
Abstract
Nipah virus (NiV) is a high-lethality RNA virus from the family of Paramyxoviridae and genus Henipavirus, classified under Biosafety Level-4 (BSL-4) pathogen due to the severity of pathogenicity and lack of medications and vaccines. Direct contacts or the body fluids of infected animals are the major factor of transmission of NiV. As it is not an airborne infection, the transmission rate is relatively low. Still, mutations of the NiV in the animal reservoir over the years, followed by zoonotic transfer, can make the deadliness of the virus manifold in upcoming years. Therefore, there is no denial of the possibility of a pandemic after COVID-19 considering the severe pathogenicity of NiV, and that is why we need to be prepared with possible drugs in upcoming days. Considering the time constraints, computational aided drug design (CADD) is an efficient way to study the virus and perform the drug design and test the HITs to lead experimentally. Therefore, this review focuses primarily on NiV target proteins (covering NiV and human), experimentally tested repurposed drug details, and latest computational studies on potential lead molecules, which can be explored as potential drug candidates. Computationally identified drug candidates, including their chemical structures, docking scores, amino acid level interaction with corresponding protein, and the platform used for the studies, are thoroughly discussed. The review will offer a one-stop study to access what had been performed and what can be performed in the CADD of NiV.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Nipah virus is a single-stranded, negative-sense RNA virus [1] which belongs to genus Henipavirus and family Paramyxoviridae [2]. First appearance of Nipah virus (NiV) was reported in 1998 in Malaysia among pig farmers. Subsequently, the virus was detected in Bangladesh [3], Singapore [4], India [5], and the Philippines [6]. Up to December 2022, all the cases followed by mortality ration with the cases and types of strain reported by the WHO is listed in Table 1. Fruit bats of the Pteropodidae family and Pteropus bat species are considered as a primary or natural reservoir of NiV. To support the claim, the NiV was isolated from urine samples of Pteropus lylei [7] in Cambodia and Pteropus hypomelanus and Pteropus vampyrus in Malaysia [8]. As per various reports, the transmission occurs from bats to humans, pigs, sick pigs, or pig’s contaminated tissue to humans; even transmission is possible from human to human as well as from the consumption of contaminated food such as raw date palm juice contaminated with saliva or urine of infected bats. Initial research indicated that pig is a primary reason for human infections [9], while some studies pointed out that dogs and cats can also be infected by this virus [10]. A report from the World Health Organization (WHO) dated 30 May 2018 indicated that the fatality rate can range from 40 to 75% and the rate of transmission as well as mortality can vary depending on the outbreak, epidemiological scrutiny, and clinical management of the governing authorities of the place [11].
NiV-infected person can experience a series of clinical symptoms, from subclinical asymptomatic infection to acute respiratory infection and fatal encephalitis followed by death. Fever can be uniformly present as indicated by the probable case diagnosis, followed by altered mental state, headache, severe weakness, cough, trouble breathing, diarrhea, myalgia, and dizziness [12]. Around 20% of patients have experienced residual neurological consequences like seizure, mental disorders, and altered personality. The recovered patients developed delayed onset encephalitis, as per WHO reports. In one study done on 2009, they estimated the R0 value (the average number of people that one infected person can pass the illness to) of NiV in rural Bangladesh, which was equal to 0.48 [13]. The tendency of NiV showed a low R0 value but high mortality. So long as all spillover virus strains have a R0 less than 1 and do not evolve within their human host, each spillover will result in only sporadic person-to-person transmission chains. However, if a strain with a R0 higher than 1 spread or if a strain infecting a person acquires a R0 higher than 1, humanity might confront its most destructive pandemic in the world [14]. The last outbreak of NiV occurred in Kozhikode, Kerala, South India, along with 18 cases and 88.8% mortality, in 2018 [15]. In the same year, NiV was considered in the 2018 Annual Review of Diseases prioritized under the Research and Development Blueprint proposed by the WHO [16]. However, there is no approved specific drug for NiV or vaccine until today.
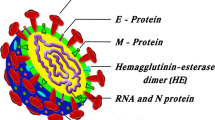
NiV encodes six structural proteins, out of which glycoprotein (NiV-G) and fusion protein (NiV-F) are surface proteins of NiV. Remaining four proteins are inner proteins comprising matrix protein (NiV-M), phosphoprotein (NiV-P), nucleoprotein (NiV-N), and the large protein or RNA polymerase protein (NiV-L). Meanwhile, the P gene also encodes to P, W, V, and C proteins. The NiV structure with six major targeted proteins with their nucleotide length is illustrated in Fig. 1. The detailed function of each protein will be explained in the next section.

NiV structure with six target proteins and their nucleotide length
Facing the large probability of a possible NiV outbreak, it offers serious attention to finding potential drugs that can treat NiV without further delay. Over the years, computer-aided drug design (CADD) has enabled scientists to reduce the amount of time spent on synthetic and biological testing [17] to speed up the drug design and discovery process for many instances. The classic function of CADD in drug development is to check out large chemical databases and/or libraries into different subsets of predicted active compounds, allowing for the optimization of lead compounds by enhancing their biological properties (such as affinity and ADMET profiling) and the construction of chemotypes from a nucleating site by incorporating fragments with optimized function [18]. There is no denying a rational amalgamation of CADD and experimental analysis is the best possible approach to find the small drug molecules for NiV.
This review discusses in detail all the NiV protein targets that can be explored for CADD and discovery to fight NiV. Although there is no approved medication available for NiV, we have tried to summarize major medications used over the years to treat or alleviate the symptoms and severity of the disease, along with their suggested dose and administered routes. Followed by the present status of computational drug research and future directions for NiV drug discovery is discussed in detail to offer researchers a complete current state of the art of Nipah research.
Potential protein targets for NiV drug discovery
In this section, we have discussed major structural features and characteristics of all available target proteins for the drug discovery of NiV covering target of NiV and human target. All available PDB ID for all NiV proteins is discussed and listed in Table 2.
Potential protein target of NiV
Nipah attachment glycoprotein (NiV-G)
NiV attachment glycoprotein (G Protein) was a critical virulent factor in charge of the host cell receptor attachment [19] including 602 amino acids [20]. The NiV-G protein, unlike most paramyxoviruses, lacks hemagglutinating and neuraminidase activity and does not attach to carbohydrate moieties [20, 21]. Research also indicated that the tyrosine 28/29 was critical for correct targeting [22].
