
Abstract
Somatic embryogenesis (SE) is a process by which somatic cells reprogram, acquire totipotency and embark on embryo formation. Although SE is a valuable tool for micropropagation in many crops, it presents specific challenges in woody species due to some bottlenecks, such as loss of embryogenic competence during subcultures and the often-low somatic embryo conversion into plantlets. Hence, great interest exists in exploring the regulatory networks involved on SE. As transcription modulators, long non-coding RNAs (lncRNAs) have been found essential in a wide range of biological processes. This work aimed to identify lncRNAs related to the embryogenic competence in Solanum betaceum Cav. (tamarillo). Nanopore® long-read sequencing was conducted in cell lines with distinct cell fates and, based on their coding potential, 60 transcripts were selected as lncRNA candidates. Similar expression patterns were found among embryogenic cell lines (EC) and cells that lost their embryogenic potential (herein long-term callus, LTC), whereas non-EC (NEC) showed differentially expressed lncRNAs. Whereas lncRNAs upregulated in EC and LTC were predicted to target embryogenesis-related genes, such as AGAMOUS-like 15 and WUSCHEL-related HOMEOBOX 2, lncRNAs upregulated in NEC were predicted to target mainly genes involved in carbohydrate metabolism, cell wall formation, auxin and ethylene signalling pathways. Altogether, these results show the involvement of lncRNA in the process of somatic embryogenesis in S. betaceum, indicating that EC present a pattern of lncRNA expression that suggests its action on genes that directly modulate the morphogenic response in vitro, while in NEC the modulation through this type of RNAs seems to be predominantly reflected in genes more related to cellular physiology.
Graphical Abstract

Key message
LncRNAs upregulated in EC/LTC were predicted to target embryogenesis-related genes, while lncRNAs upregulated in NEC were predicted to target genes involved in auxin and ethylene signalling and carbohydrate metabolism.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Long non-coding RNAs (lncRNAs) originate from a large part of the eukaryotic genome that is transcribed into RNA but not translated into proteins (Rai et al. 2019). These transcripts have been defined as longer than 200 base pairs (bp) and have a broad range of lengths and structures (Yu et al. 2019). Like mRNAs, most lncRNAs are transcribed by RNA polymerase II (Pol II) and are characterized by a 5’ cap and a 3’ poly(A) tail (Wierzbicki et al. 2021). Other three plant RNA polymerases are also involved in lncRNA transcription (Waititu et al. 2020), but they lack the poly(A) tail (Zhang et al. 2014; Budak et al. 2020). LncRNAs can be linear or circular and classified in different ways as intronic (incRNAs, on the intron) or intergenic (lincRNAs, between genes) according to genomic location, sense (same strand) or antisense (lncNATs, opposite strand) according to the strand of origin in relation to exons, and also, as enhancer lncRNAs, when emerging from an enhancer region of protein-coding genes (Rai et al. 2019; Budak et al. 2020; Waititu et al. 2020).
Besides being poorly conserved among the plant kingdom (see Palos et al. (2023), lncRNAs have a tissue-specific expression with spatiotemporal patterns of distribution (Yu et al. 2019). These ncRNAs modulate the expression of target genes, near the location where they were transcribed (cis-acting) or away from their synthesis site (trans-acting; Lee 2012), by chromatin remodelling, RNA splicing regulation, transcriptional activation or repression or miRNA-target mimicry (Waititu et al. 2020; Wierzbicki et al. 2021).
Although over 1.2 million of lncRNAs have been annotated across 80 plant species (Jin et al. 2021), their functional characterization is limited due to several reasons. Firstly, plant lncRNAs have a lower degree of sequence conservation, even in closely related species, and most of them were found genus/species-specific (Zhu et al. 2022). Thus, inferring functions based on sequence similarity is challenging. Secondly, lncRNAs have tissue-specific expression patterns in plants (Yu et al. 2019), which makes the study of their functions difficult while highlighting the importance of understanding their role in developmental processes. In addition, lncRNAs are generally lowly expressed, making their detection and study difficult (Palos et al. 2023). Furthermore, lncRNAs represent a heterogeneous class of ncRNAs, with complex and varied structures and mechanisms of action (Lucero et al. 2020). The high complexity of these molecules leads to the need for new experimental approaches for the functional characterization of lncRNAs. Therefore, the development of new technologies is imperative to better understand the molecular mechanisms of lncRNAs in developmental functions.
In plants, lncRNAs have been increasingly recognized as relevant players of gene regulatory networks, mainly in developmental and stress mechanisms (Yu et al. 2019; Wu et al. 2020b). Recently, the emerging role of these and other ncRNAs in plant cell reprogramming and in vitro regeneration was highlighted (Alves et al. 2021; Cordeiro et al. 2022; Bravo-Vázquez et al. 2023). Although the lncRNA research field is still in its infancy, some lncRNAs have been recently identified as involved in plant somatic cell reprogramming towards regeneration in Dimocarpus longan Lour. (longan, Chen et al. 2018), Picea glauca (Moench) Voss (white spruce, Gao et al. 2022), Oryza sativa L. (rice, Zhang et al. 2022) and Allium sativum L. (garlic, Bai et al. 2023). Even though, the regulatory role of lncRNAs in plant morphogenesis, such as somatic embryogenesis (SE), a stress-induced process in which somatic cells reprogram and develop into somatic embryos, remains vague. In longan, differentially expressed lncRNAs during SE were analysed, showing to be involved in the regulation of gene expression with fundamental regulatory roles in plant embryogenesis, such as five lncRNAs (LTCONS-00006334, LTCONS-00008111, LTCONS-00025525, LTCONS-00030223 and LTCONS-00055024) targeting genes related to auxin response factors (Chen et al. 2018). Likewise, three lncRNAs (MSTRG.33602.1, MSTRG.505746.1 and MSTRG.1070680.1) were pointed out as involved in auxin signal transduction and somatic embryo development in white spruce (Gao et al. 2022). In rice, OsCHENAT1709 and OsCHELIN2084, two members of a subclass of lncRNAs (the nuclear chromatin-enriched lncRNAs, cheRNAs), were found positively and negatively involved in cell dedifferentiation, respectively (Zhang et al. 2022). However, to date, there has been no analysis of the lncRNA regulation of the acquisition, expression and maintenance of the embryogenic competence in plant cells of woody species.
Solanum betaceum Cav., commonly known as tamarillo or tree tomato, is a species native to the Andean region (Acosta-Quezada et al. 2015). It bears egg-shaped fruits, with colour ranging from yellow to deep red and a distinctive tart flavour (sweet and acidic). Due to the high content of dietary fiber, vitamins, potassium and iron, and low content in calories and fat of its fruits, the economic importance of tamarillo has been increasing in the fresh fruit market and food processing industry (Ramírez and Kallarackal 2019). Besides being a non-sequenced species, this diploid (2n = 2x = 24) solanaceous (Pringle and Murray 1992) has been extensively used as an experimental model system for investigating regulatory aspects involved on indirect SE induction in woody species (Correia et al. 2011, 2012a, 2019; Correia and Canhoto 2012; Alves et al. 2017; Cordeiro et al. 2023a, b; Caeiro et al. 2022) including by the generation of proteomics data (Correia et al. 2012b) and gene expression analysis studies (Cordeiro et al. 2020). In S. betaceum, SE is efficiently induced through a well-described two-step protocol (Correia and Canhoto 2018), in which, during the induction phase, two types of calli composed of cell populations with distinct cell fates but the same genetic background are formed. While embryogenic callus (EC) can proliferate and, in specific conditions, develop embryos, non-EC (NEC) can only proliferate without showing any embryogenic aptitude. Although both calli can be maintained through subcultures for several years, the embryogenic potential of EC decreases with successive subcultures, becoming unable to develop somatic embryos (herein termed long-term callus, Currais et al. 2013). Thus, in S. betaceum it is possible to have distinct SE-induced cell lines with the same genetic background, but different morphogenic competencies. Transcriptome and epitranscriptome analysis of these cell lines are important tools to unveil the molecular mechanisms underlying plant totipotency and especially factors controlling the embryogenic competence acquisition, expression, and maintenance in woody dicot species. Accordingly, deeper knowledge of the gene expression regulation by lncRNAs would enable further modulation of key mechanisms essential for SE achievement and/or improvement. For instance, overcoming the recalcitrance of some crops to SE induction, enhancing the embryogenicity of the explants and/or increasing the stability of the embryogenic competence for continued somatic embryo development would empower such techniques with vast biotechnology applications, including clonal propagation, germplasm conservation, regeneration of genetically modified plants and embryology fundamental research (Corredoira et al. 2019).
