
Abstract
A wide range of natural products important for the engineering and drug design of pharmaceuticals comprise largely of nitrogen-based heterocycles. Fungal natural products have proven to be a rich source of the industrially-important molecules, many of which are promising drug leads. Although, natural products containing a phthalimidine core tends not to be given distant classification, but compounds containing these structures exhibit antimicrobial, anthelmintic, antimalarial and insecticidal activities, and are among the potential target for discovering new drug candidates. Intriguingly, these are primarily isolated from fungal sources and to a very lesser extent from plants or bacteria. This review surveys fungal-derived phthalimidine metabolites published until the end of 2022, isolated from both terrestrial and aquatic or marine sources with emphasis on their unique chemistry, bioactivities, biogenesis and taxonomic classification. Their unique chemistry and diverse bioactivities (including antiviral, antiproliferative, antioxidant and antimicrobial) provide a chemical library with high medicinal potential, representing a treasure trove for synthetic chemists.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A vast number of nitrogen-containing heterocyclic compounds are known to exhibit wide range of pharmacological activities including anticancer, anti-HIV, antimalarial, anti-tubercular, anti-microbial and anti-diabetic (Chaudhari et al. 2016; Kerru et al. 2020; Ma et al. 2018). Natural N-heterocyclic moieties are part of many biologically important molecules, including vitamins, nucleic acids, pharmaceuticals, antibiotics, dyes and agrochemicals. The reason behind the variety of biological activities of these compounds is that the electron-rich nitrogen atom readily accepts or donates a proton and can easily establish diverse weak interactions with other molecules, which allow N-heterocyclic compounds to bind with active sites of enzymes and receptors in biological targets. FDA databases discloses that about 75% of unique small-molecule drugs contain a nitrogen-based heterocycle, which are important in drug design and engineering of pharmaceuticals (Kerru et al. 2020).
Among such heterocycles, phthalimidines got fame in several capacities including discovery of new drug candidates (Tok et al. 2023) and exhibit antimicrobial, anthelmintic, antimalarial and insecticidal activities (Chi et al. 2022). In the synthetic chemistry field, phthalimides are reduced to phthalimidines (1,3-dihydro-2H-isoindole-1-one, A), which are a rare class of N-heterocyclic natural products. These compounds are characterized by a bicyclic nucleus derived from fusion of a benzene ring and a γ-lactam. They are a growing class of compounds attracting the interest of synthetic chemists due to their wide applications in material sciences, serving as pigments or biochemical fluorescent markers (Azumaya et al. 1991; Iqbal et al. 2009; Tsuruta and Inoue 1998). Furthermore, they are valuable building blocks for the synthesis of medicinally relevant compounds due to their diverse biological activities. Pharmaceutically important compounds with a phthalimidine skeleton include the thiazide diuretic, chlorthalidone (B); the cyclooxygenase (COX)-inhibitor, indobufen (C); and the thalidomide derivative, lenalidomide (D) (Fig. 1). Their wide spectrum of biological activities is mainly due to their diverse structural functionalisation, making them an interesting class of compounds to be explored further.

Pharmaceuticals containing a phthalimidine core structure
Interestingly, phthalimidine core is also found in nature as part of a variety of secondary metabolites, however less abundant in nature compared to their oxygen containing analogues known as phthalides. They are reported primarily from fungi and to a lesser extent from bacteria and plants. The fungal kingdom is therefore regarded as a prolific producer of phthalimidine-containing metabolites, and this review aims to illustrate the diversity of this rare and unique class of fungal metabolites. Molecules containing the phthalimidine-core tend not to be given a distinct classification and are often labeled as polyketides, meroterpenoids, alkaloids or nitrogen-containing compounds/heterocycles. To date, phthalimidines have been reported from the fungal sub-kingdom Dikayra (higher fungi), comprising the fungal phyla Ascomycota and Basidiomycota, both of which have been well-studied for their secondary metabolite (SM) production. Here fungal-derived phthalimidines from both terrestrial and aquatic marine and non-marine sources are surveyed till the end of 2022, highlighting characteristic chemical features, interesting bioactivities and insights into their biosynthesis. Grouping these compounds according to their chemical structure (e.g. simple phthalimidines, prenylated phthalimidines (meroterpenoids) or hybrid phthalimidines) proved challenging, as several fungal species were found to simultaneously produce several phthalimidines with various functionalities. Thus, we opted for a taxonomic classification, as specific classes of SMs have been widely used in fungal chemotaxonomic profiling (Frisvad et al. 2008; Helaly et al. 2018; Wang et al. 2015a).