From the experimental result, W504, E505, N557, Q530, T531, A532, and E533 are seven residues that formed an adjacent site on the surface of the globular head between the central shallow depression and the rim [23]. Also, the experimental result showed that ephrinB2 is the receptor of NiV. In the research proposed in 2018 by Ali et al., the binding pocket for NiV-G (PDB ID: 3D11) was computed by CASTp [24]. Asp219, Pro220, Pro276, Asn277, Val279, Tyr280, His281, Cys282, Tyr351, Gly352, Pro353, Pro448, Phe458, Gly489, Gln490, Gly506, Val507, Tyr508, Lys560, and Gly559 were the critical amino acids for potential inhibitor studies.
Another work on NiV-G was proposed in 2020 [25]. The set of ligands included in this study belonged to The Pathogen Box Medicine for Malaria Venture. The Autodock-Vina was used to perform virtual screening with a 60 × 60 × 60 centered in (XYZ dimension of 27.77, 6.268, 85.037) grid. Autodock performed rigid-flexible molecular docking between NiV-G protein (PDB ID:3D11) and ligands. In the GROMACS software and PRODRUG server, Gromos9643a1 was applied for molecular dynamics computation. PyMOL was used to determine the receptor-ligand interaction. Cys240 and Arg236 were proposed as two more critical amino acids for NiV-G binding pocket.
Nipah fusion glycoprotein (NiV-F)
During NiV fusing to human cells, NiV-G and NiV-F must work together [26]. NiV-F was another critical protein target for potential NiV inhibitor-based drug discovery in this case. The fusion protein was considered type I transmembrane protein, which includes 546 amino acids [27]. Before integration into new virions, the F0 precursor is broken into disulfide-linked components F1 and F2. The F1 cleavage product formed from F0 contains multiple functions and is accountable for fixing the F protein in lipid membranes [28]. Approximately 20 hydrophobic amino acids comprise fusion peptides at the N-terminal region of the F1 subunit. The fusion peptide is highly conserved across all paramyxoviruses and is essential for the biological activity of the F protein, which is inserted into the target membrane to initiate the fusion process [20, 29].
Nipah matrix protein (NiV-M)
NiV (NiV-M) matrix protein activates many cellular machineries to facilitate and regulate virion budding at the plasma membrane [30]. Matrix protein exploits cellular trafficking and ubiquitination mechanisms to achieve transitory nuclear localization. During the initial 16–20 h following infection, NiV-M was observed to be confined to the nucleus and nucleolus [31, 32]. It indicates that paramyxovirus M proteins have crucial nonstructural functions at early stage of the viral replication cycle. Still, their major and best-characterized function coordinates the assembly and budding of offspring viruses at the plasma membrane [30]. Various host pathways, including the ubiquitination mechanism, nuclear export apparatus, and vesicle sorting and trafficking components, can be used as prospective therapeutic targets by the detailed studies of key NiV-M interactions with host proteins for future drug discovery [30].
Nipah phosphoprotein (NiV-P)
The multimeric phosphoprotein (P) of the NiV attaches the viral polymerase to the nucleocapsid [33] which plays a significant role in the genome replication [34, 35]. The NiV-P multimerization domain consists of a long, parallel, tetrameric, coiled coil with an N-terminal cap and a hydrophobic core [33]. NiV-G has a very close connection and interaction with NiV-N, which will be mentioned in the NiV-N section. The NiV-P gene encodes 4 nonstructural proteins, i.e., P, W, V, and C proteins.
Nipah nucleocapsid protein (NiV-N)
The association of Nipah virus nucleocapsid protein (NiV-N) with NiV-P during nucleocapsid construction is a crucial step in the virus replication, as only the encapsulated RNA genome can be used for multiplication [36]. Their experimental result also proved that NiV N-P colocalization should be the critical step for the normal functioning of N in viral replication, as mutant N proteins lacking the capacity to form N-P complex cannot operate in minigenome tests [36]. In 2018, Ranadheera et al. proposed their experimental result for the interaction between the NiV-N and NiV-P virus replication regulations [37]. Their result indicated that viral translation and virion generation become dysfunctional in the presence of elevated levels of recombinant NiV N-HA including one or both NiV P binding domains. Furthermore, removal of both NiV P binding sites from NiV N rendered its effects null and void, indicating that contact between NiV N and NiV P is crucial for viral replication inhibition [37].
Nipah large protein or RNA polymerase protein (NiV-L)
The structure of the L protein of NiV was not identified up to date. However, research indicated that after performing the BLAST search, the L protein of Human Parainfluenza virus 5 (HPIV-5) with PDB ID: 6V85 gained the highest score of homology sequence [38]. Around 32.39% identity of the NiV-L protein sequence can be pairwise with HPIV-5.
Potential human protein target
Human cell surface receptor ephrin-B2/ephrin-B3
Several scientific articles have extensively studied and documented the targeting of ephrin-B2 and ephrin-B3 by the NiV. As a method of entrance and invasion into the host, the NiV targets ephrin-B2 and ephrin-B3 receptors in host cells [39,40,41,42]. Ephrin is a membrane-bound ligand that plays a significant role in various biological processes, including cell migration and differentiation [43]. The NiV attaches to these receptors via its spike glycoprotein (G). Once the virus connects to the ephrin-B2 and ephrin-B3 receptors, it undergoes endocytosis, which is absorbed into the host cell. This permits the virus to multiply and disseminate to other cells in the host, resulting in the symptoms of NiV infection. It has been demonstrated that soluble ephrin-B2 or ephrin-B3 proteins, as well as soluble NiV-G proteins, inhibit virus entry and cell–cell fusion (Fig. 2). However, the interference with ephrin-B function and the antigenicity of NiV-G itself restrict the practical value of these molecules as antivirals [44].

The interaction between NiV proteins and human proteins during the NiV replication cycle
Importin alpha 3
Importin alpha 3 (Imp3) is utilized by the NiV to reproduce and disseminate within host cells. Imp3 is a karyopherin that transports viral proteins and replication machinery into the nucleus of the host cell [45, 46]. According to studies, the NiV infects host cells by employing Imp3 as a mediator for its entrance into the cell nucleus [47]. NiV hijacks Imp3 upon infection and exploits it to carry its replication machinery into the host cell nucleus, where it replicates and spreads (Fig. 2). Simultaneously, research indicated that compared to all other importin molecules, the unique ARM7 and ARM8 conformation of Imp3 provided increased binding interface availability for NLS interactions [48]. Significantly, additional proteins vulnerable to nuclear transport by Imp3 and Imp4 are outcompeted by W, resulting in decreased transport and nuclear activity [49, 50].
