
Abstract
Nature of bonding in the NgBeS (Ng = Ar, Kr, Xe) molecules has been studied using topological analysis of ELF, ELI-D functions with the wave function approximated at the DFT (M062X, B3LYP + ZORA), MP2, CCSD and CASSCF level of calculations. Both Xe–Be and Be=S bonds display topological features typical for the covalent-dative bonding. The V2(Xe) attractor characterising electron density, involved in interaction with the beryllium atom, is closer to the C(Be) core than to C(Xe). The population of the respective basin ranges between 1.59e (B3LYP + ZORA) and 1.83e (CCSD). The beryllium–sulphur bond is described by the bonding disynaptic basin V(Be,S) with the population between 3.22e (CASSCF) and 3.48e (M062X). The approximate weights for the Be–S and Be=S resonance forms are 0.3 and 0.7, respectively, in all molecules. Both the NgBe and BeS bonds are highly polarised with the values of the p SBe and p NgBe polarity indices (CCSD) of 0.8 and 0.9–1.0 for all studied molecules.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The main focus of this paper, as the title suggests, is an inquiry into the nature of interatomic interactions between the noble gas and beryllium atoms and also between the beryllium and sulphur atom in the ArBeS, KrBeS and XeBeS molecules. In order to gain a deeper insight into chemical bonding, electron density in the chemical bond area needs to be examined. Two methods, collectively known as quantum chemical topology [1, 2] are commonly used: topological analysis of electron localisation function η(r) (ELF) [3] and topological analysis of electron localisability indicator (ELI-D) [4, 5].
Classical view of 2-centre, 2-electron covalent A–B bond insists that electron density is shared by both atoms. Thus, two electrons should be found somewhere between core regions that itself do not participate in the covalent bonding. Within the molecular orbital theory, a doubly occupied molecular orbital confined to the region of the A–B bond should be found. In the real (physical) space, electron density, ρ(r), can be integrated within boundaries defined by ELF-topological basin V(A,B) associated with the local maximum (attractor) V(A,B) of η(r) field corresponding to the A–B bond [6].
Molecules formed by noble gas atoms are very interesting and relatively unstudied. Frenking et al. [7] showed that HeBeO, NeBeO and ArBeO are stable towards dissociation. NgLiF, NgBN and HeLiH structures have been shown to have much weaker Ng–AB interactions. These interactions were classed as van der Waals complexes, bound by dipole-induced dipole forces. Electron density analysis revealed charge-induced dipole interactions between the noble gas atoms and BeO. There was no evidence of any covalent interaction. The attractive Ng–Be interactions were thought to be partially enhanced by HOMO–LUMO interactions. Finally the HeBeO, NeBeO and ArBeO structures were classified as unusually stable van der Waals complexes [7].
Wang and Wang [8] suggest that the Ng–Be bond in the NgBeS molecule is formed with Lewis acid–base interaction, and electron density in the Ng lone pairs is donated to vacant orbitals on the Be centre. Their calculations yielded higher dipole moments for the NgBeS molecules than for BeS due to partial electron transfer from Ng to Be. The charge transfer from Ng to Be increased in the following order: Ne < Ar < Kr < Xe, which results in the blueshift from Ne to Xe of the BeS stretching modes for NgBeS molecules. Wang and Wang also observed similarly large blueshifts for noble gas hydrides, where charge transfer from antibonding molecular orbitals occurred due to environmental effects. The AIM parameters showed the bonding in XeBeS to be covalent in nature due to a droplet-like appendix of electron concentration towards the beryllium atom exhibited by the Xe atom. In general, the shape of the noble gas valence sphere showed increasing deformation of the Ne < Ar < Kr < Xe atoms. The Laplacian distribution suggests an increase in covalent character of the Be–Ng bond along the series from Ne to Xe. Overall, the observed infrared spectroscopy absorptions were in a very good agreement with the DFT and CCSD(T) results. The BeS diatomic molecule, which is a Lewis acid, can form neutral noble gas complexes, which show strong chemical binding between the Be and Ng atoms [8].