Chemodiversity and biological activity of fungal phthalimidines
Phthalimidines from Ascomycete
Phthalimidines from Aspergillus species (phylum Ascomycota, order Eurotiales)
The fungal genus Aspergillus of Ascomycetes has been intensively investigated for its bioactive natural products. This genus has also been identified as producer of various phthalimidines-derivatives. For example the first phthalimidine, duricaulic acid (1) was isolated in 1985 from Aspergillus duricaulis (Achenbach et al. 1985). Although phthalide analogues of compound 1 from the same fungus were identified as antimicrobial, however, no such activity was reported for compound 1. Similarly 6-hydroxy-4-methoxy-5-methylphthalimidine (2) was isolated from Aspergillus silvaticus (Ageta and Ageta 1984; kawahara et al. 1988), and later it was re-isolated from Alternaria cichorii, and was named cichorine (2) (Stierle et al. 1993), but again no activity has been reported for this compound. Compounds 1 and 2 isolated as new natural products, but were never tested for their medicinal potential, however, newer analogues 8-methoxycichorine (3), 8-epi-methoxycichorine (4), and N-(4’-carboxybutyl) cichorine (5) (Liao et al. 2019), separated through genetic inactivation of cichorine biosynthetic pathway in Aspergillus nidulans strain LO8030, were identified as phytotoxic and could be candidates for herbicide development. A genetic dereplication strategy in the fungus A. nidulans also produced a hybrid metabolite aspercryptin (6) (Chiang et al. 2016), which is a cichorine-derived hexapeptide with 2-aminododecanol and 2-aminocaprylic acid residues. Other phthalimidines obtained due to epigenetic modification through histone deacetylase inhibition (HDACi) and MS/MS networking were characterized as aspercryptin analogues 7–9 (Henke et al. 2016). Unfortunately, these metabolites were also not studied for their bioactivity.
In addition to epigenetic modification techniques, the one-strain many compounds (OSMAC) approach is another powerful tool to investigate microorganisms secondary metabolome through alteration of cultivation parameters (Bode et al. 2002). This approach led to the isolation of the unique prenylated phthalimidines, aspernidine A (10) and B (11) (Scherlach et al. 2010) and F–H (12–14) from the fungus A. nidulans (Li et al. 2020), which showed moderate antiproliferative activity against various tumour cell lines, especially compounds 13 and 14 exhibited significant IC50 values i.e. in the range of 4.77–33.03 μM against HL-60, A-549, SMMC-7721, MCF-7, and SW- 480 cancer cell lines. Further, compounds 10 and 11 were re-isolated from a mangrove endophytic Aspergillus sp. (HK-ZJ). In addition, HK-ZJ species has also been reported to produce emerimidines A (15) and B (16) and the O-prenylated phthalimidines emeriphenolicin A-D (17–20) (Zhang et al. 2011). Compound 10–20 are prenylated analogues except compounds 15 and 16; these compounds exhibited moderate in-vitro inhibitory activity against anti-influenza A virus (H1N1) replication in MDCK cells, with IC50 values of 201 and 296.8 μM, respectively. It can be predicted that the absence or presence of a smaller prenyl moiety may be significant in antiviral activity. Compound 15 also showed antimicrobial activity against Bacillus subtilis, Candida albicans, Salmonella typhi and methicillin-resistant Staphylococcus aureus (MRSA), with a MIC value of 59.9 μM and Aspergillus fumigatus, Klebsiella pneumoniae and Pseudomonas aeruginosa with a MIC value of 59.8 μM (Yashavantha Rao et al. 2017). This compound has also been identified as metabolite of an endophytic fungus Aspergillus variecolor CLB38. Another endophytic Aspergillus sp., A. nidulans HDN12-249 produces N-farnesyl dicarboxylate derivatives of phthalimidines; emeriphenolicins E–G (21–23), and macrolide derivatives; emericellolides A-C (24–26), composed of an unprecedented L-glutamate fragment and a sesquiterpene moiety. All these isolates were screened for cytotoxic activity but only compound 21 showed selective activity against three human cancer cell lines; HeLa, A549, and HCT-16, with IC50 values of 4.77, 12.04, and 33.05 μM, respectively (Zhou et al. 2016). A review of their structure–activity relationship (SAR) revealed that the macrolide moiety eliminates cytotoxic activity of these compounds.