Heparan sulfate proteoglycans
NiV can utilize an attachment receptor called heparan sulfate proteoglycans to adhere to nonpermissive circulating leukocytes, facilitating viral spread inside the host [51]. Heparan sulfate proteoglycans (HSPG) consist of unbranched, negatively charged heparan sulfate (HS) polysaccharides that are linked to a variety of cell surface or extracellular matrix proteins [52]. Lacking heparan sulfate prevented cells from mediating Henipavirus trans-infection and decreased their susceptibility to infection.
Present medication status for NiV
Currently, no US FDA-approved drug or vaccine can be prescribed for NiV treatment. According to the Centers for Disease Control (CDC) [53], the treatment of NiV is narrowed down to providing treatment based on the symptoms that occurred. Based on research up to date, ribavirin [54], chloroquine [55], remdesivir [56], and favipiravir [57] were experimentally proven effective on either African green monkeys or Syrian hamsters. Ribavirin [54] reported increase of overall survival rate (36%) when tested on Nipah encephalitis patients using both oral and i.v. dose administration. However, no NiV-specific target drug can effectively treat the patients. Although CDC posted that one of the monoclonal antibodies m102.4 has passed phase 1 clinical studies and has been administered for compassionate use in 2020, the m102.4 is not approved to date [53]. Due to the lack of specific drugs for NiV at present and the possible outbreak of NiV in the future, CADD is an integral part of accelerating drug development of the Nipah fight without any doubt, in combination with experimental efforts. In Table 3, we have combined experimentally tested drugs to date where majorly existing drugs were repurposed for checking the effectiveness of the NiV along with monoclonal antibodies.
Computational drug and drug derivatives under investigating
Computational modeling through high-throughput screening and virtual screening is highly significant for selecting the best drug candidates for NiV. To speed up the traditional drug discovery process, CADD is a proven method in amalgamation with experimental efforts. Over the years, scientists worldwide have tried to find a possible cure for NiV employing molecular modeling tools like quantitative structure–activity relationships (QSARs), docking, pharmacophore, homology modeling, molecular dynamic (MD) simulations, binding energy calculations, and ADMET profiling approaches. We have tried to summarize their efforts in this section which can be a helpful resource for researchers who want to invest their time in the drug discovery of NiV.
Nipah G (NiV-G) protein target and computational drug
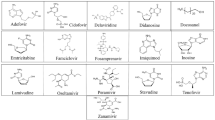
Pathania et al. come up with potential antivirals for NiV [65]. The authors used Autodock Tool to prepare NiV-G crystal structure (PDB ID:2VSM). A pool of 2327 US FDA-approved drugs were collected from the DrugBank database for virtual screening. OpenBabel program was used for ligand energy optimization with MMFF94 force. QuickVina undertook molecular docking. Simultaneously, molecular image rendering was carried out by PyMOL. Considering binding affinity of ≤ − 10 kcal/mol as threshold, 17 potential inhibitors were selected. OSIRIS Property Explorer computed chemical-protein interaction network analysis to access the risk of side effects on all these molecules, where nilotinib, deslanoside, and acetyldigitoxin were identified as top three drug candidates (Fig. 3). Compound 1 (nilotinib), compound 2 (deslanoside), and compound 3 (acetyldigitoxin) had a binding affinity of − 10.3, − 10.2, and − 10.7 kcal/mol, respectively. Nilotinib formed H-bond with Gln559; hydrophobic interaction with Ile304, Trp504, Phe458, Val507, Gln559, Ala 532, and Asp557; and salt bridge with Try581. For acetyldigitoxin, H-bonds were found with Gln559, Asn557, and Gly506, while hydrophobic interactions were found with Try581, Gln490, Trp504, and Phe458. Between deslanoside and NiV-G, H-bonds were formed with Tyr581, Ala558, Ala532, and Pro488; hydrophobic interaction with Gln490, Trp504, Leu305, His281, and Lys560; and salt bridge with Lys560 and His281. Since these three US FDA-approved drugs were identified as cardiac glycosides, they were expected to regulate cardiac inflammation during NiV infection treatment.

Copyright 2022 Taylor & Francis
Top 3 lead molecules structure (compound 1 (nilotinib), compound 2 (deslanoside), and compound 3 (acetyldigitoxin) along with their 3D interactions and docking posture with 2VSM protein. Adapted with permission from J. Biomol. Structur. Dyn. 2019, 38, 17, 5108–5125 [65].
Virtual screening was performed on NiV G-protein (PDB ID: 3D11) by Naeem et al. [19] employing 2118 blood–brain barrier (BBB −) and 189 BBB + compounds from the gold and platinum Asinex library using a total pool of among 211,620 molecules. Molecular docking, density functional theory (DFT), and MD simulations were performed. The two compounds in Fig. 4 had highest performances in each group. For compound 4 (from BBB −), − 10.4 kcal/mol binding energy was gained, and His281 and Tyr508 were found forming a conventional hydrogen bond with a distance of 2.26 Å and 2.07 Å. Carbon hydrogen bond was also found on the protein where Cys282 and His281 had a distance of 3.22 Å and 3.41 Å, respectively. Meanwhile, hydrophobic interaction existed between compound 4 and TRP504, Phe458, Pro 41, and Try508. Wile, Leu305, Tyr351, Gly506, Val507, Pro353, and Gly352 showed Van der Waals interaction. Compound 5 was from BBB + group, and a − 9.2 kcal/mol binding energy was reported. Hydrogen bond existed between compound 5 and Lys560 and Try351 with a reported distance of 2.79 Å and 1.91 Å, respectively. Simultaneously, hydrophobic interaction existed on Leu305, Trp504, Pro441, and Pro353. Van der Waal interaction was found with 12 amino acids: Tyr508, Asp219, Ser241, Cys240, Arg236, Thr218, Asp302, Ile304, Phe458, His281, Cys282, and Gly352. Ribavirin acted as a control group for the study, which reported − 6.3 kcal/mol binding energy. ELUMO and EHOMO were studied, BBB − group compound 4 gained an energy gap of 0.14382 eV, while BBB + group compound 5 had a 0.15387 eV energy gap. Both had low energy gaps and illustrate a good chemical reactivity and a low kinetic stability.

