left and centre, all n captured right

System evolution and product distribution with CT agent loading = 10 μmol dm−3 (1 eqv. vs. transition metal catalyst). Note: M(n) and CT(n) up to n = 5 illustrated left and centre, all n captured right

System evolution and product distribution with CT agent loading = 100 μmol dm−3 (10 eqv. vs. transition metal catalyst). Note: M(n) and CT(n) up to n = 5 illustrated left and centre, all n captured right
3.2.5 The Fitting Process
The preceding discussion demonstrated how Poisson distributions or Schulz–Flory distributions are still generated for first-order systems involving CCG. Section 2.1 provided the details for modelling first-order steady state oligomerisation systems, which by extension may be applied to first-order oligomerisation systems involving CCG. Section 3.1 gave a derivation to the Poisson distribution for living polymerisation systems, ending at Eqs. 31a and 31b.
When Eqs. 31a and 31b are applied to living polymerisation systems with CCG, in order to capture all intermediate alkyl chains, \(L\) is now defined as:
Furthermore, \(k_{p}\) may now be interpreted as the effective propagation rate constant for the entire system. The process for modelling living polymerisation systems with CCG is described next, along with derivations of the experimentally-relevant metrics \(M_{n}\), \(M_{w}\) and \(PDI\). For illustration, a comparison is also made between an experimentally determined result vs. the fitted data.
3.2.6 Back-Solving to Determine L and λ, and Checking for the Best Fit
In practice, the \(mol\left( n \right)_{{t_{end} }}\) values in Eq. 31a are known (i.e. mol of each oligomer produced during the reaction time), and the aim is to find \(L\) and \(\lambda\). See SI.5.4 for a discussion on the method used to determine \(\alpha\) and \(R\), as well as a checking method to determine that the best fit has been found.
3.2.7 Derivations of Further Experimentally-Relevant Metrics
For a distribution of n-alkenes, the following expressions can be derived (see SI.5.5 and SI.5.6):
As \(\lambda\) increases, the factor \(\left( {1 - e^{ - \lambda } } \right)\) approaches 1. Under such circumstances, Eqs. 35 and 37 simplify.
With high values of \(\lambda\), \(PDI\) tends towards unity, as per the literature [47].
3.2.8 Comparison of an Experimentally Determined Result vs. Fitted Data
CCG polymerisations have been performed, using ZnEt2 as the CT agent with 2,6-diacetylpyridinebis(2,6-diisopropylanil)FeCl2/MAO (experimental details, see SI.5.7). Termination of the polymerisation reaction is carried out by hydrolysis, resulting in a mixture of long-chain alkanes. Given a highly symmetric Poisson distribution of the resulting n-alkane oligomers, quantitative 13C{1H} NMR analysis may be used to estimate the mean chain length by comparing integrals for CH3 with those for CH2. For the given experiment, quantitative 13C{1H} NMR spectroscopy gives a CH3:CH2 ratio of 2:45.38, implying a mean chain length between 46 and 48 carbons (see Fig. 24).

Quantitative 13C{1H} NMR spectrum of n-alkane oligomer product (solvent: d2-1,1,2,2-tetrachloroethane/1,2,4-trichlorobenzene 50/50 v/v), 100 MHz, 100 °C
Experimental characterisation of the alkane distribution in such mixtures is non-trivial. The average molecular weight is typically too low for accurate GPC analysis, and the longer alkanes are often insufficiently volatile for conventional quantitative GC-FID analysis. Only the first part of the n-alkane oligomer distribution is usually quantifiable via GC. These signals have been used here as inputs for the fitting model. Following the procedure previously described in this sub-section, the values in Table 5 are found.

Modelled outputs (see Table 5) vs. experimental GC data
3.2.9 Model Outputs
There is a high degree of correlation between calculated and experimental values in Table 5 (goodness of fit R2 = 0.996), and the calculated mean chain length of 46.6 carbons is within the range previously specified by quantitative 13C{1H} NMR spectroscopy. This method therefore allows the entire product distribution to be determined from a subset of experimental data obtained by GC analysis.