The present study aimed to identify lncRNAs with presumable regulatory roles in gene regulatory networks involved in the embryogenic competence in S. betaceum SE-induced cell lines. A high-throughput sequencing approach was carried out using Oxford Nanopore Technologies® (ONT). This technology allows for long-read sequencing (> 4 Mb in a single read), which has been highlighted as the Method of the Year 2022 by Nature (Marx 2023), due to its high potential in the detection of complete full-length transcripts (Wang et al. 2021). Accordingly, a more complete reading of genomic information is achieved, which is extremely important for overcoming the difficult assembly in non-sequenced species, such as S. betaceum. Our findings shed light on the roles of several differentially expressed lncRNAs in cells with different embryogenic competencies and expand our knowledge of the molecular mechanisms underlying SE induction.
Materials and methods
Plant material, libraries preparation and sequencing
Proliferating S. betaceum cell lines with distinct embryogenic competencies were used in this assay, namely 2-year-old EC, 10-year-old NEC and 8-year-old LTC. These calli were induced, from leaf explants of in vitro proliferating shoots (the same individual explant-donor in the case of NEC and LTC), and maintained in Murashige and Skoog medium (Murashige and Skoog 1962; Duchefa Biochemie, Haarlem, The Netherlands), supplemented with 20 µM picloram (Sigma-Aldrich®, Missouri, USA) plus 9% (w/v) sucrose and semi-solidified with 0.25% (w/v) Phytagel™ (Sigma-Aldrich®) at pH 5.7, at 24 ± 1 °C under dark conditions, by monthly subculture, following the methodology previously described (Correia and Canhoto 2018).
Barcoded cDNA libraries (3 per cell line) were prepared and sequenced following the methodology described in Cordeiro et al. (2023b). Briefly, poly(A) RNA was purified from callus samples (100 mg) from different plates, using Dynabeads™ mRNA DIRECT™ Kit (Invitrogen™, Thermo Fisher Scientific, Massachusetts, USA, Cat. No. 61,011). Barcoded cDNA libraries were prepared using the Direct cDNA Sequencing Kit (ONT, Oxford, UK, Cat. No. SQK-DCS109) with Native Barcoding Expansion 1–12 (ONT, Cat. No. EXP-NBD104). Sequencing was carried out using MinION (ONT, MK1C), MinKnow (ONT, version 21.10.8) and flow cell R9.4.1 (ONT, Cat. No. FLO-MIN106). The datasets generated by ONT sequencing, using MinION, were deposited in the National Centre for Biotechnology Information (NCBI) repository, under BioProject ID PRJNA892465. From this BioProject, accessions SRR21981148, SRR21981149, SRR21981151, SRR21981152, SRR21981153, SRR21981155, SRR21981157, SRR21981158 and SRR21981159 were used for further analysis.
Bioinformatic data analysis
Data quality was assessed by FastQC (version 0.11.9; Andrews 2010) and a graph showing the Phred quality score per base sequence for all libraries was created using the MultiQC tool (Ewels et al. 2016). Raw reads were first filtered with a minimum read quality score of 8 and a minimum read length of 500 bp. Full-length, non-chimeric (FLNC) transcripts were determined by searching for primers at both ends of reads. Clusters of FLNC transcripts were obtained after mapping to the reference tomato (Solanum lycopersicum) genome at NCBI (GCF_000188115.5, annotation release 103) using minimap2 software (Li 2018), and consensus isoforms were obtained after polishing within each cluster by pinfish (https://github.com/nanoporetech/pinfish). Consensus sequences were mapped again to the reference genome using minimap2. Mapped reads were further collapsed by the cDNA_Cupcake package with a minimum coverage of 85% and a minimum identity of 90%. 5’ difference was not considered when collapsing redundant transcripts. Transcripts were validated against known reference transcript annotations with the gffcompare program (https://ccb.jhu.edu/software/stringtie/gffcompare.shtml; Pertea et al. 2015; Pertea and Pertea 2020).
LncRNA prediction
Transcripts with more than 200 nt in length and having more than two exons were selected as lncRNA candidates. Four computational approaches, including CPC (Kong et al. 2007), CNCI (Sun et al. 2013), CPAT (Wang et al. 2013) and Pfam (Finn et al. 2016), were combined to sort ncRNA candidates from putative protein-coding RNAs in the transcripts. Full-length reads were then mapped back to the reference genome, and quantification was performed by featureCounts (Liao et al. 2014). Inter-sample differential expression analysis of lncRNAs was performed using the DESeq2 R package (Love et al. 2014). The resulting P values were adjusted for controlling the false discovery rate (Benjamini and Hochberg 1995). Genes with a P value < 0.05 and fold change ≥ 1.5 found by DESeq2 were assigned as differentially expressed.
LncRNA-target gene prediction and annotation
The protein-coding genes near the genomic location of lncRNA transcripts/isoforms (within 100 kb upstream and downstream) were screened out as potential cis-regulatory targets. This search was performed using the window function in BEDTools software (Quinlan and Hall 2010). To identify lncRNA as trans-target genes, the Spearman correlation coefficient (PCC) between lncRNAs and mRNA was calculated (r ≥ 0.9 and P < 0.05). Gene function was annotated based on the following databases: COG/EggNOG (Clusters of Orthologous Groups of Proteins/Evolutionary Genealogy of Genes: Non-supervised Orthologous Groups; Huerta-Cepas et al. 2019), Pfam (Protein family), Swiss-Prot (Bairoch and Apweiler 1999), GO (Gene Ontology; Ashburner et al. 2000) and KEGG (Kyoto Encyclopedia of Genes and Genomes; Kanehisa et al. 2012).
Results
Callus cultures and morphology
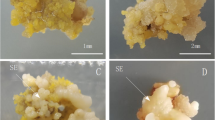
Three S. betaceum cell lines with distinct embryogenic competencies were used (Fig. 1). Despite having a similar genetic background, given that they were all induced from the red tamarillo cultivar, these calli have distinct morphologies and morphogenic competencies. NEC (10 years old; Fig. 1a) is a yellowish, friable and mucilaginous mass of undifferentiated cells with no embryogenic ability but with high proliferation capacity. In turn, EC (2 years old; Fig. 1b) is formed by whiter and more compact globular clusters, able to develop somatic embryos when transferred to an auxin-free medium (60 ± 18 somatic embryos in average per each 100 mg of callus). LTC (8 years old; Fig. 1c) was an EC forming somatic embryos, that with the subsequent long-term subcultures (after the third year) became unable to develop viable mature somatic embryos. Despite the loss of the embryogenic potential, cells continued to divide and proliferate, even at a higher proliferation rate.

S. betaceum cell lines subcultured in the proliferation medium and sampled for lncRNAseq. a Non-embryogenic callus. b Embryogenic callus. c Long-term callus. Bars:1 mm
Sequencing and identification of lncRNAs
To investigate the regulatory roles of lncRNAs associated to embryogenic competence, three barcoded cDNA libraries from the three S. betaceum cell lines were sequenced using ONT®. In the total nine libraries, 2.02 M raw reads were generated (Table S1), with an estimated average read length of N50 ~ 1.2 kb (Fig. S1) and a mean quality score over 15 (Fig. S1). An average of 305 thousand raw sequences were obtained per sample in the EC replicates, while an average of 199 and 168 thousand were obtained in the LTC and NEC, respectively.