Enantiomers of ( ±)-asperglactam (27) were isolated from the mangrove endophytic fungus Aspergillus versicolor SYSU-SKS025, using a chiral HPLC column, and were identified due to their opposite optical rotations and opposite Cotton effects at 210 nm in their circular dichroism (CD) spectra. Both enantiomers were nearly equally active (IC50 value of 50.5 and 60.1 μM, respectively) against the enzyme α-glucosidase, and although the investigators did not comment on the SAR, it is speculated that a chiral center is not necessary for enzyme inhibition activity, rather the phthalimidines core is essential (Cui et al. 2018b). The phthalimidines isolated from various Aspergillus species are presented in Fig. 2.


Phthalimidines isolated from Aspergillus species
Phthalimidines from Alternaria species (phylum Ascomycota, order Pleosporales)
Alternaria fungi are plant pathogens, since they produce phytotoxins (Logrieco et al. 2009), however, they are producing other worthy metabolites, for example mycotoxin cichorine (2), O-prenylated analogues, zinnimidine (28) and Z-hydroxyzinnimidine (29), which were isolated from the phytopathogenic fungus A. cichorii (Fig. 3). Unlike cichorine (2), the zinnimidines (28 and 29) are non-phytotoxic compounds. This indicates that prenylation could have masked the phytotoxicity by converting the phenolic function to an ether group; however, later the analogues of 28 as N-substituted compounds porritoxin (30) and porritoxin sulfonic acid (31) (Fig. 4), isolated from A. porri were also found phytotoxic. These variations in activity could be attributed to the experimental design and not the structural diversity. The fungus A. porri is known as the causal fungus of black spot disease in stone-leek and onion (Horiuchi et al. 2003; Suemitsu et al. 1995). The phytotoxic compound 30 was first reported in 1992 as a benzoxazocine derivative (Suemitsu et al. 1992), whose structure was revised a decade later through detailed 2D NMR as 2-(2′-hydroxyethyl)-4-methoxy-5-methyl-6-(3′′-methyl-2′′-butenyloxy)-2,3-dihydro-1H-isoindol-1-one (Horiuchi et al. 2002) and subsequently confirmed through total synthesis (Cornella and Kelly 2004). Compound 30 inhibited seedling growth at concentrations as low as 32.7 μM, while compound 31 exhibited weaker phytotoxic activity compared to compound 30, possibly due to sulfonation of the terminal hydroxyl group (Horiuchi et al. 2003). Porritoxin (30) belongs to the group of non-host specific phytotoxins and has the potential to be developed as a natural broad spectrum herbicide (Xu et al. 2021). In cytotoxicity assays, 28 was not active against both HeLa and KB cells (Phuwapraisirisan et al. 2009), however, its N-substituted analogue 30 showed 96% inhibition of Epstein-Barr virus early antigen (EBV-EA) activation induced by 1000 mol ratio of 12-O-tetradecanoylphorbol-13-acetate (TPA), without remarkable cytotoxicity on the viability of Raji cells. This is superior to the 91.9% inhibition of EBV-EA activation of the known anti-tumor promoter β-carotene, which makes it a potential anti-tumor agent (Horiuchi et al. 2006).