In this paper the nature of the bonding in the NgBeS (Ng = Ar, Kr, Xe) molecules is studied using the topological analysis of the electron localisation function and electron localisability indicator. Thus, the electronic structure has been analysed in the real space. In order to ensure reliability of results for the electronic properties, in addition to the DFT calculations with the B3LYP [9] and M062X [10] functionals, the wave functions approximated by Møller–Plesset perturbation theory (MP2) [11, 12] and post-Hartree–Fock numerical technique couple-cluster have been used, including single, double and triple excitations, the latter obtained at the CCSD(T) level [13]. Def2-TZVPPD basis set [14] has been used, where core electrons of the Xe atom have been replaced by effective core potential (ecp-28). Since the ecp-28 approximation may result in some ‘deformations’ of the ELF electronic structure, additional DFT(B3LYP) calculations have been performed for all-electron basis set, QZ4P [15] with relativistic effects described by zero-order regular approximation (ZORA) to the Dirac equation [16–18]. Finally, the electronic structure of XeBeS has been verified using correlated wave function approximated by single-point CASSCF(12,12)/Def2-TZVPPD//CCSD(T)/Def2-TZVPPD calculations (CASSCF—Complete Active Space Self-Consistent Field [19–24]).
Computational details
The NgBeS (Ng = Ar, Kr, Xe) geometrical structures have been optimised at the DFT(M062X)/Def2-TZVPPD, MP2/Def2-TZVPPD and CCSD(T)/Def2-TZVPPD computational levels using Gaussian 09 programme [25]. For the Xe atom, 28 electrons have been replaced by a pseudopotential. Additional optimisations for NgBeS have been performed using the DFT(B3LYP) method and all-electron QZ4P basis set for all the atoms. Relativistic effects have been incorporated through zero-order regular approximation (ZORA) to the Dirac equation as implemented in ADF modelling suit [26–28]. All geometrical structures have been fully optimised, and the minima found on the potential energy surface have been verified on the basis of harmonic infrared spectra with non-imaginary frequencies. The single-point CASSCF(12,12)/Def2-TZVPPD calculation for the XeBeS molecule has been carried out using the CCSD(T)/Def2-TZVPPD-optimised geometrical structure.
Topographical and topological analyses of the η(r) function have been performed using TopMod09 package [29] for the wave function approximated by the DFT(B3LYP + ZORA), MP2 and CCSD methods. For the wave functions approximated by the CCSD methods, the approximation based on natural orbitals and their occupancy proposed by Feixas et al. [30] has been used. For all the molecules, the topological analysis of ELF has been carried out using the parallelepipedic grid of points with step of 0.05 bohr. The topographical analysis of ELI-D function for CASSCF(12,12) and topological analysis of ELF obtained from ADF calculations have been carried out using DGrid 4.6 programme developed by Kohout [31]. The parallelepipedic grid of points with step 0.05 bohr has been used.
The graphical representations of ELF and ELI-D functions have been prepared using VMD programme.
Results and discussion
Values of the Ng–Be and Be–S bond length optimised at the DFT(M062X)/Def2-TZVPPD, DFT(B3LYP + ZORA)/QZ4P, MP2/Def2-TZVPPD and CCSD(T)/Def2-TZVPPD computational levels are collected in Table 1. All optimised geometrical structures of the NgBeS (Ng = Ar, Kr, Xe) molecules are linear. The Ng–Be and Be–S bond lengths increase from argon to xenon. The CCSD(T) calculations yield the longest Ng–Be bond, whereas the DFT(M062X) and DFT(B3LYP + ZORA) calculations the shortest ones. The differences between the Ng–Be bond lengths are: for Ar–Be calculated at CCSD(T) and DFT(M062X) levels: 0.056 Å; for Kr–Be and Xe–Be, where the shortest bond have been calculated at the DFT(B3LYP + ZORA) level these are: 0.043 and 0.030 Å, respectively. For the Be–S bond, the length varies from 1.730 Å with the M062X functional for ArBeS molecule to 1.760 Å with the CCSD(T) method. This tendency is also true for Kr and Xe. The Be–S bond lengths are the longest for CCSD(T) and the shortest for DFT(M062X) method.