Since the genome of S. betaceum has not yet been sequenced, full-length reads were mapped to the S. lycopersicum reference genome (GCF_000188115.5). Then, a combination of CPC, CNCI, CPAT and Pfam methods was used to sort the lncRNA candidates from the putative protein-coding RNAs across the transcripts, based on their coding potential. A total of 29,997 gene transcripts were detected and 60 lncRNA transcripts were predicted by all four methods (Fig. 2a, Table S2). However, quantification of transcript expression levels revealed a residual expression of some sequences, with 21 lncRNAs showing 0 fragments per kilobase million (FPKM) in all replicates (Table S2). From the other lncRNAs identified, 36 were detected in the three callus lines, whereas three were only found in two of them: TCONS_00000089 was shared among LTC and NEC, TCONS_00000158 was shared among EC and NEC, while TCONS_00000232 was shared among EC and LTC (Fig. 2b). The length of these lncRNAs was analysed and the results showed that 19 lncRNAs were 200–500 bp in length, 13 were 500–1000 bp, 6 were 1000–2000 bp and only one lncRNA was longer than 3000 bp in length (Fig. 2c).

a Combination of CPC, CNCI, CPAT and Pfam methods to sort the lncRNA candidates based on their coding potential. b Number of lncRNAs expressed in all samples (EC - embryogenic callus, LTC – long-term callus, NEC – non-EC) and tissue-specifically expressed lncRNAs. c LncRNAs length distribution in bp
Differential expressed lncRNAs
An inter-sample differential expression analysis of lncRNAs was performed (Fig. 3). While NEC vs. EC (Fig. 3a) and LTC vs. NEC (Fig. 3b) showed a similar number of significant differentially expressed lncRNAs, no significant differences were found between LTC and EC (Fig. 3c). In total, 15 and 17 lncRNAs were significantly differentially expressed in NEC vs. EC and LTC vs. NEC, respectively (Figs. 3d and 4a; Table S3). Compared with NEC, 5 lncRNAs were up-regulated in EC and the other 10 were down-regulated (Fig. 4b), whereas in LTC 6 lncRNAs were up-regulated and the other 11 were down-regulated (Fig. 4c). TCONS_00000218, 246, 483, 488 and 511 were all more expressed in EC and LTC than in NEC (Tables S2 and S3). By contrast, TCONS_00000050, 58, 157, 158, 203, 309, 360, 410, 482 and 526 were all more expressed in NEC than in EC and LTC (Tables S2 and S3). Therefore, the first lncRNAs group might be positively involved in the acquisition of embryogenic competence, whereas the second might be considered negative regulators. Despite no expression differences in NEC vs. EC, TCONS_00000491 was more expressed in NEC than in LTC, while TCONS_00000460 was specifically more expressed in LTC than in NEC (Table S2). Although TCONS_00000089 was only shared among LTC and NEC, there were neither significant differences in its expression between these two callus lines nor between them and EC. In the same way, TCONS_00000232 was only expressed in EC and LTC but there were neither significant differences in its expression between these two group samples nor between them and NEC.

Inter-sample differential expression analysis of lncRNAs, in S. betaceum SE-induced cell lines (EC - embryogenic callus, LTC – long-term callus, NEC – non-EC), using the DESeq2 R package and Benjamini and Hochberg (1995). a NEC vs. EC. b LTC vs. NEC. c LTC vs. EC. d Number of significantly differentially expressed lncRNAs (P value < 0.05 and fold change ≥ 1.5, see also Table S3)

a Clustered heatmap, with the expression values of the differentially expressed lncRNAs, in S. betaceum SE-induced cell lines (EC - embryogenic callus, LTC – long-term callus, NEC – non-EC). b and c Volcano plots representing the Log2 fold change (Log2 FC, green dots), adjusted p-value (padj, blue dots) and padj and Log2 FC (red dots) against the corresponding – Log10 P-value of all differentially expressed lncRNAs for b) NEC vs. EC and c) LTC vs. NEC
LncRNA target prediction and functional annotation
LncRNAs are known regulators of protein-coding genes that lie near their genomic locations. The target genes of the differentially expressed lncRNAs, among S. betaceum SE-induced cell lines, were predicted according to their regulatory potential. From the 17 differentially expressed lncRNAs found in this work, all were predicted to target protein-coding genes in cis, with a total of 167 cis-regulated target genes (Table S4). In trans-regulation, no predicted target genes were found for TCONS_00000460 and 488, whereas the other lncRNAs were predicted to target 1573 genes in total (Table S4). The putative function of these genes was annotated based on COG/EggNOG, Pfam, Swiss-Prot, GO and KEGG databases for each group (NEC vs. EC and LTC vs. NEC).
According to the COG method and for both groups, 97 genes were predicted to be targeted by lncRNAs in cis- and 745 in trans-regulation (Table S4). From them, 35 and 256 were from function unknown, respectively (Fig. 5). The other target genes were enriched in several pathways; for instance, 15 and 22 pathways were found in cis- and trans-regulation, respectively. The top five function classes of matched genes commonly found in cis and trans-regulation included: posttranslational modifications, protein turnover and chaperones, carbohydrate transport and metabolism, secondary metabolites biosynthesis, transport and catabolism, signal transduction mechanisms and transcription (Fig. 5).

Clusters of orthologous groups of proteins (COG) analysis of the lncRNAs predicted target genes in cis and trans-regulation
From the GO analysis, a similar number of predicted target genes were found for the differently expressed lncRNAs in NEC vs. EC and LTC vs. NEC (Table S4). In a total of 40 predicted target genes in cis, the most enriched genes, in general, were related to biological processes, mainly catabolic processes, and the highest percentage of genes were osmotic stress related (Fig. S3). In turn, from the 356 predicted target genes in trans, the most enriched genes were related with the structure/function of cellular components, mainly related to the cell wall and external encapsulating structure, with the highest percentage of genes attributed to vacuole functions.
According to KEGG pathway annotation, 13 of the 40 cis-targeted genes (Table S4) were more related to carbohydrate metabolism (Fig. 6). From the 751 trans-targeted genes, 220 were related to signal transduction and 140 genes were also related to carbohydrate metabolism (Fig. 6). For instance, FRUCTOKINASE (FRK2), GLUCOSE-6-PHOSPHATE 1-DEHYDROGENASE (G6PD) and LOC101268695, coding for beta-fructofuranosidase, insoluble isoenzyme CWINV3-like, were found as target genes of TCONS_00000058 and 158, TCONS_00000050 and 158, and TCONS_00000157, respectively (Table 1). Moreover, four genes related to sucrose metabolism and xyloglucan metabolism were also identified as lncRNA-target genes. These carbohydrate metabolism-related genes, all trans-targeted by lncRNAs upregulated in NEC, were all also found more expressed in NEC, in the transcription data (Fig. 7).

KEGG pathway annotation of the lncRNAs predicted target genes in cis- and trans-regulation
Combining lncRNA target prediction analysis and the mRNA transcript profile, some genes involved in the regulatory mechanisms described for plant regeneration processes were highlighted (Table 1). It was the case of two known key regulatory factors in plant embryogenesis were found among the predicted target genes: WUSCHEL-related HOMEOBOX 2 (WOX2) and AGAMOUS-like MADS-BOX PROTEIN 15 (AGL15). Like the lncRNAs, both genes were found upregulated in EC and LTC (Fig. 7). By contrast, AGL80 revealed a residual expression in all samples tested and was found targeted by a lncRNA upregulated in NEC.
Additionally, several genes involved in the auxin signalling pathway were identified as predicted target genes in trans-regulation, including members from AUXIN/INDOLE-3-ACETIC ACID (AUX/IAA) and AUXIN-RESPONSIVE GRETCHEN HAGEN3 (GH3) gene families (Table 1). All the auxin-related target genes, as well as their putative lncRNAs regulators, were found upregulated in NEC (Fig. 7). Other auxin-related genes were also found more expressed in NEC samples in the mRNA transcript profiling, including AUXIN RESPONSE FACTOR 1 (ARF1), ARF6, ARF8-1, IAA8 and AUXIN EFFLUX CARRIER COMPONENT 9 (PIN9). By contrast, ARF5 was found more expressed in EC and LTC.
Another group highly represented in the lncRNA target prediction includes genes related to ethylene biosynthesis and signalling pathways (Table 1; Fig. 7). All these target genes had higher expression levels in NEC compared to EC and LTC, thus suggesting a putative positive trans-regulation of these genes by lncRNAs also upregulated in NEC (Fig. 7). Contrary, targeted by a lncRNA more expressed in NEC, LOC101265054 showed a residual expression among the samples tested. Moreover, from the mRNA transcript profile, other ethylene-related genes were also found upregulated in NEC. By contrast, ACO3 a gene involved on stress tolerance was found more expressed in EC and LTC.
