
Abstract
Favipiravir (FAV) (6-fluoro-3-oxo-3,4-dihydropyrazine-2-carboxamide) is one of the most effective antiviral drugs which is cited for action against RNA-viral infections of COVID-19. In this study, density functional theory (DFT) calculations were used to investigate three nanotubes (NTs) with FAV drug as delivery systems. The encapsulated systems (ESs) consist of FAV drug inside carbon–carbon, aluminum nitride, and boron nitride. At B3LYP-D/6-31G(d,p) and CPCM/B3LYP-D/6-31G(d,p), the optimization of NTs, FAV, and its tautomeric forms and six ESs was investigated in gas and water environments. Five tautomeric forms of FAV were investigated, two keto forms (K1 and K2) and three enol forms (E1, E2, and E3). The results revealed that E3 and K2 isomeric forms represented the most stable structures in both media; thus, these two forms were encapsulated into the NTs. The stability and the synthesis feasibility of NTs have been proven by calculating their interaction energies. Non-covalent interactions (NCIs) were investigated in the ESs to show the type of NCI with the molecular voids. The binding energies, thermochemical parameters, and recovery times were investigated to understand the mechanism of FAV encapsulation and release. The encapsulated AlNNT systems are more favorable than those of BNNTs and CNTs in gas and aqueous environments with much higher binding energies. The quantum theory of atoms in molecules (QTAIM) and recovery time analysis revealed the easier releasing of E3 from AlNNT over K2 form. Based on molecular docking simulations, we found that E3 and K2 FAV forms showed a high level of resistance to SARS-CoV-6M3M/6LU7/6W9C proteases.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Nanomedicine is the application of nanotechnology to medical science and diagnostics. Engineered nanodevices and nanostructures are used to monitor, repair, construct, and control human biological systems at the molecular level [1]. Furthermore, many studies have given attention to drug delivery systems [2,3,4,5]. The drug delivery system serves as a connection between a drug and the patient that can be used for the direct delivery of therapeutic agents to the patient or for the controlled and prolonged release of these agents.
Over the past few years, researchers have focused increasingly on discovering and developing new carbon-free nanomaterials as drug delivery systems. In comparison to carbon nanotubes (CNTs), non-carbon nanotubes seem to be better suited for biological applications, like drug delivery systems [6,7,8]. Non-carbon nanotubes (NTs) can be produced with elements of groups III and V, indicating those elements are crucial materials in the production of these NTs [3,7,9]. Among, a theoretical prediction of boron nitride nanotubes was published in 1994 by Rubio et al. [10], by finding that the hexagonal phase of cubic boron nitride (BN) exhibits similar properties to those of graphite-type carbon. The first time BNNTs were synthesized was by Chopra et al. [11] by alternating nitrogen and boron atoms instead of carbon atoms. Generally, due to their unique structures and properties, BNNTs have attracted a great deal of research attention [2,12,13]. Vatanparast et al. [12] theoretically investigated the nature of the interactions between some anticancer drugs such as 6-mercaptopurine (MP), 5-fluorouracil (FU), 6-thioguanine (TG), and BN nanosheet as a drug carrier. MP and TG drugs bind more strongly to the BN nanosheet than FU drugs, according to their results. As a result of molecular dynamics, the TG/BN system exhibited a more negative interaction energy value than either of the other two drugs. The average interaction energy values at acidic pH are overall lower than those at neutral pH, allowing the drug to be separated from the carrier. Bououden et al. [14] used the DFT to study the potential of CNTs and BNNTs as drug delivery systems through the adsorption of the anticancer molecule Crizotinib (CZT) on the surfaces. Their results showed that the adsorption between CZT and the BNNTs is endothermic, while it is exothermic for CNTs. CZT/CNT was also found to be more soluble in an aqueous medium than CZT/BNNT, and CNTs were found to be a better drug delivery system for CZT molecules than BNNTs. Gholami et al. [15] explored the application of encapsulated B36N36 with alkali or alkaline metals (AAM) as a drug delivery system for anticancer β-Lapachone drug based on DFT calculations. They found that bare B36N36 has lower binding energies in comparison to AAM/B36N36 in gas and water. And, especially for B36N36 and K-encapsulated B36N36 carries, the drug could be released simply.
According to Zhao et al. [16], aluminum nitride NTs (AlNNTs) are noticeably more reactive than CNTs. AlNNTs have not yet been shown to have chirality and diameter-dependent electronic properties, especially band structures. An investigation of the interaction of the cancer-treating drug Tegafur (TG) with pristine and Si/doped C, BN, and AlN NTs at the M06-2X/6-31G(d) level of theory was conducted by Makiabadi et al. [2]. The study aimed to determine the effects of both the original and doped AlN, BN, and C NTs in terms of how well the TG drug interacts with the target human cell. It was found that complexes exhibit Van der Waals force (VWF) interactions, suggesting that TG drugs may physiosorbed on NT surfaces. Nanotubes doped with Si atoms enhance TG drug-nanotube interaction.
For the development of a novel drug detection system, with the aid of DFT/B3LYP B3LYP/6-311G(d,p) study by Hassanpour et al. [17] investigated AlNNT interaction with dopamine drug. They found that the adsorption of the drug onto -NH2 side of AlNNTs is stronger than other positions. Additionally, an increase in AlNNT electrical conductivity after drug adsorption suggests that nanotubes could be used as electrical sensors to detect drugs like DA. Furthermore, the calculations indicate that systems are typically much more stable in solvent media than in gaseous mediums. They conclude that the calculated drug recovery time of AlNNT is about 26.29 ms, indicating a relatively short recovery time.
Favipiravir drug (6-fluoro-3-hydroxy-2-pyrazinecarboxamide (T-705; Avigan; FAV (the abbreviation used throughout this study)) was first prepared in 2014 [18]. It is an antiviral derivative of the pyrazine carboxamide family; it is like T-1105 and T-1106. As one of the drugs cited for treating COVID-19 RNA viruses [19,20,21,22], some of recent studies considered FAV to be one of the most promising drugs against SARS-CoV-2 [20,23]. FAV with a broad-spectrum inhibitor (effective against many types of viruses) was originally used to treat influenza in Japan [17]. It is effective against a large number of RNA viruses including influenza [24,25], noroviruses [26], yellow fever virus [27], West Nile virus [28], Ebola virus [29], and Lassa virus [30].
Providing guidelines for designing and improving fullerene and BNNC drug delivery systems, Soliman and Abdel Aal [4] used two functionals, B3LYP and CAM-B3LYP, along with their time-dependent functionals to investigate the adsorption of FAV on both the isolated (C24 and B12N12) and doped (BC23 and CB11N12) nanocages. In their study, no deformation in the FAV was detected, and the reactivity of doped nanocages (BC23 and CB11N12) exhibits higher interacting energies compared to the undoped nanocage. Additionally, according to the negative value of the Gibbs free energies of FAV on the doped nanocages, and the weak NCI of FAV with the nanocages, they concluded that the doping process is spontaneous and can be utilized to deliver the FAV drug. A theoretical study by Asgari et al. [31] on pristine (2,2) graphene-based nanotubes (GYNT) adsorbed by using FAV and examination of the Si/doped form (Si/GYNT) and its composite with encapsulated Fe2O3 (Fe2O3/Si-GYNT) has been conducted using DFT calculations. It has been found that FAV adsorbed at the highest energy of − 19.8 and − 17.0 kcal/mol, in pure GYNT and Si-doped GYNT, respectively. Meanwhile, the Fe2O3/Si-GYNT interacts with the FAV molecule more strongly. Higher temperatures resulted in a short recovery time of less than 10 s (sec). The FAV can also be detached from the Fe2O3/Si-GYNT by proton attack, making it a potentially effective drug carrier in cancer tissues at low pH. Pari et al. [32] analyzed the electronic structure of two bimolecular structures of FAV and the boron-nitrogen-carbon cage model (BNCC) at WB97XD/6–31 + G* level. They analyzed the interactions at various positions of the atom of FAV with the BNCC surface using six models of BNCC-FAV systems. The BNCC was therefore found useful in further investigations to deliver loads of FAV. Bibi et al. [33] explored the interaction of FAV with metal-doped (Cr, Fe, and Ni) fullerene complexes as drug delivery systems. They performed DFT calculations by using M06-2X/6-31G(d) level of theory. NiC59 has the highest adsorption energy, so it is the most stable adsorbent for FAV drug. They also found that FAV adsorption occurs spontaneously and exothermically with metal-doped fullerenes. Moreover, the Ni-doped fullerene complex exhibited the maximum conductivity. They concluded that in COVID-19 treatment, Fe, Cr, and especially Ni-doped fullerene would be the best nanodrug delivery materials because of their enhanced electronic and adsorption properties.