Fundamental information on the nature of the bonding in the NgBeS (Ng = Ar, Kr, Xe) molecules can be obtained with topographical analysis of the ELF and ELI-D functions. The 2D representation of ELF for XeBeS calculated at DFT(B3LYP + ZORA)/QZ4P level is shown in Fig. 1. For the rare-gas atom region, a large valence domain surrounding the core can be found. Closer inspection shows three maxima of η(r) field. One of them is situated between core regions of the Xe and Be atoms. At the first glance, it might seem to be an indication the Xe–Be covalent bond. Two other domains correspond to three lone pairs of Xe in the Lewis formula. They display toroidal shapes in a 3D picture of ELF but appear as two maxima in a 2D slice of ELF. In the case of the BeS subunit, core regions of beryllium and sulphur and large valence domain corresponding to the valence electron density of the Be and S atoms can be distinguished. It is worth noting that the core domain of the Be atom is not contained in any larger valence domain. Such topography of η(r) has been previously observed and described for the LiH molecule [32]. For larger values of ELF, smaller valence domain situated between the core regions of Be and S is observed. According to the classification introduced by Silvi and Savin [3], its existence can indicate covalent BeS (Be=S, Be–S) binding. In the area around the sulphur atom, a valence domains corresponding to the lone pairs on S in the Lewis formula can be found. Very similar topography of ELF is observed for other studied molecules.

2D distribution of ELF for the XeBeS molecule calculated at the CCSD/Def2-TZVPPD//CCSD(T)/Def2-TZVPPD computational level
Analysis of the η(r) field topography in the XeBeS molecule poses a question: is the interaction between the rare gas and beryllium of covalent-dative character Xe → Be = S or is it a non-covalent Xeδ1+⋯ δ2−(Be=S) interaction. If the former, then a valence domain in Xe can be interpreted as a picture of the covalent Xe–Be bond. If the latter, valence bonding of Xe, situated between core domains of Xe and Be, can be a result of Ng valence shell polarisation by positively charged Beδ+ atom, with the formal charge of 2+ (assuming ionic bond of beryllium sulphide). Population analysis performed for the wave function obtained with the DFT(B3LYP + ZORA)/QZ4P method yields the Xe atomic charge of +0.29e (Mulliken), +0.37e (Hirshfeld) and +0.33e (Voronoi). Such polarisation of the Xe atom results in reorganization of valence electron density shown in ELF analysis as the local maximum. It is worth noting that interpretation of the valence domain situated between core domains as the indication of covalent bonding is not always clear-cut. For example, topographical analysis of ELF performed for molecules with ionic bonds such like LiF and LiCl also shows polarisation of halogen valence shell with two separated domains of which one is situated between core regions of X and Li atoms. Such domain is, however, related to the halogen valence shell polarisation effect, not covalent Li–F bonding. In order to confirm whether the interaction between a noble gas atom and the BeS molecule has covalent nature, further topological study is needed.