Besides the genes above mentioned targeted by TCONS_00000410, a lncRNA more expressed in NEC, another four genes were also highlighted (Table 1). While FERTILIZATION-INDEPENDENT ENDOSPERM PROTEIN (FIE) and LOC101243931 (coding for SUVH1) had higher expression levels in LTC and NEC, the late EMBRYOGENESIS ABUNDANT PROTEINS (LEA 31-like and LEA D-34-like) genes were found more expressed in EC and LTC. Similarly, another LEA gene (LOC100750252) was found highly expressed in EC and LTC, in the mRNA transcript profile, whereas LEA14 was found more expressed in NEC (Fig. 7).
Other target genes probably involved in the regulation of the embryogenic competence were also predicted to be targeted by lncRNAs upregulated in NEC (Table 1), specifically LOC101251419, LOC101262198 and LOC101244423. In addition, LOC101251256, coding for an abscisic acid receptor PYL8, and LOC101256079, coding for histone deacetylase 5, were identified as target genes of TCONS_00000158. These trans-regulated genes were all also found more expressed in NEC. In cis-regulation, LOC101263700, coding for transcription factor VIP1, was predicted to be targeted by TCONS_00000157 and 158. In this case, both the lncRNAs and the target gene were found upregulated in NEC.
No SOMATIC EMBRYOGENESIS RECEPTOR KINASE (SERK) gene was predicted as a target gene of the differentially expressed lncRNAs. Nevertheless, from the transcripts data, SERK1 and SERK3B were found more expressed in NEC than in EC and LTC (Fig. 7).

Schematic representation of differentially expressed lncRNAs and their main predicted target genes in S. betaceum cell lines (EC - embryogenic callus, LTC – long-term callus, NEC – non-EC). The solid lines represent the cis-regulation and the dot lines the trans-regulation by the lncRNAs. On the right are the genes upregulated in these cell lines, with no predicted lncRNA regulation. In green – embryogenesis-related genes, brown – carbohydrate metabolism-related genes, orange – auxin-related genes and pink – ethylene-related genes. LncRNAs are represented in red circles and numbers represent their simplified annotations, e.g. TCONS_00000XXX
Discussion
Recently, lncRNAs were pointed out as key players in the regulation of cell differentiation and development in both plants and animals (see Mattick et al. 2023). In plants, these ncRNAs are involved in gene regulatory networks in a wide range of biological processes in plant reproductive development, such as floral organs development and transition from the vegetative to the generative stage, flowering, and responses to abiotic and biotic stresses (Budak et al. 2020; Chen et al. 2020).
As the analysis of specific expression patterns can often provide important additional clues into the functional regulation of the networks involved, here, a long-read sequencing approach was conducted to detect full-length transcripts in cell populations of a non-sequenced plant species, S. betaceum. Aiming to reveal the role of the identified lncRNAs in the regulation of embryogenic competence, differential expression analysis was performed in cell lines with distinct morphogenic competencies.
To optimize the assay for high throughput sequencing, barcoded cDNA libraries enriched in poly(A) sequences were prepared for direct sequencing using ONT. As discussed in Cordeiro et al. (2023b), the method used generated reliable data for further analysis. As Pol-II transcripts, most plant lncRNAs are polyadenylated (poly(A) (Wierzbicki et al. 2021). However, poly(A) transcripts comprise only 1–5% of a total RNA extraction. Thus, isolating the poly(A) RNAs from the rRNA and non-poly(A) RNAs, during library preparation, reduces the complexity of the transcriptome sampled and enriches the poly(A) lncRNA content. Also, poly(A) RNA-seq is a highly sensitive and specific method, that can detect low-abundant RNAs with high accuracy (Ura et al. 2022). This is especially important in lncRNA sequencing, due to their low expression levels. Nevertheless, restricting the analysis to poly(A) lncRNAs might compromise the identification of a wider range of these non-coding molecules. Alternatively, libraries can be prepared from total RNA, followed by rRNA removal and direct RNA sequencing. However, barcoding kits, which allow the pooling and sequencing of several libraries in a single run, are only available for cDNA in Oxford Nanopore Technologies® (ONT). Ultimately, the choice of sequencing method will largely depend on the research purposes (Liu et al. 2017).
Using S. lycopersicum as the reference genome and given that S. betaceum is a closely related species and a model crop, a total of 60 lncRNA transcripts were predicted among S. betaceum SE-induced cell lines. From these, 39 lncRNAs showed relevant expression, and a total of 17 were found differentially expressed among samples. Such a reduced number of identified lncRNAs can be explained by the reasons pointed out above, which ultimately contribute to poor functional characterization of plant lncRNAs. In addition, as this analysis was performed with poly(A) RNA, the transcripts here identified were only the lncRNAs transcribed by Poll II. Moreover, from the distinct existing algorithms used to identify lncRNAs from RNA-sequencing data (Palos et al. 2023), the ones used might not have been the most effective for plant transcripts. For instance, CNIT exhibits more accuracy, 0.98 versus 0.85, for Arabidopsis transcripts than CNCI (Guo et al. 2019). Also, the near absence of annotations associated with lncRNAs on reference annotations impairs more effective lncRNA research in less studied plants (Simopoulos et al. 2019).
S. Betaceum lncRNAs involved in totipotency acquisition
Throughout subcultures, previously competent EC loses its embryogenic potential and increases its proliferative capacity. Hence, regardless of the morphology resemblance with competent EC, this subcultured callus (LTC), becomes morphogenically more similar to NEC. However, contrary to what was hypothesized, the present analysis revealed similar lncRNA expression patterns between EC and LTC, while significant differences were found between these cell lines compared to NEC. These results suggest that lncRNA-mediated gene regulation mechanisms are significantly involved in plant cell totipotency acquisition but likely less involved in the maintenance of the embryogenic capacity in S. betaceum-induced EC throughout subcultures. Also, in the differential expression analysis, a high number of lncRNAs was found upregulated in NEC, compared to the ones upregulated in EC and/or LTC. Thus, contrary to what was pointed out for longan, in which more lncRNA involvement was required for somatic embryo development (Chen et al. 2018), in S. betaceum, lncRNAs seem to be more required in the blocking of the embryogenic competence acquisition.
The target genes predicted in trans-regulation showed similar expression patterns of the lncRNAs for which they were predicted as targets. This is consistent with the positive regulatory role of lncRNAs on their target genes. The involvement of several of these lncRNAs in specific regulatory networks of SE will be further discussed.
LncRNAs targeting embryogenesis-related genes
In two-step SE processes, the reprogramming of somatic cells into totipotent cells is a crucial step. In this context, the involvement of several miRNAs in callus induction and plant cell dedifferentiation is well-described in crops, such as maize, rice, wheat and tomato, and other economically important plants including Camellia sinensis (L.) Kuntze (tea plant), Gossypium hirsutum L. (cotton) and Lilium plants (reviewed by Bravo-Vázquez et al. 2023). However, the role of lncRNAs in the regulation of these mechanisms is still largely unknown. To date, only two lncRNAs were functionally characterized for their role in callus induction in rice (Zhang et al. 2022).
Previous studies involving the analysis of cell lineage-specific transcriptomes identified not only several unknown transcripts of protein-coding genes but also lncRNAs involved in the cell fate specification of zygotic proembryos in Arabidopsis (Zhou et al. 2020). Thus, as lncRNAs emerge as key regulators of zygotic embryogenesis, their further study becomes imperative to elucidate the molecular pathways they are involved in and will possibly allow overcoming the recalcitrance of some species, such as trees, for experimental embryogenesis.