In this study, more beneficial NTs were identified as delivery systems for FAV drug. At CPCM/B3LYP-D/6-31G(d,p) level of theory, FAV drug isomerization and tautomerization were investigated, and the most stable tautomeric forms were encapsulated into CNT, BNNT, and AlNNT. The geometrical, electronic, optical (at TDCAM-B3LYP-D/6-31G(d,p)), and binding energies, thermodynamic parameters, non-covalent interactions, recovery times, and QTAIM of the ESs were systematically investigated and discussed. Through molecular docking simulations, the inhibition impact of the FAV forms over three different targets of SARS-CoV-2, i.e., 6M3M, 6W9C, and 6LU7, was investigated. From a theoretical perspective and based on the main results of this paper, we propose a new system for delivering FAV drugs that is more efficient and encourage much more experimental work to validate it.
Computational details
As part of our study, we carried out our calculations using the Gaussian 09 package [34]. GaussView (version 6.0.16) was used for different plots [35]. DFT calculations with the well-known B3LYP exchange–correlation functional [36] were performed to optimize the tautomers and isomers of FAV drug, NTs, and encapsulated FAV/NTs. B3LYP is a consistently used function when studying similar nanostructures [4,17,37]. A moderate size basis set (6-31G(d,p)) was employed along with B3LYP functional. Most density functionals cannot account for van der Waals interactions due to dynamical correlations among fluctuating charge distributions. B3LYP functional was associated with Grimme’s “D” dispersion correction term [38,39,40]. With dispersion corrections, accuracy can be significantly improved, and this statement was accurately validated. Since the geometry optimization without dispersion correction failed in encapsulating the drug inside the NTs, while using it, the geometry optimization of the ES led to a complete encapsulation of the drug inside the nanotube; a representative example of FAV/CNT ES is shown in Fig. 1.

Optimization of FAV/CNT a without dispersion correction and b with dispersion correction
A time-dependent coulomb-attenuating method density functional calculation (TD-CAM-B3LYP) was used to reproduce UV–Vis spectra [41], since it provides more reliable results than the B3LYP on the nature of electronic transition spectra [42,43]. Since such a process (drug delivery) takes place in an aqueous medium, to mimic the water effect, the conductor-like solvation model, as developed in the polarizable continuum model (CPCM) framework [44,45] framework, was used to model solvation. This method creates the solute cavity via a set of overlapping spheres. A variety of weak interactions, such as H-bonding, occur in ES important for molecular mechanisms. The basis set superposition error (BSSE) is well-known in quantum chemical calculations when finite basis sets are used. It is well-known that the BSSE can have a considerable artificial effect on the geometrical structure and energy stabilization of dimeric structures. The BSSE for the ESs has been corrected using the counterpoise (CP) approach [46]. The BSSE-corrected binding energies (\({E}_{\mathrm{bind}}^{\mathrm{BSSE}}\)) and uncorrected binding energies (\({\Delta E}_{\mathrm{bind}}\)) of all optimized ESs were calculated using the following equation:
where \({E}_{ES}\) is the energy of encapsulated system of the FAV drug into the nanotube, \({E}_{NT}\) is the energy of the bare nanotube, \({E}_{FAV}\) is the energy of the FAV drug, and EBSSE corresponds to the BSSE energy. It was ensured that all optimized structures corresponded to minima on the potential energy surface without negative (imaginary) frequencies in all cases by performing vibrational frequency calculations at the same theoretical level. Also, the vibrational frequency calculations allowed obtaining the thermodynamic parameters included the enthalpy change (\({\Delta H}_{\mathrm{bind}}\)), Gibbs free energy change (\({\Delta G}_{\mathrm{bind}}\)), and entropy change (\({\Delta S}_{\mathrm{bind}}\)) for the ESs according to the following equations:
The reduced density gradient (RDG) is defined as:
where \(\rho (r)\) is the electron density and \(\left|\nabla \rho \right|\) is it electron density gradient. GaussSum program [47] was used to draw density of states (DOS) plots. Non-covalent interactions (NCI) were performed by Multiwfn 3.7 software package [48] to understand the nature of interactions between FAV drug and NT. The isosurfaces of ESs were made using visual molecular dynamics (VMD) 1.9.3 [49]. QTAIM [50] was used to study the nature of the intramolecular and intermolecular interactions in the FAV tautomers and the encapsulated drugs at the bond critical points (BCPs). In this sense, the types of intramolecular interactions can be determined by topological parameters, including electron density (\({\uprho }_{(r)}\)), Laplacian of electron density (\({\nabla }_{\rho (r)}^{2}\), total energy density (H(r)), kinetic energy density (G(r)), and potential energy density (V(r)) [51,52]. QTAIM was carried out by Multiwfn 3.7 software package [48].
Results and discussion
Optimized geometries of favipiravir
DFT studies have explored the biological activities of FAV and its tautomeric forms in recent years [53,54,55]. Before performing the encapsulation of FAV into the NTs materials, the tautomerism and isomerism of FAV were performed to define the most stable forms. For FAV, as a result of the 1,3-hydrogen transfer process, five tautomers can be imagined; three of these tautomers have enol form structure, denoted as E1, E2, and E3; and the two tautomers have keto form structure, denoted as K1 and K2. Accordingly, five planar structures are obtained: three isomeric enol forms, denoted as E1, E2, and E3, and two isomeric keto forms, denoted as K1 and K2. Both E1 and E3 are tautomers to each other, and K1 and K2 are also tautomers to each other. The five forms of FAV were optimized in gas phase and aqueous medium. The optimized geometries with their corresponding relative energies at the CPCM/B3LYP/6-31G(d,p) level of theory in the aqueous solution are shown in the Fig. 2.

(Left) The relative energies of the isomeric and tautomeric forms of FAV in water at the CPCM/B3LYP/6-31G(d,p) level of theory. (Right) The molecular graphs of K2 and E3 forms with the details of the topological parameters of the bond critical point of the intramolecular hydrogen bond (O….H)
As expected, keto-enol tautomerism, due to 1,3-hydrogen transfer, could result in significant changes. For instance, inspection of Fig. 2 shows that K1 and E3 (E1) are tautomers to each other, and they have the N–C-O angle of ~ 124 \(^\circ\), whereas the two K2 and E2 forms are tautomers to each other with the N–C-O angle of 124 \(^\circ\) and 123 \(^\circ\), respectively. Table SD1 (supplementary data) compares the main geometrical parameters of the five forms of FAV drug. Our results indicate the stability of the five forms in the gas phase following the order: E3 (0.000 kJ/mol) [53] > E1 (40.987 kJ/mol) > K2 (50.417 kJ/mol) > E2 (50.740 kJ/mol) > K1 (66.422 kJ/mol) (see Table SD2). However, the stability of the tautomers increases in the water medium in which the E3 form is still the most stable form followed by K2 and then E1, E2, and K1 by 23.955, 33.924, 36.510, and 36.930 kJ/mol, respectively. Thus, the enol form E3 of the FAV drug is the dominant form in both gas and water solvent, which agrees with the previous results [53], followed by K2 form. To explain the stability of E3 over other forms, especially K2, it is an aromatic structure with intramolecular hydrogen bond with 1.664 Å with hydrogen bond strength (EHB) of 57.4 kJ/mol (see Fig. 2), whereas, K2 has a weaker intramolecular hydrogen bond with a larger distance of 1.892 Å and a hydrogen bond strength of 33.8 kJ/mol, compared to the E3 form. This might be the reason for the low stability of the K2 form than E3. Based on these results, the encapsulation of the K2 (the most stable keto form) and E3 (the most stable form among enol forms and all other forms) into different nanotubes will be only considered in the upcoming calculations.