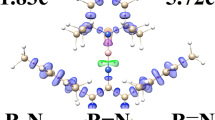
Topological analysis of ELF functions discussed in this paper is based on the calculations with the wave function approximated at the DFT(M062X)/Def2-TZVPPD, MP2/Def2-TZVPPD and CCSD/Def2-TZVPPD//CCSD(T)/Def2-TZVPPD computational levels. For all studied systems, the same number and types of seven core C(A) and valence V(A,B,…) attractors (A, B—symbol of atom) have been found, and thus only the results obtained with the CCSD/Def2-TZVPPD//CCSD(T)/Def2-TZVPPD method will be discussed. All attractors are of the point type (see Fig. 2) and are positioned on the symmetry axis. Three core attractors correspond to the core electrons of the beryllium C(Be), sulphur C(S) and noble gas C(Ng) atoms. In the BeS molecule valence space, two valence attractors are observed. One of them is the disynaptic bonding attractor V(Be,S), associated with a formal double Be=S bond or formal single Be–S bond, depending on the Lewis formula used for BeS. The other is a single monosynaptic, non-bonding attractor V(S) corresponding to two or three lone pairs of sulphur in the Lewis formula. Two attractors are also observed in the noble gas valence shell. The V2(Ng) attractor is situated between the C(Be) and C(Ng) core attractors. This attractor is associated with the valence electron density involved in binding between Ng and Be. The valence attractor, V1(Ng), corresponds to the non-bonding electron of the noble gas atom. It is classed as monosynaptic due to a single common surface with the core basin C(Ng).

Comparison of the electronic structure of the NgBeS (Ng = A, Kr, Xe) molecules, represented by the core and valence attractors of η(r) field
Interesting results has been obtained from the analysis of positions of the V2(Ng) attractor in the C(Ng)···C(Be) ‘bridge’ performed for the ArBeS, KrBeS and XeBeS molecules (see Fig. 2). In all those molecules, the V2(Ng) attractor is found at much larger distance from the core attractor C(Ng) than the V1(Ng) attractor. It reflects an essential perturbation of Ng valence electron density by beryllium and can support the hypothesis of the existence of covalent-dative bond, formed through electron pair donation to the valence region of the BeS molecule. Bearing in mind that polarisability of the noble gas atoms increases from the Ar to Xe atom [33], shortening of the distance between the V2(Ng) and C(Be) attractors from 1.27 Å in ArBeS, 1.19 Å in KrBeS to 1.14 Å in XeBeS is not surprising. For the most polarizable noble gas, xenon, perturbation of the valence shell is the largest. The relative position of V2(Ng) calculated in respect to the distance between C(Ng) and C(Be) is 0.39 (Ar), 0.47 (Kr) and 0.53 (Xe), respectively. These values show that for XeBeS the V2(Ng) attractor is situated closer to the C(Be) core attractor than to the C(Xe) one. This result is quantitatively different than the one obtained for ArBeS and KrBeS and can suggest much more advanced character of the bonding between the Xe and Be atoms.
Our next step is analysis of basin populations (\(\bar{N}\)) for all attractors found for the NgBeS molecules. The values of \(\bar{N}\) are collected in Tables 2, 3 and 4 for the wave function approximated by the DFT(M062X), MP2 and CCSD methods, respectively. Our discussion will concentrate on the results obtained at the CCSD/Def2-TZVPPD//CCSD(T)/Def2-TZVPPD level. For the covalent beryllium–sulphur bond, the basin populations of V(Be,S) are 3.31e (Ar) and 3.36e (Kr, Xe). Those results are closer to formal value of 4e (double bond) than 2e (single bond). The results confirm that representation of the bonding with the Lewis formula, containing the single Be–S bond does not provide a correct description of the binding. For the non-bonding electron density of sulphur, V(S) the basin populations lie in the range of 4.47e (Xe)–4.54e (Ar). These results are about 0.5e larger than those expected for two lone pairs. If the observed electronic structure is described by a resonance structure of two hybrids: (I) with the double Be=S and two lone pairs on S and (II) with the single Be–S bond and three lone pairs on S, their approximate weights can be calculated. These are: 0.7 (I) and 0.3 (II) for all NgBeS molecules. Thus, from the ELF perspective, the nature of the beryllium–sulphur bond in the BeS molecule has mainly character of a double bond.
The value of the basin population, obtained for the V2(Ng) basin, is much more interesting. This basin can corresponds to the binding between the Be and Ng atoms. Population for the V2(Ng) basin is smaller than 2e and varies between 1.49e (Ar) and 1.83e (Xe). Furthermore, going from argon to the most polarisable xenon, the V2(Ng) basin populations increases. If the V2(Ng) basin describes typical (represented by bonding basin in ELF picture) chemical bond, then the Ng–Be bond in both KrBeS and XeBeS molecules has a donor–acceptor character, close to a single-type bond. Such character seems to be the weakest (in term of basin population) for the ArBeS molecule.