The present work showed that lncRNAs TCONS_00000218, 483 and 511 were putatively targeting WOX2 and AGL15, in S. betaceum EC and LTC cell lines. WOX2 is a member of the WOX (WUSCHEL-related homeobox) family of transcription factors, master regulators of in vitro plant regeneration, mainly in the manifestation of cellular totipotency (Fambrini et al. 2022). Specifically, WOX2 plays an important role in the regulation of the shoot apical meristem, through a negative feedback loop involving CLAVATA (CLV) genes (Fletcher 2018). Moreover, this gene is involved in lateral organ development, such as leaves and flowers (Chung et al. 2016), and seems to have evolutionary conserved roles on the control of embryo development of both angiosperms and gymnosperms (Hassani et al. 2022). In vitro, WOX2 overexpression was found to promote SE and organogenesis in Arabidopsis (Hassani et al. 2022), cotton (Bouchabké-Coussa et al. 2013) and coffee (Arroyo-Herrera et al. 2008). AGL15 is a member of the MADS-box transcription factor family, which plays an important role in SE regulation (reviewed by Joshi et al. 2022). When ectopically expressed, it enhances the embryogenic competency in Arabidopsis (Harding et al. 2003), cotton (Yang et al. 2014) and soybean (Perry et al. 2016). Thus, since these transcription factors are well-known key regulators in embryogenesis and widely reported for several species (Wójcik et al. 2020), the results here obtained prove the reliability of this approach and, although further validation is needed, corroborate the involvement of WOX2 and AGL15 in the embryogenic potential acquisition. Moreover, as both lncRNAs and targets are upregulated in EC and LTC, these findings suggest that the TCONS_00000218, 483 and 511 lncRNAs are also positively involved in embryogenesis achievement by somatic cells.
LncRNAs targeting carbohydrate metabolism and cell wall-related genes
NEC has a higher proliferation rate when compared to EC (Alves et al. 2017), and therefore requires a rapid and enhanced energy supply, obtained through glycolysis. Accordingly, a large part of the predicted target genes of the lncRNAs found upregulated in these cells was related to carbohydrate metabolism. One example was the FRK2 transcript. Involved in starch synthesis, the enzyme encoded by this gene was also previously found significantly more expressed in NEC than EC in a comparative proteomic analysis in S. betaceum (Correia et al. 2012b). This is also consistent with the number of starch grains usually found in S. betaceum NEC (Correia et al. 2012b).
Indeed, carbohydrate metabolism-related effects are one of the most important factors in SE induction (Lipavská and Konrádová 2004; Navarro et al. 2017). For instance, in S. betaceum, 9% (w/v) sucrose is required to stimulate EC formation, and embryo development only occurs when EC is transferred to culture conditions with an accentuated decrease (to 3–4% (w/v) in sucrose (Correia et al. 2012a). In this study, G6PD transcripts were found upregulated in S. betaceum NEC and targeted by NEC-upregulated lncRNAs, sustaining the high energy requirements of such cells in active proliferation. Nevertheless, these observations are not in line with previous reports in which G6PD has been described as an indispensable regulator in embryonic development in animal and plant models (Wu et al. 2018; Ruan et al. 2022). In Arabidopsis seed development, it regulates the transcription of early embryo developmental genes and the accumulation of storage materials (Ruan et al. 2022). Since those analyses refer to embryo development, further analysis should be made to address the role of this enzyme in S. betaceum somatic embryo development stages. Nevertheless, the results here obtained suggest the involvement of some of the identified lncRNAs in the embryogenic potential acquisition by positively targeting carbohydrate metabolism-related genes, which have important roles in SE regulation.
XTH5 and XTH16 were also identified as upregulated in NEC and targeted by NEC-upregulated lncRNAs. These genes code for enzymes related to cell wall modifications, including expansion and loosening, through hemicellulose metabolism (Wu et al. 2020a). As morphogenetic processes are accompanied by changes in the cell wall, upregulation of XTH occurs in tissues undergoing SE. For instance, XTH genes were more expressed in somatic embryos than in the pro-embryogenic masses in cucumber (Malinowski et al. 2004) and longan (Ma et al. 2022a). Due to its high proliferation rate and cell morphology, changes in the cell wall of S. betaceum NEC were expected, and consequently, the upregulation of these enzymes, as also reported for rapidly diving and expanding cells in carrot cell suspensions (Hetherington and Fry 1993). Thus, besides the previous reports showing the upregulation of these proteins in S. betaceum EC compared to NEC (Alves et al. 2017), the results here obtained, related to a putative upregulation of their coding transcripts in NEC, may indicate a regulatory role of these enzymes in the non-acquisition of embryogenic competence, in which a tight control of cell expansion is required.
Regulation of auxin-related genes by lncRNAs
Several auxin signalling-related genes were identified among the putative target genes in the lncRNAs trans-regulation. Likewise, in longan, five auxin response factors (ARFs), including ARF4, IAA6, AUX22 and ABF, were also predicted as target genes positively regulated by differentially expressed lncRNAs (Chen et al. 2018). Also, recently, an auxin pathway-related network (lncRNA125175-miR393h-TIR2) was pointed out to play a major role in SE regulation (Bai et al. 2023). Indeed, auxin plays a crucial role in plant growth and development and was identified as an indispensable inducer of SE (Wójcik et al. 2020), including in S. betaceum (Caeiro et al. 2022). Thus, as lncRNAs were upregulated in NEC, it can be assumed that these molecules are involved in the negative regulation of the embryogenic potential acquisition through the activation of some auxin-responsive genes. Indeed, although most auxin-responsive genes are known to promote SE, some of them are involved in its repression. For instance, SAUR15 was found to act as a negative effector in maize EC induction from immature embryos (Wang et al. 2022). Similarly, in the present data, SAUR50 was found upregulated in S. betaceum NEC. Also, although no significant differences were found for IAA14 between S. betaceum EC and NEC in previous qPCR analysis (Caeiro et al. 2022), here this gene was found upregulated in NEC and predicted as a target gene of two lncRNAs (TCONS_00000058 and 158).
Other ARFs were found among the identified transcripts, corroborating their involvement in SE (Wójcikowska and Gaj 2017). Although ARF6 and ARF8 were found upregulated in embryogenic cultures in Arabidopsis (Gliwicka et al. 2013), these genes were found upregulated in S. betaceum NEC. This apparently not concordant result may be related to the extreme diversity of ARFs and a possible functional non-correspondence between different species.
Despite the lack of references related to the identification of lncRNAs targeting auxin-related genes in plant regeneration processes, some evidence begins to be raised. For instance, AUXIN-REGULATED PROMOTER LOOP (APOLO), one of the first lncRNAs identified in Arabidopsis, has functions in the control of auxin homeostasis (Mammarella et al. 2023), targeting auxin-responsive genes involved in lateral root development (Ma et al. 2022b).
Regulation of ethylene-related genes by lncRNAs
Another crucial plant growth regulator of in vitro regeneration processes, such as organogenesis and SE, is ethylene. These processes are influenced by ethylene modulation through stress-response signalling and ERFs expression regulation (reviewed in Neves et al. 2021). Recently, studies in tomato and apple suggest that lncRNAs could also potentially be involved in the regulation of ethylene (Wang et al. 2018; Yu et al. 2022). While ethylene has been shown to promote SE in several plant species, including soybean (Zheng et al. 2013), in others it has negative effects, such as in olive tree (Bashir et al. 2022). In preliminary studies with S. betaceum, ethylene was reported to play a fundamental role in the acquisition of embryogenic competence, potentiating somatic embryo formation (Neves 2018), and in more recent studies, ethylene inhibition led to reduced de novo shoot organogenesis and subsequent plant development (Neves et al. 2023). As the AP2/ERF (APETALA2/Ethylene Responsive Factor) represents one of the largest transcription factor families in plants, with indispensable roles in plant growth, development, hormone regulation, and especially in responses to various stresses, their functional characterization is still far from complete (Xie et al. 2019; Wu et al. 2022). Specifically, ERF8 and ERF13 were suggested as SE markers in EC and in Coffea arabica L. embryogenic cell suspension cultures (Daude et al. 2021), as in the present study different ERFs were found upregulated in S. betaceum NEC. As some ERFs respond to ethylene, promoting the activation of some genes, these findings corroborate that cell fate transitions, involving embryogenic competence acquisition, require ethylene levels to promote ERFs differential expression (or possible regulators, such as lncRNAs) and hence activate ethylene-related responses. As in the present work, ERFs were putatively found as positively regulated by some lncRNAs upregulated in NEC, it can be assumed that these potential lncRNAs participate in the negative regulation of embryogenesis through ERFs activation.