Copyright 2022 Taylor & Francis
Top 2 lead molecule structure along with their 2D and 3D interactions with 3D11 protein. Adapted with permission from J. Biomol. Structur. Dyn. 2022 [19].
Computational study related to favipiravir and its derivatives were also performed to check their effectiveness towards NiV-G protein [66]. Favipiravir shows a relatively good performance in the experimental work too [57]. Based on the computational result from Lipin et al., piperazine-substituted derivatives were found worth further study [67]. Therefore, the relationship between structure and property of favipiravir was studied in Lipin’s case. With the help of DFT calculation, a better geometrical structure of all the designed molecules was optimized. Simultaneously, ADMET and molecular docking were performed to assess the chemical properties and affinity with the G-protein of NiV. In the ADMET study, most of the designed favipiravir derivatives showed a higher pIC50. The study indicated that piperazine-substituted favipiravir derivatives have a higher ability to bind than the original favipiravir structure. James et al. studied 22 favipiravir derivatives by incorporating heterocyclic groups (moieties such as pyrazole, imidazole, and pyrazino) in favipiravir [66]. All derivatives were subjected to molecular docking for the Nipah G-protein (PDB ID:3D11) target followed by physical property evaluations and ADMET studies. The result indicated that three compounds had relatively better performances compare to control favipiravir, as shown in Fig. 5. Among these three compounds, compound 6 has the highest binding energy with a − 6.16 kcal/mol, followed by compounds 7 and 8 which have scores of − 5.50 and − 5.38 kcal/mol, respectively.

Adapted from Open access article J. Pharm. Res. Int. 2021, 33, 156–169 [66]
Top 3 favipiravir derivative chemical structure with their 2D and 3D interactions with 3D11 protein.
Meanwhile, all the designed compounds followed the Lipinski rule of five, representing that they have no issue with the oral bioavailability. Strong interaction between compound 6 and G-protein was found (hydrophobic interaction with Tyr309, Ile304, Ile401, Phe369, Tyr401, Ile408, and Leu409 and polar interaction with Thr308, Ser307, Ash306, Ser405, and Hid406). Hydrogen bond was also reported with amino acid residues Thr308 and Hid406. The result indicated that incorporating heterocyclic moieties in the favipiravir can increase the effectiveness of the designed drug compared to favipiravir against NiV.
Kalbhor et al. proposed potential inhibitors for NiV from screening large antiviral specific chemical libraries with multi-step molecular docking and MD simulation [68]. In this study, 8722 chemical entities were collected from Asinex-Antiviral Library, 3700 compounds were collected from Enamine-Antiviral Library, and 67,470 molecules were taken from ChemDiv-Antiviral Library. LigPrep prepared all the collected molecules for further docking. NiV-G (PDB ID: 2VSM) was prepared by Protein Preparation Wizard module. Multi-step docking was performed on all 79,892 molecules. At the same time, the Prime-MM-GBSA method was used for binding free energy computation. From the total pool of chemicals, 299 compounds reached the stipulated binding energy of − 6.00 kcal/mol of docking score and a − 40.00 kcal/mol MM-GBSA score. Followed by 207 molecules were further selected employing ADME study on SwissADME webserver. Based on TOPKAT toxicity prediction assessment, 14 non-toxic compounds were resulted. Combining the results from total study, 5 molecules were selected which is demonstrated in Fig. 6. Compound 9 reported highest binding energy and better interactions with the studied NiV-G protein. It made H-bonds with amino acid residues Trp504 and Tyr508 and formed hydrophobic interactions with Pro441, Pro448, Val507, and Lys 560 followed by pi-stacking with Phe458 and Trp504. The TPSA scores for compounds 9 to 13 were between 61.44 and 137.32 Å which indicated that all the mentioned compounds could potentially be orally active in nature.

Copyright 2022 Elsevier
Top 5 potential lead molecule’s chemical structure with their 2D and 3D interactions with NiV-G 2VSM protein. Adapted with permission from Biophys. Chem. 2021, 270, 106,537 [68].
NiV fusion glycoprotein (NiV-F) target drugs
During the process of Nipah virus fuse to human cells, NiV-G and NiV-F must work together [26]. NiV-F was another critical protein target for potential NiV inhibitor discovery in this case. Niedermeier et al. designed 18 quinolone derivatives targeting of NiV-F (PDB 1WP7) [69]. Schrödinger Protein Preparation Guide was used to prepare the protein before docking performed. LigPrep was used for the ligand’s preparation, and molecular docking was performed by Schrödinger Glide XP. The compound 14 in Fig. 6 showed the highest score in both computational study and experimental studies. The experiment anti-NiV EC50 with a value of 1.5 was identified. Computational modeling revealed that compound 14 fits well into a specific protein cavity on the NiV-F protein that is essential for the fusion process. Simultaneously, hydrophobic interactions were found between I474, L481, and V 484 in the ligand (marked in green) in Fig. 7 and the hydrophobic pocket in protein surface. Besides computational design, experimental work has been also executed by authors for compound 14 where authors checked inhibition of NiV envelope protein-induced cell fusion followed by cytotoxicity evaluation. With a result of 1.5 μM of anti-NiV EC50, more than 20 μM of CC50, and more than 13 μM of SI (CC50/EC50), compound 14 showed a highest activity and a lowest cytotoxicity among throughout the twenty-three compounds.

Copyright 2022 American Chemical Society
Top quinolone derivative’s 2D chemical structure and molecular binding position with the NiV-F protein 1WP7. Adapted with permission from J. Med. Chem. 2009, 52, 4257–4265 [69].
Verma et al. proposed three phytochemicals named natural 6-gingerol (compound 15), 4-hyroxypanduratin A (compound 16), and Luteolin (compound 17) as potential Niv-F inhibitors [70]. 4-Hyroxypanduratin A is a food component in many Asian countries, 6-gingerol is an active component from Zingiber officinale, and luteolin is a kind of Chinese traditional medicine. These three molecules.sdf file were converted to PDB format by Open Babel. AutoDock4 was used for molecular docking and binding scores computed, while BOVIA Drug Discovery Studio was used to visualize major interactions between ligands and the NiV-F. The best binding energy score − 4.83 kcal/mol is reported by compound 16, while compounds 17 and 15 (Fig. 8) showed binding energy of − 4.58 kcal/mol and − 3.98 kcal/mol, respectively.