Phthalimidines isolated from Alternaria species

Phthalimidine-type triprenyl phenols isolated from Stachybotrys spp
Phthalimidines from Stachybotrys species (phylum Ascomycota, order Hypocreales)
Stachybotrys spp. are prolific producers of SMs and their metabolites were comprehensively reviewed till the year 2014 by Wang et al. (Wang et al. 2015a). An important class of Stachybotrys metabolites are the triprenyl phenols (TPPs) consisting of a multitude of compounds, of which those incorporating a phthalimidine nucleus will be discussed here in detail.
The first phthalimidine-type TPPs, stachybotrins A (32) and B (33), were reported in culture of Stachybotrys sp. (CS-710–1) collected from brackish water in Florida (Xu et al. 1992). Later stachybotrin C (34) was isolated from the liquid culture broth of Stachybotrys parvispora F4708 (Nozawa et al. 1997a, 1997b). Compounds 32–34 possess a unique pyrano-isoindolinone ring system, with compound 34 having an additional ethyl phenol moiety. Compounds 32 and 33 showed antibacterial activity against B. subtilis (ATCC 6051) and antifungal activity against Ascobolus furfuraceus (NRRL 6460) and Sordaria fimicola (NRRL 6459) (Xu et al. 1992). Interestingly, compound 34 demonstrated promising neuritogenic activity, where it induced significant neurite outgrowths in PC12 cells and showed cell survival activity in the primary culture of cerebral cortical neurons. This protective effect on neuronal cell damage assumes it functions as a neurotrophic factor in cerebral neurons, and is thus expected to prevent hypoxic neuronal injury caused by ischemia and could therefore be a strong candidate for the treatment of neurodegenerative diseases (Nozawa et al. 1997b). Later, the total synthesis of compound 34 and its four stereoisomers helped to establish the absolute configurations of the stereocenters C-8 and C-9, with the relative configuration of 34 initially assigned as (8S*, 9S*) based on NOESY analysis. This was revised to (8S, 9R), and was further confirmed through X-ray diffraction analysis of its 4-bromobenzyl ether derivative (Jacolot et al. 2013; Kuroda et al. 2018).
Stachybotrys microspora IFO30018 has been reported to produce plasminogen activators, staplabin (35) and its analogues Stachybotrys microspora triprenyl phenols (SMTPs) (36–43). These natural products were named after the producing organisms S. microspora TPP (Hasumi et al. 1998; Hu et al. 2000; Kohyama et al. 1997; Shinohara et al. 1996). The staplabin/SMTP molecule consists of a tricyclic γ-lactam moiety, an isoprene side chain and an N-linked side chain, which only differs among congeners. Compounds 35–41 are single-unit congeners with an amino acid as an N-linked side chain, while compounds 42 and 43 are two-unit congeners with two core SMTP structures bridged by ornithine and lysine, respectively. The two-unit congeners 42 and 43 have been reported as the most potent in enhancing plasminogen-fibrin binding, urokinase-catalyzed activation of plasminogen, and urokinase- and plasminogen-mediated fibrin degradation (Hu et al. 2000). In particular the efficacy of 42 (orniplabin) in treating several types of ischemic strokes (including thrombotic and embolic strokes) in rodents and primates models was studied (Sawada et al. 2014; Suzuki et al. 2018). Another marine fungus Stachybotrys longispora FG216 was found to produce fungi fibrinolytic compound 1 (FGFC1), with the same structural features to that of 42 and likewise showed in-vitro and in-vivo fibrinolytic activity (Wang et al. 2015b). Fungi fibrinolytic compound 2 (FGFC2) (44) was later identified from the same fungal strain and being a one-unit congener, displayed moderate in-vitro fibrinolytic activity (Guo et al. 2016).