Conclusion
Using long-read sequencing, 60 lncRNAs were identified in cell lines, induced by SE and with different embryogenic competencies, of the non-sequenced species S. betaceum. Despite the competence loss observed in LTC, lncRNAs maintain their expression patterns in these cell lines similar to the EC. In turn, differences were found between these two lines and NEC, in which a significantly higher expression of lncRNAs was revealed. From target prediction and function annotation, the lncRNAs identified in this study revealed to putatively target described embryogenesis-related genes and genes related to carbohydrate and cell wall metabolism, auxin and ethylene signalling pathways. Further functional characterization of these putative lncRNAs and their predicted targets is required, so such knowledge can be effectively used in breeding programs, to modulate and optimize plant regeneration processes in different species. Nevertheless, the present work represents the first analysis of the lncRNA-mediated regulation of the embryogenic competence acquisition, expression and maintenance in plant cells from a woody non-sequenced species. Thus, altogether, these results are a step further in understanding plant cell reprogramming toward totipotency achievement.
Data availability
The datasets generated during the current study are available in the National Centre for Biotechnology Information (NCBI) repository with the submission ID SUB12178214 (BioProject ID PRJNA892465; Biosample Accessions SAMN31382762, SAMN31382763 and SAMN31382764).
References
Acosta-Quezada PG, Raigón MD, Riofrío-Cuenca T et al (2015) Diversity for chemical composition in a collection of different varietal types of tree tomato (Solanum betaceum Cav.), an andean exotic fruit. Food Chem 169:327–335. https://doi.org/10.1016/j.foodchem.2014.07.152
Alves A, Caeiro A, Correia SI et al (2017) Establishment and biochemical characterization of tamarillo (Solanum betaceum Cav.) Embryogenic cell suspension cultures. Vitro Cell Dev Biology - Plant 53:606–618. https://doi.org/10.1007/s11627-017-9864-z
Alves A, Cordeiro D, Correia S, Miguel C (2021) Small non-coding RNAs at the crossroads of regulatory pathways controlling somatic embryogenesis in seed plants. Plants 10:504. https://doi.org/10.3390/plants10030504
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 4 Apr 2024
Arroyo-Herrera A, Ku Gonzalez A, Canche Moo R et al (2008) Expression of WUSCHEL in Coffea canephora causes ectopic morphogenesis and increases somatic embryogenesis. Plant Cell Tissue Organ Cult 94:171–180. https://doi.org/10.1007/s11240-008-9401-1
Ashburner M, Ball CA, Blake JA et al (2000) Gene ontology: tool for the unification of biology. Nat Genet 25:25–29. https://doi.org/10.1038/75556
Bai Y, Liu M, Zhou R et al (2023) Construction of ceRNA networks at different stages of somatic embryogenesis in garlic. Int J Mol Sci 24:5311. https://doi.org/10.3390/ijms24065311
Bairoch A, Apweiler R (1999) The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic Acids Res 27:49–54. https://doi.org/10.1093/nar/27.1.49
Bashir MA, Silvestri C, Salimonti A et al (2022) Can ethylene inhibitors enhance the success of olive somatic embryogenesis? Plants 11:168. https://doi.org/10.3390/plants11020168
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc: Ser B (Methodol) 57:289–300. https://doi.org/10.1111/j.2517-6161.1995.tb02031.x
Bouchabké-Coussa O, Obellianne M, Linderme D et al (2013) Wuschel overexpression promotes somatic embryogenesis and induces organogenesis in cotton (Gossypium hirsutum L.) tissues cultured in vitro. Plant Cell Rep 32:675–686. https://doi.org/10.1007/s00299-013-1402-9
Bravo-Vázquez LA, AnguloBejarano PI, Bandyopadhyay A et al (2023) Regulatory roles of noncoding RNAs in callus induction and plant cell dedifferentiation. Plant Cell Rep. https://doi.org/10.1007/S00299-023-02992-0
Budak H, Kaya SB, Cagirici HB (2020) Long non-coding RNA in plants in the era of reference sequences. Front Plant Sci 11:276. https://doi.org/10.3389/fpls.2020.00276
Caeiro A, Caeiro S, Correia S, Canhoto J (2022) Induction of somatic embryogenesis in tamarillo (Solanum betaceum Cav.) Involves increases in the endogenous auxin indole-3-acetic acid. Plants 11:1347. https://doi.org/10.3390/plants11101347
Chen Y, Li X, Su L et al (2018) Genome-wide identification and characterization of long non-coding RNAs involved in the early somatic embryogenesis in Dimocarpus longan lour. BMC Genomics 19:805. https://doi.org/10.1186/s12864-018-5158-z
Chen L, Zhu QH, Kaufmann K (2020) Long non-coding RNAs in plants: emerging modulators of gene activity in development and stress responses. Planta 252:1–14. https://doi.org/10.1007/s00425-020-03480-5
Chung KM, Sakamoto S, Mitsuda N et al (2016) WUSCHEL-RELATED HOMEOBOX 2 is a transcriptional repressor involved in lateral organ formation and separation in Arabidopsis. Plant Biotechnol 33:245–253. https://doi.org/10.5511/plantbiotechnology.16.0202a
Cordeiro D, Rito M, Borges F et al (2020) Selection and validation of reference genes for qPCR analysis of miRNAs and their targets during somatic embryogenesis in tamarillo (Solanum betaceum Cav). Plant Cell Tissue Organ Cult 143:109–120. https://doi.org/10.1007/s11240-020-01901-7
Cordeiro D, Canhoto J, Correia S (2022) Regulatory non-coding RNAs: emerging roles during plant cell reprogramming and in vitro regeneration. Front Plant Sci 13:1049631. https://doi.org/10.3389/fpls.2022.1049631
Cordeiro D, Alves A, Ferraz R et al (2023a) An efficient Agrobacterium-mediated genetic transformation method for Solanum betaceum Cav. Embryogenic callus. Plants 12:1202. https://doi.org/10.3390/plants12051202
Cordeiro D, Camelo A, Pedrosa AC et al (2023b) An efficient method to prepare barcoded cDNA libraries from plant callus for long-read sequencing. Methods Protoc 6:31. https://doi.org/10.3390/mps6020031
Corredoira E, Merkle SA, Martínez MT et al (2019) Non-zygotic embryogenesis in hardwood species. CRC Crit Rev Plant Sci 38:29–97. https://doi.org/10.1080/07352689.2018.1551122
Correia SI, Canhoto JM (2012) Biotechnology of tamarillo (Cyphomandra betacea): from in vitro cloning to genetic transformation. Sci Hortic 148:161–168. https://doi.org/10.1016/j.scienta.2012.09.037
Correia S, Canhoto JM (2018) Somatic embryogenesis of tamarillo (Solanum betaceum Cav). In: Jain SM, Gupta PK (eds) Step wise protocols for somatic embryogenesis of important woody plants. Springer, Cham, Switzerland, pp 171–179
Correia S, Lopes ML, Canhoto JM (2011) Somatic embryogenesis induction system for cloning an adult Cyphomandra betacea (cav.) Sendt. (tamarillo). Trees 25:1009–1020. https://doi.org/10.1007/s00468-011-0575-5