Chemical structure of three potential phytochemicals as NiV-G inhibitor
NiV multi-protein target drug
Randhawa et al. [71] proposed Potential Multitarget computational drug discovery study where three databases were curated to find potential inhibitors for NiV-G, NiV-F, and NiV-N. A total pool of 1231 compounds were considered, where 142 plant-derived was retrieved form SerpentinaDB, 868 molecules from Phytochemica, and 221 ligands from Phytochemical and Drug Target Database (PDTDB). All the molecules were optimized by Merck Molecular Force Field (MMFF94) in the OpenBabel v 2.4.0 software. For NiV-G, NiV-F, and NiV-N following proteins were chosen from PDB: 2VSM, 5EVM, and 4CO6, respectively, for molecular docking study. Based on docking binding energy profile, 8 potential inhibitors were selected with the requirement of binding to all three proteins and have a higher negative value than − 7.8 kcal/mol binding affinity on each protein. Remdesivir was used as a control drug with a reported − 7.5 kcal/mol binding energy to NiV-G, − 6.6 kcal/mol to NiV-F, and − 5.6 kcal/mol to NiV-N. Based on ADMETlab, ADME and pharmacokinetic properties were computed for the selected compounds to check their PK/PD profiling. Z-score used to evaluate pharmacokinetic properties and top three compounds 18, 19, and 20 (Fig. 9) reported positive score. Simultaneously, gene expression induction was also accessed by authors for the top identified molecules and found that only a small portion of protein could be induced. MD simulations of three molecules were performed by Groningen Machine for Chemical Simulation (GROMACS). Compounds 18 and 19 were stable in three proteins with average RMSDs equal to ~ 0.12 nm in NiV-G, ~ 0.35 nm in NiV-F, and 0.12 nm in NiV-G. Meanwhile, the docking complexes were found more stable than the free apo-proteins. Compound 20 had a stable performance in NiV-G and NiV-F with a similar RMSD value as compounds 18 and 19.

Adapted from Open access article Microorganisms 2022, 10, 1181 [71]
Chemical structure of compounds 18 (blue), 19 (pink), and 20 (bisque) followed by their docked position to the active binding sites (red) of NiV-F (blue) (PDB ID: 5EVM), NiV-G (PDB ID: 2VSM) (green), and NiV-N (PDB ID: 4CO6) (gold).
Sen et al. studied all targeted proteins of NiV and proposed 4 putative peptide inhibitors and 146 small molecule inhibitors [72]. A total of 22,685 ligands from ZINC database and NiV Malaysian strain AY029768.1 were used in this study. ModPipe was used for pipeline homology modeling, and MODELLER was used for building the multimeric complexes. Using both sequence-sequence and profile-sequence search strategies, the homology modeling templates were identified by authors. AutoDock4 and DOCK6.8 were used for molecular docking studies; meanwhile, AMBER was used to determine the charge on atoms of protein during this process. Active pocket and binding sites were searched for all the proteins employing DEPTH server. The docking results indicated that a known drug ZINC04829362 could bind N protein, a potential drug molecule for depression and Parkinson’s disease. All the ligand and protein used in this study along with their ranking, binding energy, and RMSD between AutoDock and Dock poses can be accessed at http://cospi.iiserpune.ac.in/Nipah.
New proposed strategies for drug discovery of NiV
A new strategy named “Drug-target-drug network-based approach” was proposed in 2021 [73]. Nipah virus and 13 more viruses were tested with the existing US FDA-approved drugs using drug-target-drug network analysis. All the US FDA-approved drugs were evaluated with a confidence score through the computational study as a repurposed theory. Authors have found 16 repurposed drugs between hepatitis E virus (HEV) and NiV. Further, molecular docking is used to validate this strategy’s credibility and identify the best possible candidates for the specific viruses.
Rajput et al. offered the “anti-Nipah” web source in 2019 [74]. The data bank contained 313 chemicals where the QSAR model was used to estimate the prediction of the inhibitory effect. Based on the model of integrating recursive regression, any untested or unknown compound could be predicted through the web service with a 0.82 Pearson’s correlation coefficient (PCC) which is highly robust.
Conclusion
NiV is a highly lethal but weakly transmissible virus, classified as BSL-4 [75] due to its characteristics. The current situation against a possible NiV outbreak is not optimistic. The world has not developed any US FDA-approved drugs that can be used in humans, and some drugs showed excellent in vitro suppression of viruses but did not work in animals. In this case, computational drug discovery will be a perfect way to accelerate drug development. Combining all our statistical protein structures and existing papers on computational drug development, we realized that most drug development focuses on glycoprotein and fusion protein. However, four other proteins (NiV-P/M/N/L) are also critical to the NiV. Future scientific research can gradually fill in the gaps in drug development for other potential protein targets.