In addition to epigenetic modification, biotransformation (culture supplements) is another potential tool to obtain diverse fungal metabolites. Based on this technique, the supply of amino compounds to S. microspora and S. longispora FG216 cultures provided more than 50 SMTP congeners, which led to establish a detailed insight into their Severe Acute Respiratory Syndrome (SARs) (Hasegawa et al. 2010; Hasumi and Suzuki 2021; Koide et al. 2012; Matsumoto et al. 2015; Yin et al. 2017). This revealed that the N-linked side chain is crucial for their plasminogen-modulating activity. In fact, SMTP-0, the most basic congener lacking the N-linked side chain, showed no plasminogen modulator activity (Hasumi et al. 2007). Thus, SMTPs are regarded as prospective candidates for the development of a drug useful for ischemic diseases that are associated with inflammation, such as stroke. Stachybotrys sp. phthalimidine-type TPPs are illustrated in Fig. 4.
Phenylspirodrimanes (spirocyclic drimanes) are another important group of structurally diverse Stachybotrys TPPs, featuring a drimane skeleton-containing sesquiterpene with a benzene ring attached through a spirofuran. In accordance with the focus of this review, only phthalimidine-type phenylspirodrimanes have been reviewed and included. The first member of this class has been reported to be stachybotramide (45) from the aspen-tree fungus Stachybotrys cylindrospora (Ayer and Miao 1993). It was also found in the culture of the black toxic mold Stachybotrys chartarum S-17. The same fungus and other species of this group, S. chartarum MRC 1422 and Egypt 1, were found to produce 45 and the structural analogues; stachybotrylactam (46), stachybotrylactam acetate (47) and 2α-acetoxy stachybotrylactam (48) (Jarvis et al. 1995). Although no activity has been reported for these metabolites, due to their structural diversity, they are important targets for a medicinal chemist.
In 1997, Kamalov et al. isolated a nitrogen-containing compound from cultures of S. chartarum, established the structure and named as stachybotrin (Kamalov et al. 1997), which in fact was incorrectly assigned, however, a year later the structure was revised and confirmed by single X-ray crystallography, revealing it possessed the same structure and stereochemistry as compound 45 (Kamalov et al. 1998). The same fungus also produces a methylated analogue of 45, which was coined stachybotrin A (49), not to be confused with the precedent stachybotrin A (32) (Kamalov et al. 1999). In another study on the Stachybotrys strain F-1839 (closely related to S. chartarum), seven more congeners designated as F1839-A, -B, -C, -D, -E, -F, and -J (50–56) were isolated. They exhibited moderate pancreatic cholesterol esterase activity (Sakai et al. 1995).
The stachybocins A-C (57–59) are phenylspirodrimane dimers connected by a lysine residue and only differing in the hydroxyl group substitution in the drimane skeleton. They are produced by the soil-derived Stachybotrys sp. M6222 taxonomically related to S. chartarum (Nakamura et al. 1995; Ogawa et al. 1995). These compounds are reported as endothelin (ET) receptor antagonists with no in-vivo or in-vitro toxicity, making them potential candidates for the treatment of some cardiovascular diseases. They are commercialized as dual enzyme inhibitors inhibiting the binding of ET-1 to human ET-subtype A and ET-subtype B receptors in ligand binding assays.