Optimized geometries and density of states of nanotubes
Each NT has a chirality of (10,0) by a tube length of 15 Å, whereas the values of the bond lengths are 1.830, 1.470, and 1.421 Å for Al-N, B-N, and C–C bonds, respectively. Within the hexagonal rings, the two Al-N-Al bond angles were identified and are almost equal to 119° and for B-N-B between 118° and 116° and for C–C-C bond angle between 120.7° and 111°. Moreover, to minimize the boundary effect, H atoms are saturated into the model nanotubes [56], with a total number of atoms of 120, 140, and 180 atoms for the AlN, BN, and C NTs, respectively. The optimized geometries of AlNNT, BNNT, and CNT and their total density of states (TDOS) and partial density of state (PDOS) plots analysis as calculated within the water at the CPCM/B3LYP/6-31G(d,p) level of theory are represented in Fig. 3. To estimate the stability of the NT geometries, the interaction energy (\({\mathrm{E}}_{\mathrm{int}}\)) was calculated using the following equation:
where \(\sum E(atoms)\) is the summation of energies of all atoms within the NT and \(E(NT)\) is the energy of NT. The interaction energies as measure of the formation of NTs are all negative indicating an exothermic process and thus a stability and the feasibility of synthesis of these NTs. The calculated interaction energies (listed in Fig. 3) are − 531.162, − 694.850, and − 729.621 kJ/mol for AlNNT, BNNT, and CNT, respectively. The obtained \({E}_{\mathrm{int}}\) values are compared to those of other NTs reported in literature (− 653.21 kJ/mol Be12O12 nanocage [57], − 660.538 kJ/mol for Be12O12 nanosheet, − 682.15 kJ/mol for wurtzite BeO [58], and − 502.164 kJ/mol for GaN nanocage [59]). The stability order of these three NTs is AlNNT < BNNT < CNT.

The optimized geometry, TDOS, and PDOS plots of different NTs in water
Density of states (DOS) analysis for the NTs has conducted to assist the orbital analysis, Fig. 3. The TDOS plots show that the HOMO levels are about − 6.140, − 6.267, and − 4.068 eV for AlNNT, BNNT, and CNT, respectively, while their LUMO levels are − 1.496, − 0.530, and − 3.373 eV, respectively (see also Table 1). Thus, their energy gaps are 4.644, 5.737, and 0.695 eV, respectively. NT with a larger energy gap is reported to be more stable and feasible to synthesize [60]. The resulted \({E}_{g}\) value of AlNNT equals 4.644 eV; this is quite close to the band gap of AlNNT that found by Zhao et al. [16], where they found that the theoretical band gaps of AlNNTs—differ in their chirality and diameters—are ranging from 2.84 to 3.95 eV, making them semiconductors in nature. On the other hand, the reported band gaps of CNTs are small ranging from 0.2 to 2.0 eV, while BNNTs have much wider band gaps ranging 5.0–6.0 eV [61,62]. There is good agreement between our calculated energy gaps and those calculated previously. The \({E}_{g}\) of AlNNT is larger and smaller than those of CNT and BNNT, respectively, so BNNT > AlNNT > CNT is the order in which the \({E}_{g}\) values of bare nanotubes are decreasing.
The PDOS plot of AlNNT showed that the most contribution is related to the lone pairs of the N and H atoms to the occupied molecular orbitals (included the HOMO) and small contribution is noticed from the Al atoms, while all atoms (N, H, and Al) are contributed to the virtual (unoccupied) molecular orbitals (included the LUMO). The PDOS plots of BN indicate a similar trend to that of AlN in which the N and H atoms are mostly contributed to HOMO and other occupied orbitals, while all atoms (B, H, and N atoms) are contributed to the LUMO and other unoccupied orbitals, whereas the H and C atoms are equally contributed to both orbitals in CNT. This result indicates the effect of atom’s electronegativity on its contribution to HOMOs or LUMOs. Since the least electronegative (electron-deficient) B and Al atoms are contributing to LUMOs more significantly, N atoms—the most electronegative (electron-rich)—are contributing to HOMOs.
Non-covalent interaction, binding energies, and thermodynamics of encapsulated nanotubes
In this work, six different ESs were obtained, and each system was examined individually (Fig. 4). The structural and thermodynamic behavior of the encapsulated FAV forms (E3 and K2) into AlN, BN, and C NTs has been studied to detect the most stable system. Table 2 summarizes the calculated binding energies in aqueous solution, while the results obtained in the gas phase are listed in Table SD3. Since the ES (weakly bonded configuration) involves close intermolecular distances, basis functions from one molecule may inadvertently define the other, a phenomenon known as basis set superposition error (BSSE). Thus, a predicted structure and binding energy will be affected by artificial stabilization. BSSE can be removed by Boys and Bernardi counterpoise correction (CP) [46]. In all cases, the binding energies with CP corrected the BSSE (\({\mathrm{E}}_{\mathrm{bind}}^{\mathrm{BSSE}}\)) are smaller—by not less 72.048 kJ/mol and not more than 101.128 kJ/mol—than those without the BSSE correction (\({E}_{\mathrm{bind}}\)). The differences between the two sets of data are significant and indicate the necessity of including the CP corrected the BSSE.

Optimized geometries, NCI,and RDG plots of the drug molecule FAV and different NTs in water. Note: numbers in parenthesis represent the ranges of vibrational frequencies
All ESs have negative binding energies in both the gas phase and water solvent. The lowest negative binding energy signifies the most favorable contact between the drug and NT and indicates that the drug is strongly encapsulated. In water medium, \({E}_{\mathrm{bind}}^{\mathrm{BSSE}}\) values range from − 67.572 to − 308.821 kJ/mol, while in the gas phase, \({E}_{\mathrm{bind}}^{\mathrm{BSSE}}\) values range from − 95.772 to − 232.965 kJ/mol. Generally, the ESs are more stable in the gas phase than in aqueous medium, the exceptions are K2/ and E3/AlNNTs. Keep in mind that the differences in stabilities of ESs in gas and water are very small and do not exceed 3.012 kJ/mol; however, for the two exceptional systems (K2/ and E3/AlNNTs), their stability in water is much higher than that in the gas phase, for instance, \({E}_{\mathrm{bind}}^{\mathrm{BSSE}}\) − 232.965 and − 308.821 kJ/mol for K2/AlNNT in the gas phase and water, respectively, with a difference equal 75.856 kJ/mol. Therefore, these two systems show a significant extra stability in water medium. The AlNNTs are the most favorable systems in both the gas phase and water and can present higher structural stability. Moreover, the stability of encapsulated BNNTs is greater than CNTs in the gas phase and water.
Since AlNNT has a higher stability, it would accept a higher thermal conductivity and a lower coefficient of thermal expansion [63]; the reason for this may be correlated to the hydrophilicity and polarity of AlNNTs when compared with those of BNNTs and CNTs due to the difference in electronegativities of C, B, and Al atoms. However, it seems that the stability of encapsulated system depends on the tautomeric form of FAV, since K2/NTs is more favorable than E3/NTs. Regarding the relative stability of the ES to each other and since the drug delivery process is carried out in enriched aqueous medium, we will consider the results in this medium only. Inspection of Table 2 indicates that the stability of ESs can be arranged in the following order: K2/AlNNT > E3/AlNNT > K2/BNNT > E3/BNNT > K2/CNT > E3/CNT. For a comparison reason, the binding energy of the most stable ES system (K2/AlNNT) obtained from this study is compared with the binding or adsorption energies (from encapsulation or adsorption on surface mode of interaction, respectively) of FAV drug with different carriers reported literature values (Table 3).