4 Conclusions and Summary
A variety of different distributions—including Schulz–Flory, alternating, selective-LAO, and Poisson distributions—of linear hydrocarbon products can be obtained from ethylene oligomerisation and polymerisation processes. Oligomerisation processes are steady-state in nature, and can be mathematically described using recurrence relations. First order oligomerisation processes propagate via sequential single-insertions of ethylene, whereas second order oligomerisation processes propagate via sequential single- and double-insertions of ethylene. From an analytical point of view, an important consideration with regards to second order processes is the degree of alternation exhibited by a given distribution, which is generally more pronounced at higher ethylene pressure. The degree of alternation for a particular distribution may be defined as the relative ratio of alternating vs. Schulz–Flory character. An overview of the key analytical results from first order and second order processes with constant probabilities of propagation is given in Table 6.
Examples of first order and second order oligomerisation processes which involve varying probabilities of propagation have been identified from the literature. Such systems include selective oligomerisation catalysts such as the well-known chromium-based tetramerisation catalysts. An analytical method is available for handling first order processes with varying probabilities of propagation. However, there is a theoretical limitation to a similar approach for analogous second order processes.
The connection between distributions from first order steady-state oligomerisations (Schulz–Flory) and first order non-steady-state polymerisations (Poisson) is made through a consideration of kinetic differential equations. The addition of a chain transfer agent (e.g. ZnEt2) to such systems results in a slowing down of the overall propagation rate. Accurate modelling of a CCG polymerisation system featuring ZnEt2 as the CT agent has been achieved, allowing for the derivation of several key mechanistic parameters.
The mathematics of ethylene oligomerisation presented here can be easily applied to other systems where chain growth proceeds through an oligomerisation process. Examples of such systems would be the oligomerisation of higher α-olefins to poly(α-olefin) oils, the dehydropolymerisation of aminoboranes, chain growth reactions in the Fischer–Tropsch process or the formation of oligonucleotides. By fitting the experimental results to the formulas described herein for the various oligomerisation scenarios, valuable information may be extracted regarding the underlying reaction mechanisms of the oligomerisation process.
References
Breuil P-AR, Magna L, Olivier-Bourbigou H (2015) Catal Lett 145(1):173–192
Forestière A, Olivier-Bourbigou H, Saussine L (2009) Oil Gas Sci. Technol Rev l’IFP 64(6):649–667
Olivier-Bourbigou H, Forestière A, Saussine L, Magna L, Favre F, Hugues F (2010) Oil Gas Eur Mag 36:97
Ring K-L, DeGuzman M (2017) Chemical economics handbook: linear alpha-olefins, IHS Chemical
Process Economics Program (PEP) (2016) Wide range linear alpha olefin processes & on-purpose linear alpha olefin processes, PEP CR001 & CR002 Prospectus, IHS Chemical
Sydora O (2019) Organometallics 38:997–1010
Keim W (2013) Angew Chem Int Ed 52(48):12492–12496
Greiner EOC, Inoguchi Y (2010) Chemical economics handbook: linear-alpha olefins, IHS Chemical