Analysis of covariance of ELF basin populations helps in obtaining more information on electron density distribution in the NgBeS. It is a measure of electron density delocalisation between basins [34]. For the V(Be,S) basin, electron density is mainly delocalised with a lone pair, localised on sulphur, V(S) (cov[V(Be,S),V(S)] = −0.97(Ar), −0.96 (Kr), −0.95 (Xe)) and to much lesser extent with electron density of the atomic cores localised on sulphur and beryllium (cov[V(Be,S), C(S)] = −0.16(Ar), −0.17(Kr), −0.17(Xe) and cov[V(Be,S), C(Be)] = −0.09(Ar, Kr, Xe)). Analysis of electron density delocalisation in the V2(Ng) basin, involved in the binding with the BeS molecule, shows main delocalisation within the valence space of the noble gas atom, i.e. with the second valence basin V1(Ng) (cov[V1(Ng), V2(Ng)] = −0.66(Ar), −0.65(Kr), −0.61(Xe)). It is worth noting that delocalisation with electrons of the core basin C(Be) is very small (−0.05(Ar), −0.02(Kr) and −0.05(Xe)).
Polarity of chemical bond, A–B, can be quantitatively analysed using Raub and Jansen approach [35]. Topological partitioning of ρ(r) and η(r) fields are combined yielding the atomic contributions V(A,B)|A, V(A,B)|B and subsequently measure of the bond polarity—the polarity index, p AB. The value of p AB ranges between 0 for homopolar bonds and 1 for idealised ionic bonds. It is worth noting that for HF, HCl and HBr molecules p XH are as follows: 0.62, 0.14 and 0.04 [35].
Contribution of the noble gas atomic basins for beryllium and sulphur to ELF basins and the p SBe and p NgBe polarity indices are shown in Tables 2, 3 and 4. Populations of the atomic basins for the Be and S atoms, obtained from topological analysis of ρ(r) field (AIM), are 2.40e (Ar), 2.42e (Kr), 2.46e (Xe) and 17.55e (Ar), 17.54e (Kr, Xe), respectively. Those values show essentially polarised beryllium–sulphur bond with the topological charges of Be1.6 S−1.6. Contributions of the Be atomic basin to the V(Be,S) basin, V(Be,S)|Be, is very small (about 0.3e). However, the contribution of the S atomic basin, V(Be,S)|S, is about 10 times larger (3e). The value of the p SBe, polarity index, has the same value for all the molecules (0.8). Thus the covalent beryllium–sulphur bond is essentially polarised by sulphur and about 91 % of its electron population comes from the S atom. In the case of the V2(Ng) basin, involved in the interaction with the Be atom, almost all electron density is donated by the Ng atom, and the contribution of the Be atom is less than 0.1e. The value of the p NgBe index, larger than 0.9, shows that electron density residing between the C(Ng) and C(Be) cores comes almost entirely from the noble gas atom. This result supports the view that in the Ng–Be interaction there is no sharing of valence electrons from the Ng and Be atoms in the V2(Ng) basin, and the interaction has features of a donor–acceptor character.
In order to verify whether our interpretation of the nature of the Ng–Be and BeS bonding depends on the pseudopotential approximation applied for the xenon atom (Def2-TZVPPD), the topological analysis has been repeated for XeBeS using the wave function obtained from the DFT(B3LYP) calculations within the all-electron basis set QZ4P with relativistic effects described by ZORA (see “Computational details” section). The total number (7) and type of core (3) and valence (4) attractors localised in NgBeS is the same as obtained using other computational methods. 2D and 3D representations of ELF for XeBeS are shown in Fig. 3.