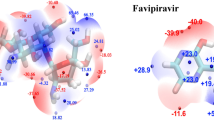
To explain the observed trend in the binding energies of the ESs, the non-covalent interactions (NCI) between the two monomers inside the ES will be applied. The NCI method, also known as reduced density gradients (RDGs), is one of the most commonly used methods to study weak interactions [70]. According to the NCI analysis, an index is derived from the electron density and its derivatives [71]. Colors are assigned to RDGs based on the value of \(\mathrm{sin}{\uplambda }_{2}\uprho\), which is a significant indicator of interaction strength. Dipole–dipole/hydrogen bonding interactions have large, negative values of \(\mathrm{sin}{\uplambda }_{2}\uprho\), steric repulsion has large, and positive values of \(\mathrm{sin}{\uplambda }_{2}\uprho\), while very weak like van der Waals (VW) interactions have values near zero. The plots of RDG and (\(\mathrm{sin\lambda }\) 2 \(\uprho\)) and 3D visual representation of NCIs between the FAV and AlNNTs, BNNTs, and CNTs are shown in Fig. 4. As seen from Fig. 4, the three types of interactions are present inside the NTs, which presented by red, green, and blue spikes. Apparently, AlNNTs possess the most steric effect interaction inside the rings in the structure of NT. Now by a close look at Fig. 4, the VW dominates the interaction between the two forms of FAV and BN and CNTs, indicated by a green circle surrounded the drug form inside the NT. In the case of AlNNT, the VWs are less indicated by the half circle colored region between the FAV form and NT. However, E3/ and K2/AlNNT are the most stable ES indicating that other factor than VW is affecting the stability of these systems such as the H-bond formation as we will discuss afterwards and in Section "QTAIM study". During the encapsulation process, planarity of the K2 and E3 structures inside the NTs did not change, whereas small changes in the bond lengths and bond angles were detected. For example, the H–O bond lengths from 1.001 to 1.032, 1.020, and 1.023 Å for E3/BNNT, E3/AlNNT, and E3/CNT, respectively. The C-O–H angle (105.39°) broads to 107.8°, 107.0°, and 105.7° in CNT, BNNT, and AlNNT system, respectively, to 107.827°. However, the encapsulation of K2 form into AlNNT affects the shape of AlNNT in the way that the proton of the N–H in the K2 form that belongs to the pyrazole ring becomes near to one of the N atoms of AlNNT resulted in a formation of a hydrogen-bond with a bond distance of 1.703 Å indicated by blue colored of this region (blue circle); thus, the stability of K2/AlNNT increases. While the O atom of the carbonyl group has become in front of one of Al atoms of AlNNT which might have repulsive force, this is evident from NCI plot indicated by the red colored of this region (red circle). Due to that, the energy of this encapsulated system significantly decreases and the stability of the system K2/AlNNT overcome those of other ESs. In contrast, a small deformation in the shape of the NT occurs when K2 is encapsulated in BN and CNTs compared to that in the case of K2/AlNNT. In conclusion, the NCI interaction of FAV within AlNNT is most significant due to H-bond formation in K2 form with the tube.
The obtained thermodynamic parameters of the ESs, enthalpy change (\({\Delta H}_{\mathrm{bind}}\)), Gibbs free energy change (\({\Delta G}_{\mathrm{bind}}\)), and entropy change (\({\Delta S}_{\mathrm{bind}}\)) are evaluated in the water medium (Table SD4), and the relative values are listed in Table 4. The values of \({\Delta H}_{\mathrm{bind}}\), \({\Delta G}_{\mathrm{bind}}\), and \({\Delta S}_{\mathrm{bind}}\) are all negative showing that the encapsulation process is favorable. It appears that the \({\Delta G}_{\mathrm{bind}}\) values are less negative compared to those of \({\Delta H}_{\mathrm{bind}}\) values. The negative \({\Delta G}_{\mathrm{bind}}\) and \({\Delta H}_{\mathrm{bind}}\) values indicate that the encapsulation processes in all systems are spontaneous and exothermic, respectively. \({\Delta \mathrm{S}}_{\mathrm{bind}}\) values are all negative, indicating a decrease of disorder of ESs. However, among all systems, E3/AlNNT and K2/AlNNT systems with the maximum binding energies have also the highest negative values of the thermochemical parameters (except \({\Delta \mathrm{S}}_{\mathrm{bind}}\)), followed by K2/ and E3/CNTs, and finally K2/ and E3/BNNT. It can be concluded that the encapsulation of drug into AlNNT systems is more favorable as the process is more spontaneous and more exothermic. It is evident that AlNNT has advanced capabilities as a FAV carrier.
Recovery time
Recovery time (\(\uptau\)) is an important characteristic for detecting drug, and it represents how difficult it is for drugs to unbind from the drug carrier. It should be neither too short nor too long to facilitate drug desorption processes. Also, it can be calculated experimentally, by exposing the sample of drug to the UV–Vis light or by heating it to higher temperatures [72,73]. The following formula, based on the transition state theory, was used to calculate the recovery time for the encapsulation process:
where \({v}_{0}\) is the attempt frequency, \({K}_{B}\) is the Boltzmann constant (8.318 × 10−3 kJ/mol.K), and T is the temperature. Since \({E}_{\mathrm{bind}}^{\mathrm{BSSE}}\) serves as a barrier for unbinding, therefore, they are identical, and it was used in this equation. As can be noted from the above equation, recovery time is mostly influenced by unbinding barrier potential and light responsiveness for drug release, while temperature has a little impact, i.e., as the detection and unbinding processes of drug are easier as the recovery time becomes shorter. In this study, and to analyze the effect of those factors on recovery time, the attempt frequency was set to \({10}^{16}\), \({10}^{14}\), and \({10}^{12}\) Hz at ultraviolet (UV), visible (Vis), and infrared (IR) regions, respectively. Since COVID-19 leads to an increase in temperature of patient human body, the recovery time for FAV into different NTs is calculated at 310.15 (37 °C, normal body temperature) and 313.15 K (40 °C, a temperature the human body can reach upon fever) and listed in Table 1.
The more negative the binding energy value, the longer the recovery time [74]. In all cases, the recovery time of E3 form/NTs is much shorter than those of K2/NTs, which means the unbinding process (drug release) of E3 form from NTs is easier than K2 form. At 310.15 K, the shortest recovery time, less than 2 s, correspond to E3/BNNT, K2/CNT, and E3/CNT in UV region in which they have the least negative binding energies (− 96 to − 67 kJ/mol). On the other hand, raising temperature to 313.15 K caused a slight reduction in the recovery time by about 0.50–1.36 s in the same region. However, K2/BNNT provided recovery time of about 6 and 4 h (corresponding to 2.10 \(\times {10}^{4}\) and 1.34 s\(\times {10}^{4}\)) in UV region at 310.15 and 313.15, respectively. Thus, the unbinding process for K2/BNNT will be difficult in comparison to E3/BNNT, K2/CNT, and E3/CNT, which make these systems suitable for repeated use many times; this result supports the superiority of such NTs as drug carriers as also evident in [17]. In contrast, the unbinding process for AlNNTs is predicted to be extremely slow and incredibly difficult due to the very long recovery time they recorded, since their binding energy is equal and less negative than − 167.611 kJ/mol.
Based on above results, our suggestion here is that the ES can perform better if there is a balance between the two values: (1) the binding energy and (2) the recovery time. Since the binding energy should have been an appropriate value, not too large because it will be combined by longer recovery time means the drug cannot escape from the system to the target human cells, and not too small (although this will be combined by shorter recovery time and thus successful drug releasing process), since it may be insufficient to bind the drug firmly inside the NT. Let us assume here, to obtain a suitable recovery time (less than 10 s), the binding energy should be equal or less negative than \(\approx\)− 96 kJ/mol in the UV region or − 68 kJ/mol in Vis and IR regions.
Additionally, similar results are found in some studies. Asgari et al. [31] used M05 and B3LYP functionals to study FAV adsorption on pristine and Si-doped graphyne NTs. The binding energies of FAV@Si-doped graphyne were − 67.781 kJ/mol by B3LYP and − 84.517 kJ/mol by M05 DFT functional. It was noted that the calculated recovery time in IR region produced by B3LYP is better than M05 functional which be \(\approx\) 10 s and \(\approx\) 0.03 s at 273 and 473 K, respectively. According to other work [75], favipiravir’s recovery time from polyamidoamine and polyester surfaces is 9.2 \({\times 10}^{4}\) and 4.2 s\({\times 10}^{4}\), respectively, at UV region and 298 K. The longer recovery time is a result of their adsorption energies of − 114.05 and − 112.13 kJ/mol using M06-2X/6-31G(d,p). Also, in study of Gholami et al. [76], when the adsorption energies ranged from \(\approx\) ‒ 20 to ‒ 10 kcal/mol, they provide recovery time from 0.001 to 0.3 s at NIR (780 to 1700 nm) in body temperature, 310.15 K.
Electronic properties
To understand the structure and reactivity of considered systems, the frontier molecular orbitals included HOMO, LUMO, and \({E}_{g}\), and some quantum chemical parameters included chemical potential (μ), global hardness (η), and the electrophilicity index (ω) for the FAV forms, and bare and encapsulated nanotubes are calculated and listed in Table 5.