McGuinness DS (2011) Chem Rev 111(3):2321–2341
Van Leeuwen PWNM, Clément ND, Tschan MJL (2011) Coord Chem Rev 255(13–14):1499–1517
Chen Y, Zuo W, Hao P, Zhang S, Gao K, Sun W-H (2008) J Organomet Chem 693(4):750–762
Luo W, Li A, Liu S, Ye H, Li Z (2016) Organometallics 35(17):3045–3050
Tenza K, Hanton MJ, Slawin AMZ (2009) Organometallics 28(16):4852–4867
Liu S, Pattacini R, Braunstein P (2011) Organometallics 30(13):3549–3558
Tomov AK, Chirinos JJ, Long RJ, Gibson VC, Elsegood MRJ (2006) J Am Chem Soc 128(24):7704–7705
Britovsek GJP, Malinowski R, McGuinness DS, Nobbs D, Tomov AK, Wadsley AW, Young CT (2015) ACS Catal 5(11):6922–6925
Tomov AK, Nobbs JD, Chirinos JJ, Saini PK, Malinowski R, Ho SKY, Young CT, McGuinness DS, White AJP, Elsegood MRJ, Britovsek GJP (2017) Organometallics 36(3):510–522
Schulz GVZ (1935) Phys Chem Abt B 1935(30):379–398
Flory PJ (1936) J Am Chem Soc 58(7):1877–1885
Britovsek GJP, Mastroianni S, Solan GA, Baugh SPD, Redshaw C, Gibson VC, White AJP, Williams DJ, Elsegood MRJ (2000) Chem. A 6(12):2221–2231
Briggs JR (1989) J Chem Soc Chem Commun 11:674
Tomov AK, Gibson VC, Britovsek GJP, Long RJ, Van Meurs M, Jones DJ, Tellmann KP, Chirinos JJ (2009) Organometallics 28(24):7033–7040
Agapie T, Labinger JA, Bercaw JE (2007) J Am Chem Soc 129(46):14281–14295
Tomov AK, Chirinos JJ, Jones DJ, Long RJ, Gibson VC (2005) J Am Chem Soc 127(29):10166–10167
Ystenes M (1991) J Catal 129:383
Marques MM, Dias AR, Costa C, Lemos F, Ramôa Ribeira F (1997) Polym Int 43:77–85
Matos I, Zhang Y, Lemos MANDA, Freire F, Fonseca IF, Marques MM, Lemos FJ (2004) Polym Sci A 42(14):3464–3472
Mark Kelly W, Wang S, Collins S (1997) Macromolecules 30(11):3151–3158
McGuinness DS, Suttil JA, Gardiner MG, Davies NW (2008) Organometallics 27(16):4238–4247
Britovsek GJP, McGuinness DS, Wierenga TS, Young CT (2015) ACS Catal 5(7):4152–4166
McGuinness DS, Chan B, Britovsek GJP, Yates BF (2014) Aust J Chem 67(10):1481–1490
Britovsek GJP, McGuinness DS, Tomov AK (2016) Catal Sci Technol 6:8234–8241
Britovsek GJP, McGuinness DS (2016) Chem-Eur J 22:16891–16896
Flory PJ (1940) J Am Chem Soc 62(6):1561–1565
Ziegler K, Gellert HG, Kühlhorn H, Martin H, Meyer K, Nagel K, Sauer H, Zosel K (1952) Angew Chem 64:323–329
Ziegler K, Gellert HG, Holzkamp E, Wilke G (1954) Brennstoff-Chem 35:321–352
Ziegler K, Holzkamp E, Breil H, Martin H (1955) Angew Chem Int Ed 67:541–547
Sauter DW, Taoufik M, Boisson C (2017) Polymers 9:185
Lehmkuhl H, Olbrysch O (1975) Eur J Org Chem 6:1162–1175
Samsel EG (1993) EP 0539876
Samsel EG, Eisenberg DC (1993) EP 0574854
Pelletier JF, Bujadoux K, Olonde X, Adisson E, Mortreux A, Chenal T (1998) US 5779942
Valente A, Mortreux A, Visseaux M, Zinck P (2013) Chem Rev 113:3836–3857
Britovsek GJP, Cohen SA, Gibson VC, Maddox PJ, van Meurs M (2002) Angew Chem Int Ed 41(3):489–491
Britovsek GJP, Cohen SA, Gibson VC, van Meurs M (2004) J Am Chem Soc 126(34):10701–10712
Sita L (2009) Angew Chem Int Ed 48(14):2464–2472
Tobita H (2016) Ullmann’s polymers and plastics: products and processes, vol 1. Wiley, Hoboken, p 265
Acknowledgements
We are grateful to INEOS for financial support. We thank Dr. Andrew Wadsley for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Young, C.T., von Goetze, R., Tomov, A.K. et al. The Mathematics of Ethylene Oligomerisation and Polymerisation. Top Catal 63, 294–318 (2020). https://doi.org/10.1007/s11244-019-01210-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-019-01210-0