Correia S, Cunha AE, Salgueiro L, Canhoto JM (2012a) Somatic embryogenesis in tamarillo (Cyphomandra betacea): approaches to increase efficiency of embryo formation and plant development. Plant Cell Tissue Organ Cult 109:143–152. https://doi.org/10.1007/s11240-011-0082-9
Correia S, Vinhas R, Manadas B et al (2012b) Comparative proteomic analysis of auxin-induced embryogenic and nonembryogenic tissues of the solanaceous tree Cyphomandra betacea (tamarillo). J Proteome Res 11:1666–1675. https://doi.org/10.1021/pr200856w
Correia S, Alhinho AT, Casimiro B et al (2019) NEP-TC a rRNA methyltransferase involved on somatic embryogenesis of tamarillo (Solanum betaceum Cav). Front Plant Sci 10:438. https://doi.org/10.3389/fpls.2019.00438
Currais L, Loureiro J, Santos C, Canhoto JM (2013) Ploidy stability in embryogenic cultures and regenerated plantlets of tamarillo. Plant Cell Tissue Organ Cult 114:149–159. https://doi.org/10.1007/s11240-013-0311-5
Daude MM, Freitas NC, Ságio SA et al (2021) Molecular analysis of ERF subfamily genes during coffee somatic embryogenesis. Vitro Cell Dev Biology - Plant 57:128–142. https://doi.org/10.1007/s11627-020-10120-w
Ewels P, Magnusson M, Lundin S, Käller M (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32:3047–3048. https://doi.org/10.1093/BIOINFORMATICS/BTW354
Fambrini M, Usai G, Pugliesi C (2022) Induction of somatic embryogenesis in plants: different players and focus on WUSCHEL and WUS-RELATED HOMEOBOX (WOX) transcription factors. Int J Mol Sci 23:15950. https://doi.org/10.3390/ijms232415950
Finn RD, Coggill P, Eberhardt RY et al (2016) The pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44:D279–D285. https://doi.org/10.1093/nar/gkv1344
Fletcher JC (2018) The CLV-WUS stem cell signaling pathway: a roadmap to crop yield optimization. Plants 7:87. https://doi.org/10.3390/plants7040087
Gao Y, Cui Y, Zhao R et al (2022) Cryo-treatment enhances the embryogenicity of mature somatic embryos via the lncRNA-miRNA-mRNA network in white spruce. Int J Mol Sci 23:1111. https://doi.org/10.3390/ijms23031111
Gliwicka M, Nowak K, Balazadeh S et al (2013) Extensive modulation of the transcription factor transcriptome during somatic embryogenesis in Arabidopsis thaliana. PLoS ONE 8:69261. https://doi.org/10.1371/journal.pone.0069261
Guo JC, Fang SS, Wu Y et al (2019) CNIT: a fast and accurate web tool for identifying protein-coding and long non-coding transcripts based on intrinsic sequence composition. Nucleic Acids Res 47:W516–W522. https://doi.org/10.1093/nar/gkz400
Harding EW, Tang W, Nichols KW et al (2003) Expression and maintenance of embryogenic potential is enhanced through constitutive expression of AGAMOUS-like 15. Plant Physiol 133:653–663. https://doi.org/10.1104/pp.103.023499
Hassani SB, Trontin JF, Raschke J et al (2022) Constitutive overexpression of a conifer WOX2 homolog affects somatic embryo development in Pinus pinaster and promotes somatic embryogenesis and organogenesis in Arabidopsis seedlings. Front Plant Sci 13:838421. https://doi.org/10.3389/fpls.2022.838421
Hetherington PR, Fry SC (1993) Xyloglucan endotransglycosylase activity in carrot cell suspensions during cell elongation and somatic embryogenesis. Plant Physiol 103:987–992. https://doi.org/10.1104/pp.103.3.987
Huerta-Cepas J, Szklarczyk D, Heller D et al (2019) eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 47:D309–D314. https://doi.org/10.1093/nar/gky1085
Jin J, Lu P, Xu Y et al (2021) PLncDB V2.0: a comprehensive encyclopedia of plant long noncoding RNAs. Nucleic Acids Res 49:D1489–D1495. https://doi.org/10.1093/nar/gkaa910
Joshi S, Paul P, Hartman JM, Perry SE (2022) AGL15 promotion of somatic embryogenesis: role and molecular mechanism. Front Plant Sci 13:770. https://doi.org/10.3389/fpls.2022.861556
Kanehisa M, Goto S, Sato Y et al (2012) KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40:D109–D114. https://doi.org/10.1093/nar/gkr988
Kong L, Zhang Y, Ye ZQ et al (2007) CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res 35. https://doi.org/10.1093/nar/gkm391
Lee JT (2012) Epigenetic regulation by long noncoding RNAs. Science 338, 1435–1439. https://doi.org/10.1126/science.1231776
Li H (2018) Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34:3094–3100. https://doi.org/10.1093/bioinformatics/bty191
Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30:923–930. https://doi.org/10.1093/bioinformatics/btt656
Lipavská H, Konrádová H (2004) Somatic embryogenesis in conifers: the role of carbohydrate metabolism. Vitro Cell Dev Biology - Plant 40:23–30. https://doi.org/10.1079/ivp2003482
Liu D, Mewalal R, Hu R et al (2017) New technologies accelerate the exploration of non-coding RNAs in horticultural plants. Hortic Res 4:17031. https://doi.org/10.1038/hortres.2017.31
Love MI, Huber W, Anders S (2014) Moderated estimation of Fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. https://doi.org/10.1186/s13059-014-0550-8
Lucero L, Fonouni-Farde C, Crespi M, Ariel F (2020) Long noncoding RNAs shape transcription in plants. Transcription 11:160–171. https://doi.org/10.1080/21541264.2020.1764312
Ma X, Chen Y, Liu M et al (2022a) Genome-wide analysis of the XTH gene family and functional analysis of DlXTH23.5/25 during early longan somatic embryogenesis. Front Plant Sci 13:1043464. https://doi.org/10.3389/fpls.2022.1043464
Ma X, Zhao F, Zhou B (2022b) The characters of non-coding RNAs and their biological roles in plant development and abiotic stress response. Int J Mol Sci 23:4124. https://doi.org/10.3390/ijms23084124
Malinowski R, Filipecki M, Tagashira N et al (2004) Xyloglucan endotransglucosylase/hydrolase genes in cucumber (Cucumis sativus)– differential expression during somatic embryogenesis. Physiol Plant 120:678–685. https://doi.org/10.1111/j.0031-9317.2004.0289.x
Mammarella MF, Lucero L, Hussain N et al (2023) Long noncoding RNA-mediated epigenetic regulation of auxin-related genes controlling shade avoidance syndrome in Arabidopsis thaliana. bioRxiv 2023.03.06.531280. https://doi.org/10.1101/2023.03.06.531280
Marx V (2023) Method of the year: long-read sequencing. Nat Methods 20:6–11. https://doi.org/10.1038/s41592-022-01759-x
Mattick JS, Amaral PP, Carninci P et al (2023) Long non-coding RNAs: definitions, functions, challenges and recommendations. Nature Reviews Molecular Cell Biology 2023 17:1–17. https://doi.org/10.1038/s41580-022-00566-8
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15:473–497. https://doi.org/10.1111/j.1399-3054.1962.tb08052.x
Navarro BV, Elbl P, De Souza AP et al (2017) Carbohydrate-mediated responses during zygotic and early somatic embryogenesis in the endangered conifer, Araucaria angustifolia. PLoS ONE 12:e0180051. https://doi.org/10.1371/journal.pone.0180051
Neves M (2018) Effect of ethylene on somatic embryogenesis of tamarillo (Solanum betaceum Cav.)