The HOMO energies of bare nanotubes are in the range from ‒ 4.068 to ‒ 6.276 eV, while the LUMO energies are between ‒ 0.530 and ‒ 3.373 eV confirming the obtained data from PDOS and TDOS plots (see Fig. 3 and Section "Optimized geometries and density of states of nanotubes"). The HOMO levels of NTs become destabilized (become less negative), while LUMO levels become significantly stabilized (more negative) after the encapsulation process. The destabilization of HOMOs leads to smaller ionization potentials (IP; \(\mathrm{IP}=-{\mathrm{E}}_{\mathrm{HOMO}}\)) and hence easier transfer of electrons due to encapsulation, while the stabilization of LUMOs due to encapsulation leads to larger electron affinities (EA; \(\mathrm{EA}=-{\mathrm{E}}_{\mathrm{LUMO}}\)), and thus, the electron acceptance ability will increase. On the other hand, the values of \({E}_{g}\) of AlNNT, BNNT, and CNT equal 4.644, 5.735, and 0.695 eV, respectively, and reduced considerably due to encapsulation of E3 and K2 forms of FAV drug; similar observation was found in literature [16,61,62]; the difference is not less than 1.11, 2.33, and 0.08 eV, respectively. However, since the current study used the zig-zag tube (10,0), \({E}_{g}\) values of BNNT and CNT are compared to those of the armchair tubes (5.5), (5.81), and (0.41) eV, respectively, of the study [80]. In other words, the decrease in energy gaps can be expressed by the term \(\left(\frac{\Delta {E}_{g}}{{E}_{g}}\right)\) (see Table 5). \(\left(\frac{\Delta {E}_{g}}{{E}_{g}}\right)\) values are in the range from 0.111 to 0.488 eV. The electrical conductivity of nanostructured is related to its energy gap through the following equation:
where \(\upsigma\) is the electrical conductivity and \({K}_{B}\) is the Boltzmann’s constant). NT with smaller energy gap has more tendency to transfer electrons across the conduction band and hence better electrical conductivity. Therefore, the encapsulated NTs have much better electrical conductivity than bare NTs. The higher value of energy gap belongs to the E3/BNNT encapsulated system, and the smaller value is for K2/CNT. The encapsulation process reduces the \({E}_{g}\) values of E3 and K2 forms. The differences in the \({E}_{g}\) values of E3 form and AlNNT, BNNT, and CNT are 1.541, 1.143, and 3.899 eV, respectively, while the encapsulation of K2 into AlNNT, BNNT, and CNT decreases the \({E}_{g}\) value by 1.413, 1.037, and 3.364 eV, respectively. As for the case of isolated NTs, similar conclusion can be drawn; the electrical conductivities of ESs were enhanced because of reduction in their energy gaps in comparison to those of isolated FAV forms.
The energy gap also describes the softness/hardness, chemical potential [78], electrophilicity, and dipole moment. A significant conclusion arises from the dipole moment data. Table 5 shows the dipole moment values of bare NTs and their systems. Since the drug delivery process occurs in the high polar water solvent, larger dipole moment of ESs resulted in better solubility and hence better properties of the carrier. The encapsulation of E3 form into AlNNT and BNNT resulted in the elevation of dipole moments by 0.144 and 0.726 Debye, while the encapsulation of K2 form into AlNNT and BNNT resulted in noticeable reduction of dipole moments by 5.430 and 3.364 Debye. On the other hand, the encapsulation of E3 and K2 into CNTs resulted in a significant increase in the dipole moments compared to their isolated CNTs. Hence, the E3/AlNNT, E3/BNNT, E3/CNT, and K2/CNT have high polarizability and thus high solubility in polar solvent (water) compared to the remaining ESs (K2/AlNNT and K2/BNNT).
The ESs are much softer/less hard than their isolated monomers (Table SD2 and Table 5). Furthermore, the trend observed for the variation of global hardness values when compared to isolated NTs is the same as that observed for energy gaps, confirming Pearson’s maximum hardness rule [77]. In Fig. SD1, a linear relationship with an excellent correlation coefficient (\({R}^{2}=1.000\)) is shown between these two sets of data. Upon encapsulation, the system K2/AlNNT has become softer than the bare AlNNT, and the other systems—except for CNT systems—indicate a better reactivity which is in a good agreement with Muz et al. [81]. The ESs can be arranged in order of increasing/decreasing in their softness/hardness values, respectively, as follows: K2/CNT > E3/CNT > K2/AlNNT > K2/BNNT > E3/AlNNT > E3/BNNT.
The electrophilicity index (ω) as suggested by Parr et al. [79] is a measure of energy reduction due to the maximal electron flow between donors and acceptors. The higher values of ω, the higher electrophilicity of the geometry. The values of electrophilicity index of all considered systems are larger in comparison to those of the bare NTs. The electrophilicity indices of the bare NTs are 3.138, 2.013, and 19.906 eV for AlNNT, BNNT, and CNT, respectively, while due to the encapsulation process, these values increased. Since the electrophilic indices for AlNNT and BNNT ESs are in the range from 6.316 to 8.384 eV, for CNT ESs, they exceeded 21.616 eV. On the other hand, the electrophilicities of E3 inside AlN and BN NTs are smaller than those of K2 inside the same NTs, while the opposite is true in the case of CNTs.
The more the negative value of chemical potential (µ) the more the stability is, and among the three nanotubes, AlNNT has the most negative value of (\(\upmu\) =\(-\) 3.818 eV). However, the encapsulation affects the µ values in the way they become more negative for AlNNT and BNNT ES (ranging between − 4.746 and − 4.577 eV) and less negative for CNT systems and equal \(\sim\)‒ 3.6 eV. The stability of CNTs encapsulated with E3 and K2 forms is smaller than those of other encapsulated NTs; this is maybe beneficial for the drug release (drug unbinding) process from the NTs (see Section "Recovery time").
Ultraviolet absorption spectra
ESs and FAV drugs were examined for their excited state properties using UV and Vis spectra. Drug concentrations in the body must be monitored, and the appropriate carrier must be developed to ensure that the drug molecules are delivered effectively without causing toxic side effects [82]. UV–Vis detectors can be used as a continuous monitors in drug delivery process, and this is done by measuring the amount of light absorbed and wavelength position [83]. For a deeper understanding of the electronic absorption of FAV two forms, bare NTs, and ESs in aqueous medium, the calculations are performed with the TD-CAM-B3LYP-D/6-31G(d,p) and are listed in Table 6.
The absorption peaks of FAV forms were observed at 93.73 and 111.08 nm, with oscillator strengths 0.3725 and 0.356 for E3 and K2, respectively. The bare NTs showed sharpest peaks, located at wavelengths 197.13,173.56, and 599.78 nm with 1.265, 5.983, and 2.316 of oscillator strengths, for AlNNT, BNNT, and CNT, respectively. Among the three NTs, AlNNT exhibits the intense absorption at 197.13 nm in the UV region (100–300 nm) which is comparable with a theoretical study in [81] (188–194 nm).
After encapsulation, the absorption peaks of the systems become much longer when compared to those of the bare NTs, except for CNT systems. Hence, the encapsulation causes a blue shift for CNT only, while it causes a redshift for both AlNNT and BNNT. For instance, the encapsulation of CNT with E3 and K2 shifted the absorption from 599.78 nm to 404.24 and 407.44 nm for E3/CNT and K2/CNT, respectively. In contrast, the absorption of K2/AlNNT and K2/BNNT at wavelengths of 341.01 and 180.40 nm is longer than that of bare AlNNT (197.13 nm) and bare BNNT (173.56 nm), respectively. Moreover, E3/AlNNT and E3/BNNT contain transitions at 300.99 and 184.62 nm with oscillator strength equal to 0.0815 and 0.1613, respectively. The encapsulation of NTs—except for BNNT—with K2 resulted in that the main absorption peaks are shifted to longer wavelengths \({\uplambda }_{\mathrm{max}}\) when compared to the encapsulation of E3. Therefore, the significant modification of the absorption spectra (red/blueshift) of ESs compared to their monomers can be used to online monitoring of the concentration of FAV forms in the human body.
QTAIM study
QTAIM was carried out at the bond critical point (BCP), to reveal the nature of the intermolecular interactions in the E3/AlNNT, K2/AlNNT, E3/BNNT, K2/BNNT, E3/CNNT, and K2/CNNT complexes. The topological parameters such as electron density at the BCP (\({\uprho }_{\mathrm{BCP}}(r)\)), Laplacian of the electron density (\({\nabla }_{\uprho }^{2}(r)\)), total energy density (H(r)), kinetic energy density (K(r)), and potential energy density (V(r)) can be used to identify the nature of the intermolecular interactions with the encapsulated drugs. In addition, the strength of the intermolecular hydrogen bond (EHB = -V(r)/2) can be also estimated. The molecular graphs of the E3 form of the FAV drug encapsulated inside the bulk of the AlNNT, BNT, and CNT are graphically shown in Fig. 5. Careful inspection of Fig. 5 shows that there are several important bond critical points due to the interactions between the FAV and the NTs. In this study, the intermolecular hydrogen bonds are investigated. The topological parameters of the selected interactions are summarized Table 7.