References
Hino K, Sato H, Sugai A, Kato M, Yoneda M, Kai C (2013) Downregulation of Nipah virus N mRNA occurs through interaction between its 3′ untranslated region and hnRNP D. J Virol 87:6582–6588
Eaton BT, Broder CC, Middleton D, Wang L-F (2006) Hendra and Nipah viruses: different and dangerous. Nat Rev Microbiol 4:23–35
Rahman MA, Hossain MJ, Sultana S, Homaira N, Khan SU, Rahman M, Gurley ES, Rollin PE, Lo MK, Comer JA, Lowe L, Rota PA, Ksiazek TG, Kenah E, Sharker Y, Luby SP (2012) Date palm sap linked to Nipah virus outbreak in Bangladesh, 2008. Vector Borne Zoonotic Dis 12:65–72
Paton NI, Leo YS, Zaki SR, Auchus AP, Lee KE, Ling AE, Chew SK, Ang B, Rollin PE, Umapathi T, Sng I, Lee CC, Lim E, Ksiazek TG (1999) Outbreak of Nipah-virus infection among abattoir workers in Singapore. The Lancet 354:1253–1256
Yadav PD, Sahay RR, Balakrishnan A, Mohandas S, Radhakrishnan C, Gokhale MD, Balasubramanian R, Abraham P, Gupta N, Sugunan AP, Khobragade R, George K, Shete A, Patil S, Thankappan UP, Dighe H, Koshy J, Vijay V, Gayathri R, Keerthi KV (2022) Nipah VIRUS OUTBREAK IN Kerala State, India amidst of COVID-19 pandemic. Public Health Front 10
Ching PK, de los Reyes, VC, Sucaldito, MN, Tayag, E, Columna-Vingno, AB, Malbas, FF, Jr., Bolo, GC, Jr., Sejvar, JJ, Eagles, D, Playford, G, Dueger, E, Kaku, Y, Morikawa, S, Kuroda, M, Marsh, GA, McCullough, S, & Foxwell, AR, (2015) Outbreak of henipavirus infection, Philippines, 2014. Emerg Infect Dis 21:328–331
Reynes JM, Counor D, Ong S, Faure C, Seng V, Molia S, Walston J, Georges-Courbot MC, Deubel V, Sarthou JL (2005) Nipah virus in lyle’s flying foxes, Cambodia. Emerg Infect Dis 11:1042–1047
Chua KB, Lek Koh C, Hooi PS, Wee KF, Khong JH, Chua BH, Chan YP, Lim ME, Lam SK (2002) Isolation of Nipah virus from Malaysian Island flying-foxes. Microbes Infect 4:145–151
Parashar, UD, Sunn, LM, Ong, F, Mounts, AW, Arif, MT, Ksiazek, TG, Kamaluddin, MA, Mustafa, AN, Kaur, H, Ding, LM, Othman, G, Radzi, HM, Kitsutani, PT, Stockton, PC, Arokiasamy, J, Gary, HE, Jr, Anderson, LJ, & Team, ftNEOI (2000) Case-control study of risk factors for human infection with a new zoonotic paramyxovirus, Nipah virus, during a 1998–1999 outbreak of severe encephalitis in Malaysia. J Infect Dis 181:1755–1759
Sahani, M, Parashar, UD, Ali, R, Das, P, Lye, M, Isa, MM, Arif, MT, Ksiazek, TG, Sivamoorthy, M, & Group, TNEOI (2001) Nipah virus infection among abattoir workers in Malaysia, 1998–1999. Int J Epidemiol 30:1017–1020
WHO (2018) Nipah virus. Retrieved Dec 16 from https://www.who.int/news-room/fact-sheets/detail/nipah-virus
Hossain MJ, Gurley ES, Montgomery JM, Bell M, Carroll DS, Hsu VP, Formenty P, Croisier A, Bertherat E, Faiz MA, Azad AK, Islam R, Molla MAR, Ksiazek TG, Rota PA, Comer JA, Rollin PE, Luby SP, Breiman RF (2008) Clinical presentation of Nipah virus infection in Bangladesh. Clin Infect Dis 46:977–984
Luby SP, Hossain MJ, Gurley ES, Ahmed BN, Banu S, Khan SU, Homaira N, Rota PA, Rollin PE, Comer JA, Kenah E, Ksiazek TG, Rahman M (2009) Recurrent zoonotic transmission of Nipah virus into humans, Bangladesh, 2001–2007. Emerg Infect Dis 15:1229–1235
Luby SP (2013) The pandemic potential of Nipah virus. Antiviral Res 100:38–43
Thomas B, Chandran P, Lilabi MP, George B, Sivakumar CP, Jayadev VK, Bindu V, Rajasi RS, Vijayan B, Mohandas A, Hafeez N (2019) Nipah virus infection in Kozhikode, Kerala, South India, in 2018: epidemiology of an outbreak of an emerging disease. Indian J Community Med 44:383–387
2018 Annual review of diseases prioritized under the Research and Development Blueprint (2018) Retrieved Nov 11 from https://www.who.int/news-room/events/detail/2018/02/06/default-calendar/2018-annual-review-of-diseases-prioritized-under-the-research-anddevelopment-blueprint
Tomar V, Mazumder M, Chandra R, Yang J, Sakharkar MK (2019). Small molecule drug design. In S. Ranganathan, M. Gribskov, K. Nakai, & C. Schönbach (Eds.). Encyclopedia of Bioinformatics and Computational Biology (pp. 741–760). Academic Press. https://doi.org/10.1016/B978-0-12-809633-8.20157-X
Osakwe O (2016) Chapter 5 - The significance of discovery screening and structure optimization studies. In O. Osakwe & S. A. A. Rizvi (Eds.). Social Aspects of Drug Discovery, Development and Commercialization (pp. 109–128). Academic Press. https://doi.org/10.1016/B978-0-12-802220-7.00005-3
Naeem I, Mateen RM, Sibtul Hassan S, Tariq A, Parveen R, Saqib MAN, Fareed MI, Hussain M, Afzal MS (2022) In silico identification of potential drug-like molecules against G glycoprotein of Nipah virus by molecular docking, DFT studies, and molecular dynamic simulation. J Biomol Struct Dyn 1–15
Diederich S, Maisner A (2007) Molecular characteristics of the Nipah virus glycoproteins. Ann N Y Acad Sci 1102:39–50
Eaton BT, Wright PJ, Wang LF, Sergeyev O, Michalski WP, Bossart KN, Broder CC (2004) Henipaviruses: recent observations on regulation of transcription and the nature of the cell receptor. Arch Virol Suppl 122–131
Erbar S, Maisner A (2010) Nipah virus infection and glycoprotein targeting in endothelial cells. Virol J 7:305
Guillaume V, Aslan H, Ainouze M, Guerbois M, Wild TF, Buckland R, Langedijk JPM (2006) Evidence of a potential receptor-binding site on the Nipah virus G protein (NiV-G): identification of globular head residues with a role in fusion promotion and their localization on an NiV-G structural model. J Virol 80:7546–7554
Ali H, Anwar S, Roy PK, Ashrafuzzaman M (2018) Virtual screening for identification of small lead compound inhibitors of Nipah virus attachment glycoprotein. J Pharmacog Pharmacopro 9