2D and 3D representations of electron localisation function (ELF) in XeBeS. Calculations performed at DFT(B3LYP + ZORA)/QZ4P computational level
Analysis of the ELF topography (2D) shows a valence region with high values of electron localisation (η (3,−3) = 0.88 Ar, 0.86 Kr, 0.85 Xe), situated between a large core region with visible shell structure of Xe and the core region of Be with equally distributed ELF values close to 1 characterising very high pairing in the K-shell. Furthermore, a relatively large region of very small ELF values (≈0.05), surrounding the core region of Be separating valence electrons of Xe from valence electrons of the BeS fragment, can be noticed. Values of basin populations are shown in Table 5. Population of the V(Be,S) basin is 3.38e (3.37e). This result is very similar to that obtained at the CCSD level. In the case of the V2(Ng) basin, the values of \(\bar{N}\) differ by 0.10e, 0.28e and 0.24e for ArBeS, KrBeS and XeBeS, respectively, from that calculated by CCSD method. The results of topological analysis performed using all-electron basis set with ZORA are in a good agreement with those obtained using the pseudopotential approximation.
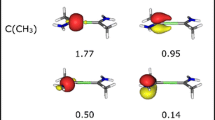
Finally, the electronic structure of the XeBeS molecule has been studied for the wave function calculated using the CASSCF(12,12)/Def2-TZVPPD method with the CCSD(T)/Def2-TZVPPD-optimised geometrical structures. The results of topological analysis of electron localisability indicator (ELI-D) field, displayed in a form of the core and valence attractors, are illustrated in Fig. 4. The number and type (core, valence) of attractors are the same as for the DFT and CCSD calculations, and thus both methods correctly describe electronic structures of studied molecules. The only difference lies in the type of valence attractors V(Xe), V(Be,S) and V(S), which are circular in comparison to the point-type attractors found using other methods. The circular attractor, V(Be,S), can suggest spatially more diffused Be=S bond, similar to π-bonding. Basin populations, also presented in Fig. 4, indicate 3.22e for the beryllium–sulphur bond.

Core and valence attractors localised for the ELI-D field in the XeBeS molecule. Calculations performed at the CASSCF(12,12)/Def2-TZVPPD//CCSD(T)/Def2-TZVPPD computational level
Conclusions
Three methods of quantum chemical topology, namely topological analysis of electron localisation function, electron localisability index, and electron density (used for calculation of polarity index) have been used to investigate the electronic structure of the NgBeS (Ng = Ar, Kr, Xe) molecules. Calculations performed for wave functions, approximated by the DFT(M062X, B3LYP + ZORA), MP2, CCSD and CASSCF//CCSD computational methods yielded qualitatively very similar results. Both pseudopotential approximation (ecp-28) and ZORA, used to account for relativistic effects, also yielded very similar topology of ELF.
The NgBeS (Ng = Ar, Kr, Xe) molecules consist of the noble gas atom and the beryllium sulphide molecule, bound by the Ng–Be interaction with topological features resembling covalent-dative bonding. The perturbation of the noble gas valence shell at the beryllium end is dramatic in comparison with the part not involved in an interaction. It is the most profound for xenon which has the largest polarisability. The V2(Xe) attractor is localised closer to C(Be) than to C(Xe) core thus the Xe–Be interaction differs from the Ar–Be and Kr–Be interactions. The population of the V2(Xe) basin is in a range of 1.59e (B3LYP + ZORA)–1.83e (CCSD).
The beryllium sulphide molecule is bound by a very polarised covalent bond, with 91 % of the electron density donated by the sulphur atom. Formally this bond could be considered ionic, Be2+S2−. However, analysis of the ELF-results shows all typical features of the covalent-dative bonding. The V(Be,S) attractor is a valence disynaptic attractor, situated approximately in the geometrical centre of the bond and the basin population values are between 3.22e (CASSCF) and 3.48e (M062X). When two resonance structures, Be=S and Be–S, are taken into account, their partial contribution in a mesomeric equilibrium yield 0.7 and 0.3 values. In summary, the topological analysis of ELF confirms that the XeBeS molecules are bound by two covalent-dative bonds.