Neves M, Correia S, Cavaleiro C, Canhoto J (2021) Modulation of organogenesis and somatic embryogenesis by ethylene: an overview. Plants 10:1208. https://doi.org/10.3390/plants10061208
Neves M, Correia S, Canhoto J (2023) Ethylene inhibition reduces de novo shoot organogenesis and subsequent plant development from leaf explants of Solanum betaceum Cav. Plants 12:1854. https://doi.org/10.3390/plants12091854
Palos K, Yu L, Railey CE et al (2023) Linking discoveries, mechanisms, and technologies to develop a clearer perspective on plant long noncoding RNAs. https://doi.org/10.1093/plcell/koad027. Plant Cell koad027
Perry SE, Zheng Q, Zheng Y (2016) Transcriptome analysis indicates that GmAGAMOUS-Like 15 may enhance somatic embryogenesis by promoting a dedifferentiated state. Plant Signal Behav 11:e1197463. https://doi.org/10.1080/15592324.2016.1197463
Pertea G, Pertea M (2020) GFF utilities: GffRead and GffCompare. https://doi.org/10.12688/F1000RESEARCH.23297.2/DOI. F1000Res 9:
Pertea M, Pertea GM, Antonescu CM et al (2015) StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology 2015 33:3 33:290–295. https://doi.org/10.1038/nbt.3122
Pringle GJ, Murray BG (1992) Polyploidy and aneuploidy in the tamarillo, Cyphomandra betacea (Cav.) Sendt. (Solanaceae). Plant Breeding 108:132–138. https://doi.org/10.1111/J.1439-0523.1992.TB00112.X
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. https://doi.org/10.1093/BIOINFORMATICS/BTQ033
Rai MI, Alam M, Lightfoot DA et al (2019) Classification and experimental identification of plant long non-coding RNAs. Genomics 111:997–1005. https://doi.org/10.1016/j.ygeno.2018.04.014
Ramírez F, Kallarackal J (2019) Tree tomato (Solanum betaceum Cav.) Reproductive physiology: a review. Sci Hortic 248:206–215. https://doi.org/10.1016/j.scienta.2019.01.019
Ruan M, He W, Sun H et al (2022) Cytosolic glucose-6-phosphate dehydrogenases play a pivotal role in Arabidopsis seed development. Plant Physiol Biochem 186:207–219. https://doi.org/10.1016/j.plaphy.2022.07.017
Simopoulos CMA, Weretilnyk EA, Golding GB (2019) Molecular traits of long non-protein coding RNAs from diverse plant species show little evidence of phylogenetic relationships. G3 Genes|Genomes|Genetics 9:2511–2520. https://doi.org/10.1534/g3.119.400201
Sun L, Luo H, Bu D et al (2013) Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res 41:e166. https://doi.org/10.1093/nar/gkt646
Ura H, Togi S, Niida Y (2022) Poly(A) capture full length cDNA sequencing improves the accuracy and detection ability of transcript quantification and alternative splicing events. Sci Rep 12:10599. https://doi.org/10.1038/s41598-022-14902-7
Waititu JK, Zhang C, Liu J, Wang H (2020) Plant non-coding RNAs: origin, biogenesis, mode of action and their roles in abiotic stress. Int J Mol Sci 21:8401. https://doi.org/10.3390/ijms21218401
Wang L, Park HJ, Dasari S et al (2013) CPAT: coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res 41:e74. https://doi.org/10.1093/nar/gkt006
Wang Y, Gao L, Li J et al (2018) Analysis of long-non-coding RNAs associated with ethylene in tomato. Gene 674:151–160. https://doi.org/10.1016/j.gene.2018.06.089
Wang Y, Zhao Y, Bollas A et al (2021) Nanopore sequencing technology, bioinformatics and applications. Nat Biotechnol 39:1348–1365. https://doi.org/10.1038/s41587-021-01108-x
Wang Y, He S, Long Y et al (2022) Genetic variations in ZmSAUR15 contribute to the formation of immature embryo-derived embryonic calluses in maize. Plant J 109:980–991. https://doi.org/10.1111/tpj.15609
Wierzbicki AT, Blevins T, Swiezewski S (2021) Long noncoding RNAs in plants. Annu Rev Plant Biol 72:245–271. https://doi.org/10.1146/annurev-arplant-093020-035446
Wójcik AM, Wójcikowska B, Gaj MD (2020) Current perspectives on the auxin-mediated genetic network that controls the induction of somatic embryogenesis in plants. Int J Mol Sci 21:1333. https://doi.org/10.3390/ijms21041333
Wójcikowska B, Gaj MD (2017) Expression profiling of AUXIN RESPONSE FACTOR genes during somatic embryogenesis induction in Arabidopsis. Plant Cell Rep 36:843–858. https://doi.org/10.1007/s00299-017-2114-3
Wu YH, Lee YH, Shih HY et al (2018) Glucose-6-phosphate dehydrogenase is indispensable in embryonic development by modulation of epithelial-mesenchymal transition via the NOX/Smad3/miR-200b axis. Cell Death Disease 2018 9(1 9):1–14. https://doi.org/10.1038/s41419-017-0005-8
Wu D, Liu A, Qu X et al (2020a) Genome-wide identification, and phylogenetic and expression profiling analyses, of XTH gene families in Brassica rapa L. and Brassica oleracea L. BMC Genomics 21:782. https://doi.org/10.1186/s12864-020-07153-1
Wu L, Liu S, Qi H et al (2020b) Research progress on plant long non-coding RNA. Plants 9:408. https://doi.org/10.3390/plants9040408
Wu Y, Li X, Zhang J et al (2022) ERF subfamily transcription factors and their function in plant responses to abiotic stresses. Front Plant Sci 13:4845. https://doi.org/10.3389/fpls.2022.1042084
Xie Z, Nolan TM, Jiang H, Yin Y (2019) AP2/ERF transcription factor regulatory networks in hormone and abiotic stress responses in Arabidopsis. Front Plant Sci 10:228. https://doi.org/10.3389/fpls.2019.00228
Yang Z, Li C, Wang Y et al (2014) GhAGL15s, preferentially expressed during somatic embryogenesis, promote embryogenic callus formation in cotton (Gossypium hirsutum L). Mol Genet Genomics 289:873–883. https://doi.org/10.1007/s00438-014-0856-y
Yu Y, Zhang Y, Chen X, Chen Y (2019) Plant noncoding RNAs: hidden players in development and stress responses. Annu Rev Cell Dev Biol 35:407–431. https://doi.org/10.1146/annurev-cellbio-100818-125218
Yu J, Qiu K, Sun W et al (2022) A long noncoding RNA functions in high-light-induced anthocyanin accumulation in apple by activating ethylene synthesis. Plant Physiol 189:66–83. https://doi.org/10.1093/plphys/kiac049
Zhang Y, Yang L, Chen LL (2014) Life without a tail: new formats of long noncoding RNAs. Int J Biochem Cell Biol 54:338–349. https://doi.org/10.1016/j.biocel.2013.10.009
Zhang YC, Zhou YF, Cheng Y et al (2022) Genome-wide analysis and functional annotation of chromatin-enriched noncoding RNAs in rice during somatic cell regeneration. Genome Biol 23:28. https://doi.org/10.1186/s13059-022-02608-y
Zheng Q, Zheng Y, Perry SE (2013) AGAMOUS-Like15 promotes somatic embryogenesis in Arabidopsis and soybean in part by the control of ethylene biosynthesis and response. Plant Physiol 161:2113–2127. https://doi.org/10.1104/pp.113.216275
Zhou X, Liu Z, Shen K et al (2020) Cell lineage-specific transcriptome analysis for interpreting cell fate specification of proembryos. Nat Commun 11:1366. https://doi.org/10.1038/s41467-020-15189-w
Zhu Y, Chen L, Hong X et al (2022) Revealing the novel complexity of plant long non-coding RNA by strand-specific and whole transcriptome sequencing for evolutionarily representative plant species. BMC Genomics 23:381. https://doi.org/10.1186/s12864-022-08602-9
Acknowledgements
The authors acknowledge CD Genomics and Ricardo Ramiro and Ehsan Valiollahi from InnovPlantProtect CoLab for the bioinformatic analysis.
Funding
Open access funding provided by FCT|FCCN (b-on). The Foundation for Science and Technology (Portugal) supported this work through PhD fellowship SFRH/BD/136925/2018 and COVID/BD/152914/2023 awarded to Daniela Cordeiro. This work was carried out at the R&D Unit Center for Functional Ecology—Science for People and the Planet (UIDB/04004/2020), financed by FCT/MCTES through national funds (PIDDAC), and at CATAA, this work was made in the scope of CULTIVAR project (CENTRO-01-0145-FEDER-000020), co-financed by the Regional Operational Programme Centro 2020, Portugal 2020, and the European Union, through the European Fund for Regional Development (ERDF), Collaborative Project.
Open access funding provided by FCT|FCCN (b-on).
Author information
Authors and Affiliations
Contributions
Conceptualization– DC, CES and SC; investigation– DC, AC, CES and SC; methodology– DC, AC, ACP, IB, CES and SC; data curation– DC and CES; supervision– CES and SC; funding acquisition and resources– CES, JC and SC; formal analysis, visualization and writing - original draft preparation– DC; validation– DC and SC; writing - review and editing– DC, CES, JC and SC. All authors have read and agreed to the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Communicated by Joyce Van Eck.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cordeiro, D., Camelo, A., Pedrosa, A.C. et al. Analysis of the potential involvement of lncRNAs in embryogenic competence of Solanum betaceum Cav. (tamarillo) calli. Plant Cell Tiss Organ Cult 157, 67 (2024). https://doi.org/10.1007/s11240-024-02779-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11240-024-02779-5