Ropón-Palacios G, Chenet-Zuta ME, Olivos-Ramirez GE, Otazu K, Acurio-Saavedra J, Camps I (2020) Potential novel inhibitors against emerging zoonotic pathogen Nipah virus: a virtual screening and molecular dynamics approach. J Biomol Struct Dyn 38:3225–3234
Weis M, Maisner A (2015) Nipah virus fusion protein: importance of the cytoplasmic tail for endosomal trafficking and bioactivity. Eur J Cell Biol 94:316–322
Harcourt BH, Tamin A, Ksiazek TG, Rollin PE, Anderson LJ, Bellini WJ, Rota PA (2000) Molecular characterization of Nipah virus, a newly emergent paramyxovirus. Virology 271:334–349
Vogt C, Eickmann M, Diederich S, Moll M, Maisner A (2005) Endocytosis of the Nipah virus glycoproteins. J Virol 79:3865–3872
Epand RM (2003) Fusion peptides and the mechanism of viral fusion. Biochim Biophys Acta 1614:116–121
Watkinson RE, Lee B (2016) Nipah virus matrix protein: expert hacker of cellular machines. FEBS Lett 590:2494–2511
Pentecost M, Vashisht AA, Lester T, Voros T, Beaty SM, Park A, Wang YE, Yun TE, Freiberg AN, Wohlschlegel JA, Lee B (2015) Evidence for ubiquitin-regulated nuclear and subnuclear trafficking among Paramyxovirinae matrix proteins. PLoS Pathog 11:e1004739
Wang YE, Park A, Lake M, Pentecost M, Torres B, Yun TE, Wolf MC, Holbrook MR, Freiberg AN, Lee B (2010) Ubiquitin-regulated nuclear-cytoplasmic trafficking of the Nipah virus matrix protein is important for viral budding. PLoS Pathog 6:e1001186
Bruhn JF, Barnett KC, Bibby J, Thomas JM, Keegan RM, Rigden DJ, Bornholdt ZA, Saphire EO (2014) Crystal structure of the nipah virus phosphoprotein tetramerization domain. J Virol 88:758–762
Chen M, Ogino T, Banerjee AK (2006) Mapping and functional role of the self-association domain of vesicular stomatitis virus phosphoprotein. J Virol 80:9511–9518
Schneider U, Blechschmidt K, Schwemmle M, Staeheli P (2004) Overlap of interaction domains indicates a central role of the P protein in assembly and regulation of the Borna disease virus polymerase complex. J Biol Chem 279:55290–55296
Omi-Furutani M, Yoneda M, Fujita K, Ikeda F, Kai C (2010) Novel phosphoprotein-interacting region in Nipah virus nucleocapsid protein and its involvement in viral replication. J Virol 84:9793–9799
Ranadheera C, Proulx R, Chaiyakul M, Jones S, Grolla A, Leung A, Rutherford J, Kobasa D, Carpenter M, Czub M (2018) The interaction between the Nipah virus nucleocapsid protein and phosphoprotein regulates virus replication. Sci Rep 8:15994
Abduljalil JM, Elfiky AA, Sayed E-STA, AlKhazindar MM (2022) In silico structural elucidation of Nipah virus L protein and targeting RNA-dependent RNA polymerase domain by nucleoside analogs. J Biomol Struct Dyn 1–15
Bowden TA, Aricescu AR, Gilbert RJC, Grimes JM, Jones EY, Stuart DI (2008) Structural basis of Nipah and Hendra virus attachment to their cell-surface receptor ephrin-B2. Nat Struct Mol Biol 15:567–572
Negrete OA, Levroney EL, Aguilar HC, Bertolotti-Ciarlet A, Nazarian R, Tajyar S, Lee B (2005) EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature 436:401–405
Negrete OA, Wolf MC, Aguilar HC, Enterlein S, Wang W, Muhlberger E, Su SV, Bertolotti-Ciarlet A, Flick R, Lee B (2006) Two key residues in ephrinB3 are critical for its use as an alternative receptor for Nipah virus. PLoS Pathog 2:78–86
Bonaparte MI, Dimitrov AS, Bossart KN, Crameri G, Mungal BA, Bishop KA, Choudhry V, Dimitrov DS, Wang LF, Eaton BT, Broder CC (2005) Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus. Proc Natl Acad Sci USA 102:10652–10657
Darling TK, Lamb TJ (2019) Emerging roles for Eph receptors and ephrin ligands in immunity. Front Immunol 10
Aguilar HC, Lee B (2011) Emerging paramyxoviruses: molecular mechanisms and antiviral strategies. Expert Rev Mol Med 13:e6
Stewart M (2007) Molecular mechanism of the nuclear protein import cycle. Nat Rev Mol Cell Biol 8:195–208
Bednenko J, Cingolani G, Gerace L (2003) Nucleocytoplasmic transport: navigating the channel. Traffic 4:127–135
Smith KM, Tsimbalyuk S, Edwards MR, Cross EM, Batra J, Soares da Costa TP, Aragão D, Basler CF, Forwood JK (2018) Structural basis for importin alpha 3 specificity of W proteins in Hendra and Nipah viruses. Nat Commun 9:3703
Tsimbalyuk, S, Cross EM, Hoad M, Donnelly CM, Roby JA, Forwood JK (2020) The intrinsically disordered W protein is multifunctional during Henipavirus infection, disrupting host signalling pathways and nuclear import. Cells 9
Kumar KP, McBride KM, Weaver BK, Dingwall C, Reich NC (2000) Regulated nuclear-cytoplasmic localization of interferon regulatory factor 3, a subunit of double-stranded RNA-activated factor 1. Mol Cell Biol 20:4159–4168
Zhu M, Fang T, Li S, Meng K, Guo D (2015) Bipartite nuclear localization signal controls nuclear import and DNA-binding activity of IFN regulatory factor 3. J Immunol 195:289–297
Mathieu C, Dhondt KP, Châlons M, Mély S, Raoul H, Negre D, Cosset F-L, Gerlier D, Vivès RR, Horvat B (2015) Heparan sulfate-dependent enhancement of Henipavirus infection. mBio 6:e02427–02414
Cagno V, Tseligka ED, Jones ST, Tapparel C (2019) Heparan sulfate proteoglycans and viral attachment: true receptors or adaptation bias? Viruses 11
CDC (2020) Nipah virus(NiV) treatment. Retrieved Dec 10 from https://www.cdc.gov/vhf/nipah/treatment/index.html
Chong H-T, Kamarulzaman A, Tan C-T, Goh K-J, Thayaparan T, Kunjapan SR, Chew N-K, Chua K-B, Lam S-K (2001) Treatment of acute Nipah encephalitis with ribavirin. Ann Neurol 49:810–813