Optimized structures (left column in a and b) and molecular graphs (right column in a and b) of E3/K2, E3/K2 and E3/K2 inside AlNNT, BNNT, and CNT. The superimposed numbers correspond to the most important intermolecular interactions. Details of these interactions are given in Table 7
As known, the value of \({\rho }_{(r)}\) at the BCP indicates the strength of a chemical bond. Previous studies reported that the system with higher value of \({\rho }_{(r)}\) has more interaction and vice versa. Our results reveal that several intermolecular hydrogen bonds have been detected in the investigated encapsulated systems, for example, three intermolecular hydrogen bonds in case of E3/AlNNT system and one intermolecular hydrogen bond in case of K2/AlNNT, K2/BNNT, and E3/BNNT. There is no any intermolecular hydrogen bond in case of K2/CNT and E3/CNT systems. As can be seen in Table 7, the enol form E3 bounds with the AlNNT and BNNT by three and one intermolecular hydrogen bonds, respectively, whereas the keto form K2 bounds with the AlNNT and BNNT systems with only one intermolecular hydrogen bond. Additionally, the intermolecular hydrogen bonds that result between the E3 form and the AlNNT are weaker than those in case of BNNT, indicating that these interactions can be easily recovered. In addition, it is found that the interactions between the keto form and the NTs are stronger than those in case of E3 form, which is similar to the study [84]; they found that the interactions between acetaminophen (ACM) and Fe-BN nanocones (FBN) are stronger as keto/thio than enol/thiol interactions.
Among all the encapsulated systems under probe, the strongest interactions between the NTs and the FAV drug belong to the K2/AlNNT with electron density (\({\rho }_{(r)}\)) of 0.0567, a dO-H bond distance of 1.704 Å and EHB of 53.67 kJ/mol, while the weakest interactions correspond to E3/AlNNT with (\({\rho }_{(r)}\)) = 0.0076, dO-H = 2.777 Å, and EHB of 5.16 kJ/mol. Except to K2/AlNNT system, the positive values of both \({\nabla }_{\uprho (\mathrm{r})}^{2}\) and H(r) suggest the intermediate interactions. Another important descriptor is − G(r)/V(r), which can be used to categorize the nature of the interactions between the drug and the NTs as follows: (1) 0.5 < − G(r)/V(r) < 0 → covalent interactions, (2) 1 < − G(r)/V(r) < 0.5 → electrostatic and partially covalent, and (3) − G(r)/V(r) > 1.0 → non-covalent interaction. As reported in Table 7, except to K2/AlNNT, the − G(r)/V(r) values are ≥ 1.0, suggesting that the interaction is a weak non-covalent interaction. These findings strongly suggest that the delivery of the E3 form of the E3/AlNNT is much easier than that of the other NTs, and the recovery of E3 form is much faster than the K2 form. These results agree with the above-mentioned results of the recovery time. The findings from QTAIM support to a great extent the values obtained for the binding energies of ESs. Wherever there is a strong intermolecular interaction between the drug form and NT (especially the intermolecular H-bonding), the ES will be combined with higher binding energy and thus larger stability. In other words, the most stable ESs consist of K2/ and E3/AlNNT and /BNNT combined by one/more intermolecular H-bonds, while the least stable ESs consisting of K2 & E3/CNT show no evidence of this type of H-bonds.
Molecular docking simulations
The molecular docking technique is used to predict the binding affinity between drugs and target proteins at the atomic level. This study carried out using Autodock software [85] more than one protein, since three target proteins of SARS-CoV2 were studied. The investigated target proteins included RNA binding domain of nucleocapsid protein (6M3M) [86], papain-like protease (6W9C) [87], and main protease (6LU7) [87]. Their crystal structures were downloaded from the Worldwide Protein Data Bank (https://www.rcsb.org). The main role of 6M3M protein is to facilitate transcribing and replicating subgenomic viruses, while 6W9C and 6LU7 proteins have a basic responsibility to cleave nitrogen and carbon termini of polyproteins into a functional product, respectively. The two forms of FAV drug were docked with the three selected proteases from SARS-CoV2 infection. The structures of 6M3M, 6W9C, and 6LU7 proteins were formed by removing all water molecules and ligands; activity sites of protein were predicted by Pymol software [88]. In order to validate the docking protocol, ligands were removed from the targets; then, they were re-docked. PyMOL software was used to superimpose the re-docked complexes onto the native complexes and determine the root mean square deviation (RMSD). It found that RMSD between of re-docking complexes and native complexes are ≤ 2.14 Å. For the purpose of demonstrating the docking procedure, K2-6M3M is taken as an example. The binding energy of re-docking K2-6M3M is − 19.163 kJ/mol, and RMSD is 0.13 Å with a difference of 0.970 kJ/mol and 0.13 Å between re-docking and native complex. Figure 6 illustrates the superimposed 2-dimensional structure.

Superimposition of re-docked K2-6M3M (green) onto native complex (orange) in the active site using PyMOL
Table 8 and Fig. 7 present the obtained binding energies, root mean square deviation (RMSD), the number of hydrogen bonds, and residues of the targets interacting with the FAV forms. The RMSD describes the displacement of atoms at a point in time relative to a reference structure, which is usually the first frame of the simulation [89]. The obtained RMSD results showed no deviation in the protein. Discover Studio 2021 from Biovia was used to visualize the 2-dimensional ligand interaction with the protein.

Molecular docking simulations of the interaction between K2 and E3 forms and the selected proteins
The calculated binding energies between K2/E3 forms and 6M3M protein are significantly high and equal − 18.912 and − 18.033 kJ/mole, respectively. As shown in Fig. 7, five H-bonding is found between the two H atoms of the amino group (NH2) of the K2 and 6M3M by two active sites which are GLN71 and ALA135 residues. While the E3 form interacts with the O atom and the hydroxyl group (OH) to PHE67, while amine to GLN71 of 6M3M protein.
On the other hand, the binding energies of K2/E3 with 6W9C are − 12.217 and − 21.254 kJ/mol, respectively. One active site was found for K2 form to interact with the O atoms of residues in 6W9C: TYR72. In contrast, three active sites for E3 interact with ASN13, TYR56, and ALA131. The binding energy of K2/E3 exhibited the least values with 6LU7 protein, and they are − 11.966 and − 17.238 kJ/mol, respectively. The H atom of the amino group in K2 interacts with the N atom of LEU282 and PHE3 residues, while E3 interacts with four active sites GLN74, PHE66, VAL77, and HIS64 residues. In conclusion, among the two FAV forms, E3 exhibited better binding energy (− 21.254 kJ/mol) in comparison to K2; hence, E3 might be more anti-COVID19. Based on these findings, the feasibility of FAV in the prevention and treatment of SARS-CoV2 should be further investigated.
The calculated binding energies between K2/E3 forms and 6M3M protein are significantly high and equal − 18.912 and − 18.033 kJ/mole, respectively. As shown in Fig. 7, two H-bonding are found between the two H atoms of the amino group (NH2) of the K2 and 6M3M by two active sites which are GLN71 and ALA135 residues with bond lengths of 2.194 and 1.747 Å, respectively, while the E3 form interacts with the O atom to GLN71 and ARG69 and with the hydroxyl group (OH) to TYR124 of 6M3M protein with bond lengths of 1.734, 1.859, and 1.956 Å, respectively.
On the other hand, the binding energies of K2/E3 with 6W9C are − 12.217 and − 21.254 kJ/mol, respectively. One active site was found for K2 form to interact with the O atoms of residues in 6W9C: ILE14 with a bond distance of 2.173 Å for K2. In contrast, three active sites for E3 interact with ASP76 and LEU80 by bond lengths 1.971, 2.014, and 2.039 Å, respectively. The binding energy of K2/E3 exhibited the least values with 6LU7 protein, and they are − 11.966 and − 17.238 kJ/mol, respectively. The H atom of the amino group in K2 interacts with the N atom of LYS5 residues with a bond length of 2.068 Å, while E3 interacts with two active sites TRP207 and LEU282 residues at 2.051 and 2.103 Å, respectively. In conclusion, among the two FAV forms, E3 exhibited better binding energy (− 21.254 kJ/mol) in comparison to K2; hence, E3 might be more anti-COVID19. Based on these findings, the feasibility of FAV in the prevention and treatment of SARS-CoV2 should be further investigated.