References
Popelier PLA (2005) In: Structure and bonding. Intermolecular forces and clusters. Springer, Heidelberg
Popelier PLA, Brémond ÉAG (2009) Int J Quantum Chem 109:2542–2553
Silvi B, Savin A (1994) Nature (London, UK) 371:683–686
Kohout M (2004) Int J Quantum Chem 97:658
Kohout M (2007) Faraday Discuss 135:43–54
Fradera X, Austen MA, Bader RFW (1999) J Phys Chem A 103:304–314
Frenking G, Koch W, Gauss J, Cremer D (1988) J Am Chem Soc 110:8007–8016
Wang Q, Wang X (2013) J Phys Chem A 117:1508–1513
Becke AD (1993) J Chem Phys 98:5648–5652
Zhao Y, Truhlar DG (2008) Theor Chem Acc 120:215–241
Møller C, Plesset MS (1934) Phys Rev 46:618–622
Head-Gordon M, Pople JA, Frisch MJ (1998) Chem Phys Lett 153:503–506
Pople JA, Head-Gordon M, Raghavachari K (1987) J Chem Phys 87:5968–7595
Rappoport D, Furche F (2010) J Chem Phys 133:134105–134111
van Lenthe E, Baerends EJ (2003) J Comput Chem 24:1142–1156
van Lenthe E, Baerends EJ, Snijders JG (1993) J Chem Phys 99:4597–4610
van Lenthe E, Baerends EJ, Snijders JG (1994) J Chem Phys 101:9783–9792
van Lenthe E, Ehlers AE, Baerends EJ (1999) J Chem Phys 110:8943–8953
Hegarty D, Robb MA (1979) Mol Phys 38:1795–1812
Eade RHA, Robb MA (1981) Chem Phys Lett 83:362–368
Schlegel HB, Robb MA (1982) Chem Phys Lett 93:43–46
Bernardi F, Bottini A, McDougall JJW, Robb MA, Schlegel HB (1984) Symp Chem Soc 19:137–147
Frisch MJ, Ragazos IN, Robb MA, Schlegel HB (1992) Chem Phys Lett 189:524–528
Yamamoto N, Vreven T, Robb MA, Frisch MJ, Schlegel HB (1996) Chem Phys Lett 250:373–378
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA et al (2010) Gaussian 09, revision B.01. Gaussian, Inc., Wallingford, CT
te Velde G, Bickelhaupt FM, van Gisbergen SJA, Fonseca Guerra C, Baerends EJ, Snijders JG, Ziegler T (2001) J Comput Chem 22:931–967
Fonseca Guerra C, Snijders JG, te Velde G, Baerends EJ (1998) Theor Chem Acc 99:391–403
ADF2013 (2013), SCM. Theoretical chemistry. Vrije Universiteit, Amsterdam, http://www.scm.com
Noury S, Krokidis X, Fuster F, Silvi B (2009) Topmod09. Université Pierre et Marie Curie and CNRS, Paris
Feixas F, Matito E, Duran M, Solà M, Silvi B (2011) J Chem Theory Comput 7:1231
Kohout M (2011) DGrid, version 4.6, User’s Guide, Radebeul
Savin A, Silvi B, Coionna F (1996) Can J Chem 74:1088–1096
Anslyn EV, Doughert DA (2006) Modern physical organic chemistry. University Science Book, Sausalito, CA
Silvi B (2004) Phys Chem Chem Phys 6:256–260
Raub S, Jansen G (2001) Theor Chem Acc 106:223–232
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Makarewicz, E., Gordon, A.J. & Berski, S. How many electrons form chemical bonds in the NgBeS (Ng = Ar, Kr, Xe) molecules? Topological study using the electron localisation function (ELF) and electron localisability indicator (ELI-D). Struct Chem 27, 57–64 (2016). https://doi.org/10.1007/s11224-015-0719-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-015-0719-0