Porotto M, Orefice G, Yokoyama CC, Mungall BA, Realubit R, Sganga ML, Aljofan M, Whitt M, Glickman F, Moscona A (2009) Simulating Henipavirus multicycle replication in a screening assay leads to identification of a promising candidate for therapy. J Virol 83:5148–5155
Lo MK, Feldmann F, Gary JM, Jordan R, Bannister R, Cronin J, Patel NR, Klena JD, Nichol ST, Cihlar T, Zaki SR, Feldmann H, Spiropoulou CF, de Wit E (2019) Remdesivir (GS-5734) protects African green monkeys from Nipah virus challenge. Sci Transl Med 11:eaau9242
Dawes BE, Kalveram B, Ikegami T, Juelich T, Smith JK, Zhang LH, Park A, Lee B, Komeno T, Furuta Y, Freiberg AN (2018) Favipiravir (T-705) protects against Nipah virus infection in the hamster model. Sci Rep 8
Georges-Courbot MC, Contamin H, Faure C, Loth P, Baize S, Leyssen P, Neyts J, Deubel V (2006) Poly(I)-Poly(C<sub>12</sub>U) but not ribavirin prevents death in a hamster model of Nipah virus infection. Antimicrob Agents Chemother 50:1768–1772
Freiberg AN, Worthy MN, Lee B, Holbrook MR (2010) Combined chloroquine and ribavirin treatment does not prevent death in a hamster model of Nipah and Hendra virus infection. J Gen Virol 91:765–772
Pallister J, Middleton D, Crameri G, Yamada M, Klein R, Hancock TJ, Foord A, Shiell B, Michalski W, Broder CC, Wang L-F (2009) Chloroquine administration does not prevent Nipah virus infection and disease in ferrets. J Virol 83:11979–11982
Welch SR, Spengler JR, Harmon JR, Coleman-McCray JD, Scholte FEM, Genzer SC, Lo MK, Montgomery JM, Nichol ST, Spiropoulou CF (2022) Defective interfering viral particle treatment reduces clinical signs and protects hamsters from lethal Nipah virus disease. mBio 13:e03294–03221
Geisbert TW, Mire CE, Geisbert JB, Chan Y-P, Agans KN, Feldmann F, Fenton KA, Zhu Z, Dimitrov DS, Scott DP, Bossart KN, Feldmann H, Broder CC (2014) Therapeutic treatment of Nipah virus infection in nonhuman primates with a neutralizing human monoclonal antibody. Sci Transl Med 6:242ra282–242ra282
Mire CE, Chan Y-P, Borisevich V, Cross RW, Yan L, Agans KN, Dang HV, Veesler D, Fenton KA, Geisbert TW, Broder CC (2019) A cross-reactive humanized monoclonal antibody targeting fusion glycoprotein function protects ferrets against lethal Nipah virus and Hendra virus infection. J Infect Dis 221:S471–S479
Lo MK, Spengler JR, Krumpe LRH, Welch SR, Chattopadhyay A, Harmon JR, Coleman-McCray JD, Scholte FEM, Hotard AL, Fuqua JL, Rose JK, Nichol ST, Palmer KE, O’Keefe BR, Spiropoulou CF (2020) Griffithsin inhibits Nipah virus entry and fusion and can protect Syrian golden hamsters from lethal Nipah virus challenge. J Infect Dis 221:S480–S492
Pathania S, Randhawa V, Kumar M (2020) Identifying potential entry inhibitors for emerging Nipah virus by molecular docking and chemical-protein interaction network. J Biomol Struct Dyn 38:5108–5125
James JP, Apoorva M, SR, Sukesh, KB, & Varun, A (2021) Design and identification of lead compounds targeting Nipah G attachment glycoprotein by in silico approaches. J Pharm Res Int 33:156–169
Lipin R, Dhanabalan AK, Gunasekaran K, Solomon RV (2021) Piperazine-substituted derivatives of favipiravir for Nipah virus inhibition: what do in silico studies unravel? Sn Applied Sciences 3
Kalbhor MS, Bhowmick S, Alanazi AM, Patil PC, Islam MA (2021) Multi-step molecular docking and dynamics simulation-based screening of large antiviral specific chemical libraries for identification of Nipah virus glycoprotein inhibitors. Biophys Chem 270:106537
Niedermeier S, Singethan K, Rohrer SG, Matz M, Kossner M, Diederich S, Maisner A, Schmitz J, Hiltensperger G, Baumann K, Holzgrabe U, Schneider-Schaulies J (2009) A small-molecule inhibitor of Nipah virus envelope protein-mediated membrane fusion. J Med Chem 52:4257–4265
Verma A, Mittal P, Pander MS, Trivedi N (2021) In silico drug discovery of 4-hyroxypanduratin A, 6-gingerol and luteolin targeting Nipah virus. J Pharm Res Int 33:477–484
Randhawa V, Pathania S, Kumar M (2022) Computational identification of potential multitarget inhibitors of Nipah virus by molecular docking and molecular dynamics. Microorganisms 10:1181
Sen N, Kanitkar TR, Roy AA, Soni N, Amritkar K, Supekar S, Nair S, Singh G, Madhusudhan MS (2019) Predicting and designing therapeutics against the Nipah virus. Plos Neglected Tropical Diseases 13
Rajput A, Thakur A, Rastogi A, Choudhury S, Kumar M (2021) Computational identification of repurposed drugs against viruses causing epidemics and pandemics via drug-target network analysis. Comput Biol Med 136
Rajput A, Kumar A, Kumar M (2019) Computational identification of inhibitors using QSAR approach against Nipah virus. Front pharmacol 10
Tigabu B, Rasmussen L, White EL, Tower N, Saeed M, Bukreyev A, Rockx B, Leduc JW, Noah JW (2014) A BSL-4 high-throughput screen identifies sulfonamide inhibitors of Nipah virus. Assay Drug Dev Technol 12:155–161
Acknowledgements
SY and SK want to thank the administration of Dorothy and George Hennings College of Science, Mathematics and Technology (HCSMT) of Kean University for providing research opportunities through research release time and resources.
Author information
Authors and Affiliations
Contributions
S.Y. (reviewing literatures, initial draft writing, editing) and S.K. (concept, reviewing, editing).
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yang, S., Kar, S. Are we ready to fight the Nipah virus pandemic? An overview of drug targets, current medications, and potential leads. Struct Chem 34, 2119–2137 (2023). https://doi.org/10.1007/s11224-023-02148-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-023-02148-6