Conclusions
Using DFT calculations, at B3LYP-D/6-31G(d,p) and TDCAM-B3LYP-D/6-31G(d,p) levels of theory in gas and with CPCM in water, the FAV drug isomers and tautomers were studied. The feasibility of three NTs (CNT, BNNT, and AlNNT) was proved from their interaction energies. The most stable tautomeric forms (E3 and K2) were encapsulated into these NTs. The following conclusions can be drawn:
-
Among the six ESs, encapsulation inside AlNNT resulted the most stable ESs in both gas and water; AlNNT provides the highest binding energies (using CP correcting BSSE), the most spontaneous and exothermic process, followed by the process inside BNNT and then CNT.
-
In comparison to both bare NTs and drug forms, the encapsulated NTs have much lower energy gaps and thus better electrical conductivity, are softer, and thus have better reactivity and better electron-donating and accepting abilities due to the destabilization and stabilization of HOMO and LUMO levels, respectively.
-
The stability of CNTs encapsulated with E3 and K2 forms is smaller than those of other encapsulated NTs as evident from the chemical potential values; this is supporting the drug release process in this type of NTs.
-
Recovery times of \(\sim\) 10 s put a boundary limit on binding energies. Since the binding energies should be \(\le\)− 96 kJ/mol in the UV region or \(\le\)− 68 kJ/mol in Vis and IR regions. This results not in the advantage of AlNNTs because they have much larger binding energies and extremely longer recovery times. On the other hand, this result validates CNTs as better carriers, followed by BNNT (recovery times reached days), those allow better drug release process over AlNNT.
-
In all cases, the recovery times of E3 form/NTs are much shorter than those of K2/NTs, which means the unbinding process (drug release) of E3 form from NTs is easier than K2 form.
-
The absorption of the bare NTs shifted after encapsulation from the UV region to the Vis region in the range 300–407 nm. Thus, ESs have different absorption properties compared to the bare NTs, and thus, monitoring the drug concentration in human body through UV–Vis spectroscopic technique is feasible.
Finally, molecular docking simulations demonstrated a good inhibitory effect of FAV forms (K2 and E3) over SARS-CoV-2, implicating FAV drug resistance to SARS-CoV-6M3M/6LU7/6W9C proteases, and E3 exhibits better inhibitory activity than K2 form.
Availability of data
The authors confirm that the data supporting this study’s findings are available within the article and its supplementary data.
References
Kumari A et al (2014) Nanoencapsulation for drug delivery EXCLI journal 13:265
Makiabadi B, Zakarianezhad M, Hosseini SS (2021) Investigation and comparison of pristine/doped BN, AlN, and CN nanotubes as drug delivery systems for Tegafur drug: a theoretical study. Struct Chem 32(3):1019–1037
Mirsalari H et al (2021) Investigation of the pristine and functionalized carbon nanotubes as a delivery system for the anticancer drug dacarbazine: drug encapsulation. J Pharm Sci 110(5):2005–2016
Soliman KA, Aal SA (2021) Theoretical investigation of favipiravir antiviral drug based on fullerene and boron nitride nanocages. Diam Relat Mater 117:108458
Wazzan N, Soliman KA, Halim W (2019) Theoretical study of gallium nitride nanocage as a carrier for 5-fluorouracil anticancer drug. J Mol Model 25(9):1–19
Vatanparast M, Shariatinia Z (2018) AlN and AlP doped graphene quantum dots as novel drug delivery systems for 5-fluorouracil drug: theoretical studies. J Fluorine Chem 211:81–93
Al-Zuhairy SA et al (2022) Study to molecular insight into the role of aluminum nitride nanotubes on to deliver of 5-fluorouracil (5FU) drug in smart drug delivery. Inorg Chem Commun 142:109617
Saleh RO et al (2022) Application of aluminum nitride nanotubes as a promising nanocarriers for anticancer drug 5-aminosalicylic acid in drug delivery system. J Mol Liq 352:118676
Nafiu S et al (2022) Boron nitride nanotubes for curcumin delivery as an anticancer drug: a DFT investigation. Appl Sci 12(2):879
Rubio A, Corkill JL, Cohen ML (1994) Theory of graphitic boron nitride nanotubes. Phys Rev B 49(7):5081
Chopra NG et al (1995) Boron nitride nanotubes. science 269(5226):966–967
Vatanparast M, Shariatinia Z (2019) Hexagonal boron nitride nanosheet as novel drug delivery system for anticancer drugs: insights from DFT calculations and molecular dynamics simulations. J Mol Graph Model 89:50–59
Shakerzadeh E et al (2022) Pristine and alkali and alkaline Earth metals encapsulated B36N36 nanoclusters as prospective delivery agents and detectors for 5-fluorouracil anticancer drug. Appl Organomet Chem 36(7):e6721
Bououden W et al (2021) Surface adsorption of crizotinib on carbon and boron nitride nanotubes as anti-cancer drug carriers: COSMO-RS and DFT molecular insights. J Mol Liq 338:116666
Gholami A, Shakerzadeh E, Anota EC (2023) Exploring the potential use of pristine and metal-encapsulated B36N36 fullerenes in delivery of β-lapachone anticancer drug: DFT approach. Polyhedron 116295
Zhao M et al (2003) Stability and electronic structure of AlN nanotubes. Phys Rev B 68(23):235415
Hassanpour A et al (2022) Dopamine drug adsorption on the aluminum nitride single-wall nanotube: ab initio study. Arab J Sci Eng 47(1):477–484
Shi F et al (2014) Synthesis and crystal structure of 6-fluoro-3-hydroxypyrazine-2-carboxamide. Drug Discoveries & Therapeutics 8(3):117–120
Dabbous HM et al (2021) Efficacy of favipiravir in COVID-19 treatment: a multi-center randomized study. Adv Virol 166(3):949–954
Cai Q et al (2020) Experimental treatment with favipiravir for COVID-19: an open-label control study. Engineering 6(10):1192–1198
Joshi S et al (2021) Role of favipiravir in the treatment of COVID-19. Int J Infect Dis 102:501–508
Doi Y et al (2020) A prospective, randomized, open-label trial of early versus late favipiravir therapy in hospitalized patients with COVID-19. Antimicrob Agents Chemother 64(12):e01897–e1920
Kamali A et al (2023) Evaluation of the effect of favipiravir on patients with COVID-19. J Family Med Prim Care 12(2):242–245
McKee DL et al (2020) Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol Res 157:104859
Furuta Y et al (2013) Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antiviral Res 100(2):446–454
Rocha-Pereira J et al (2012) Favipiravir (T-705) inhibits in vitro norovirus replication. Biochem Biophys Res Commun 424(4):777–780
Julander JG et al (2009) Activity of T-705 in a hamster model of yellow fever virus infection in comparison with that of a chemically related compound, T-1106. Antimicrob Agents Chemother 53(1):202–209
Morrey JD et al (2008) Efficacy of orally administered T-705 pyrazine analog on lethal West Nile virus infection in rodents. Antiviral Res 80(3):377–379
Zhang T et al (2017) Recent progress on the treatment of Ebola virus disease with Favipiravir and other related strategies. Bioorg Med Chem Lett 27(11):2364–2368
Safronetz D et al (2015) The broad-spectrum antiviral favipiravir protects guinea pigs from lethal Lassa virus infection post-disease onset. Sci Rep 5(1):1–11
Asgari MA, Bahmani N (2022) Synergistic effect of Si-doping and Fe2O3-encapsulation on drug delivery and sensor applications of γ-graphyne nanotube toward favipiravir as an antiviral for COVID-19: A DFT study. J Indian Chem Soc 99(9):100666
Pari AA, Yousefi M (2022) Interactions between favipiravir and a BNC cage towards drug delivery applications. Struct Chem 33(1):159–167
Bibi S et al (2021) Metal doped fullerene complexes as promising drug delivery materials against COVID-19. Chem Pap 75(12):6487–6497
Frisch MJ (2009) gaussian09. https://www.gaussian.com
Dennington R, Keith T, Millam JG (2009) Version 5. Semichem Inc., Shawnee Mission, KS, USA
Kohn W, Becke AD, Parr RG (1996) Density functional theory of electronic structure. J Phys Chem 100(31):12974–12980
Abbas G et al (2022) Synthesis and investigation of anti-COVID19 ability of ferrocene Schiff base derivatives by quantum chemical and molecular docking. J Mol Struct 1253:132242
Becke AD (1993) Density‐functional thermochemistry. III. The role of exact exchange. J Chem Phys 98(7):5648–5652
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37(2):785–789
Hazrati MK, Javanshir Z, Bagheri Z (2017) B24N24 fullerene as a carrier for 5-fluorouracil anti-cancer drug delivery: DFT studies. J Mol Graph Model 77:17–24
Yanai T, Tew DP, Handy NC (2004) A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem Phys Lett 393(1):51–57
Petrushenko IK, Petrushenko KB (2018) Effect of methyl substituents on the electronic transitions in simple meso-aniline-BODIPY based dyes: RI-CC2 and TD-CAM-B3LYP computational investigation. Spectrochim Acta Part A Mol Biomol Spectrosc 190:239–245
Yanai T, Tew DP, Handy NC (2004) A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem Phys Lett 393(1–3):51–57
Cossi M et al (2003) Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J Comput Chem 24(6):669–681
Barone V, Cossi M (1998) Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J Phys Chem A 102(11):1995–2001
Boys SF, Bernardi F (1970) The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys 19(4):553–566
O'boyle NM, Tenderholt AL, Langner KM (2008) Cclib: a library for package‐independent computational chemistry algorithms. J Comput Chem 29(5):839–845
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33(5):580–592
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14(1):33–38
Matta CF, Boyd RJ (2007) An introduction to the quantum theory of atoms in molecules. The quantum theory of atoms in molecules: from solid state to DNA and drug design
Bader RF (1985) Atoms in molecules. Acc Chem Res 18(1):9–15
Bader RF (1991) A quantum theory of molecular structure and its applications. Chem Rev 91(5):893–928
Antonov L (2020) Favipiravir tautomerism: a theoretical insight. Theoret Chem Acc 139(8):1–7
Harismah K, Mirzaei M (2020) Favipiravir: structural analysis and activity against COVID-19. Adv J Chem B 2(2):55–60
Assis LC et al (2021) Theoretical insights into the effect of halogenated substituent on the electronic structure and spectroscopic properties of the favipiravir tautomeric forms and its implications for the treatment of COVID-19. RSC Adv 11(56):35228–35244
Bredas J-L (2014) Mind the gap! Mater Horiz 1(1):17–19
Beheshtian J, Ravaei I (2016) Hydrogen storage by BeO nano-cage: a DFT study. Appl Surf Sci 368:76–81
Wu W et al (2011) Electronic and magnetic properties and structural stability of BeO sheet and nanoribbons. ACS Appl Mater Interfaces 3(12):4787–4795
Wazzan N, Soliman KA, Halim WSA (2019) Theoretical study of gallium nitride nanocage as a carrier for 5-fluorouracil anticancer drug. J Mol Model 25(9):265
Zerenturk A, Berber S (2013) Stability of GaN nanocages. Europhys Lett 103(1):16003
Zhao J-X, Dai B-Q (2004) DFT studies of electro-conductivity of carbon-doped boron nitride nanotube. Mater Chem Phys 88(2–3):244–249
Dai BQ, Zhang GL, Zhao JX (2003) A DFT/B3LYP computational study of boron-nitride nanotubes. J Chin Chem Soc 50(3B):525–528
Yim W et al (1973) Epitaxially grown AlN and its optical band gap. J Appl Phys 44(1):292–296
Hasan MM et al (2022) The computational quantum mechanical investigation of the functionalized boron nitride nanocage as the smart carriers for favipiravir drug delivery: a DFT and QTAIM analysis. J Biomol Struct Dyn 40(23):13190–13206
Ibrahim MA et al (2023) Investigation of aluminum nitride nanocarrier for drug delivery process of favipiravir: a DFT study. J Mol Liq 21209
Bazyari-Delavar S et al (2021) PAMAM and polyester dendrimers as favipiravir nanocarriers: a comparative study using DFT method. J Nanopart Res 23:1–12
Muz İ, Göktaş F, Kurban M (2022) A density functional theory study on favipiravir drug interaction with BN-doped C60 heterofullerene. Physica E 135:114950
Alver Ö et al (2019) DFT/QTAIM analysis of favipiravir adsorption on pristine and silicon doped C20 fullerenes. Main Group Met Chem 42(1):143–149
Harismah K et al (2022) Favipiravir attachment to a conical nanocarbon: DFT assessments of the drug delivery approach. Comput Theor Chem 1216:113866
Johnson ER et al (2010) Revealing noncovalent interactions. J Am Chem Soc 132(18):6498–6506
Contreras-García J et al (2011) NCIPLOT: a program for plotting noncovalent interaction regions. J Chem Theory Comput 7(3):625–632
Li J et al (2003) Carbon nanotube sensors for gas and organic vapor detection. Nano Lett 3(7):929–933
Bagheri R et al (2018) Si-doped phagraphene as a drug carrier for adrucil anti-cancer drug: DFT studies. Inorg Chem Commun 90:8–14
Yang H et al (2019) Adsorption behavior of nucleobases on doped MoS2 monolayer: a DFT study. The Journal of Physical Chemistry C 123(51):30949–30957
Bazyari-Delavar S et al (2021) PAMAM and polyester dendrimers as favipiravir nanocarriers: a comparative study using DFT method. J Nanopart Res 23(10):231
Gholami A, Shakerzadeh E, Anota EC (2023) Exploring the potential use of pristine and metal-encapsulated B36N36 fullerenes in delivery of β-lapachone anticancer drug: DFT approach. Polyhedron 232 116295
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105(26):7512–7516
Pearson RG (1992) The electronic chemical potential and chemical hardness. J Mol Struct (Thoechem) 255:261–270
Parr RG, Szentpály LV, Liu S (1999) Electrophilicity index. J Am Chem Soc 121(9):1922–1924
Juárez AR et al (2017) Stability and electronic properties of armchair boron nitride/carbon nanotubes. Fullerenes, Nanotubes, Carbon Nanostruct 25(12):716–725
Muz İ, Kurban H, Kurban M (2021) A DFT study on stability and electronic structure of AlN nanotubes. Mater Today Commun 26:102118
Bibi S et al (2022) Investigation of the adsorption properties of gemcitabine anticancer drug with metal-doped boron nitride fullerenes as a drug-delivery carrier: a DFT study. RSC Adv 12(5):2873–2887
Hosseinzadeh E, Foroumadi A, Firoozpour L (2023) A DFT study on the transition metal doped BN and AlN nanocages as a drug delivery vehicle for the cladribine drug. J Mol Liq 374:121262
Saadh M et al (2023) Sensing functions of an iron-doped boron nitride nanocone towards acetaminophen and its thio/thiol analogs: A DFT outlook. Diam Relat Mater 133:109749
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461
Kang S et al (2020) Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm Sin B 10(7):1228–1238
Jin Z et al (2020) Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582(7811):289–293
Seeliger D, de Groot BL (2010) Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des 24(5):417–422
Martínez L (2015) Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis. PLoS ONE 10(3):e0119264
Acknowledgements
This project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under grant No. (KEP-PhD: 93-247-1443); the authors, therefore, gratefully acknowledge DSR technical and financial support. The authors gratefully acknowledge King Abdulaziz University’s High-Performance Computing Centre (Aziz Supercomputer) (http://hpc.kau.edu.sa) for assisting the calculations for the work of this paper.
Funding
This project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under grant No. (KEP-PhD: 93–247-1443); the authors, therefore, gratefully acknowledge DSR technical and financial support.
Author information
Authors and Affiliations
Contributions
R. A. conceptualized, designed, computed, analyzed, and interpreted these data, and drafted/wrote the manuscript related to this research as part of her PhD’s thesis, and N. W. has been a supervisor of this study and provided guidance on the concept, design, and draft development, and Z. S. and O. S. A. are collaborators and authors of the present work. All authors have participated in (a) conception and design or analysis and interpretation of the data, (b) drafting the article or revising it critically for important intellectual content, and (c) approval of the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Albarakati, R., Al-Qurashi, O., Safi, Z. et al. A dispersion-corrected DFT calculation on encapsulation of favipiravir drug used as antiviral against COVID-19 into carbon-, boron-, and aluminum-nitride nanotubes for optimal drug delivery systems combined with molecular docking simulations. Struct Chem 35, 277–295 (2024). https://doi.org/10.1007/s11224-023-02182-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-023-02182-4