Spirodihydrobenzofuranlactams I (46), II (45), III (60), IV (61) and VI (57) were identified from cultures of two different Stachybotrys strains, DSM 8767 and F11402. Similar to the stachybocins (57–59), they showed antagonistic effects in the ET receptor binding assay and additionally inhibited HIV-1 protease. These are regioisomers to the already reported phenylspirodrimanes. Intrigued by the similar ET receptor antagonist activity to stachybocins (57–59) (Roggo et al. 1996a, 1996b) and to assign the absolute configuration, however, their structures were revised through synthesis (Deng et al. 2003; Kende et al. 2003). In fact, spirodihydrobenzofuranlactams I has the same structure as compound 46, spirodihydrobenzofuranlactams II has the same structure as compound 45, and spirodihydrobenzofuranlactams VI has the same structure as compound 57. The most active of these compounds was spirodihydrobenzofuranlactams VI (57), with IC50 values of 1.5 μM in the ET-subtype A receptor binding assay and 11 μM in the HIV-1 protease inhibition assay (ROGGO et al. 1996b).
Tyrosine kinase receptors with immunoglobulin and epidermal growth factor (EGF) homology domains (Tie2) and their ligands are important in angiogenesis and small molecule inhibitors of Tie2 activity are of therapeutic interest. Vázquez et al. found that extracts of a soil-derived isolate S. chartarum inhibited the Tie2 kinase receptor, from which seven K-76 phenylspirodrimane derivatives (62–68) differing in the N-substituted side chain were isolated (Vázquez et al. 2004). All the seven compounds 62–68 reproducibly inhibited Tie2 kinase receptor, with 65 showing the highest inhibition (IC50 = 25 μM). The reported inhibitory potential of each compound and structural features are not helpful to comment on SAR.
Stachybotrys spp. derived from marine sources further enriched the growing array of phthalimidine-type phenylspirodrimanes, since the endophytic fungus S. chartarum MXH-X73 associated with the sponge Xestospongia testudinaris, produced stachybotrins D-F (69–71), the phenylspirodrimane dimers stachybocins E (72) and F (73) and compound 62 (Ma et al. 2013). In antiviral assays against HIV-1, only compound 69 displayed inhibitory effects on wild-type and five non-nucleoside reverse transcriptase (RT) inhibitors (NNRTI)-resistant HIV-1 strains with no cytotoxicity. EC50 values for inhibitory effects on HIV-1 replication were between 6.2 and 23.8 μM. Compound 69 also inhibited HIV-1 RT RNA-dependent DNA polymerase activity in a dose-dependent manner with an EC50 of 50 μM, providing a new chemical class of novel NNRTIs (Minagawa et al. 2002c). SAR could suggest that the nature of the N-alkyl chain is important, which revealed a keto-function and overall hydrophobicity could be the activity determining factors. Ma et al. characterized stachybotrin G (74) (Ma et al. 2015), which does not belong to the isoindolinones but is rather a cyclised isoindolinimine with farnesylated decahydropyrrolo[1,2-α][1,3]diazocine skeleton. A year earlier, Zhou et al. isolated compound 45 and a derivative stachybotrin G (75) with a 2-piperidinone as the N-substituent, from the soil-derived fungus S. parvispora HS-FG-843 (Zhou et al. 2015). Compounds 45 and 75 were reported to possess in-vitro cytotoxic activity against the human lung carcinoma A549 cell line with IC50 values of 22.1 and 36.1 μM, respectively. This was the first report of any bioactivity for compound 45, specifically cytotoxicity, which is contrary to the non-cytotoxic activity of the structurally related marine-derived compounds 69–71, when tested at a concentration of 10 μM (Ma et al. 2013). Stachybotrys spp. phthalimidine-type phenylspirodrimanes are illustrated in Fig. 5.

Phthalimidine-type phenylspirodrimanes isolated from Stachybotrys spp
Another marine sponge associated fungus S. chartarum strain WGC-25C-6 produced chartarlactams A-P (76–91), in addition to the compounds 45–48, 50, 53 and 54 (Fig. 6). An interesting aspect of this work is that compound 79 possess the same phenylspirodrimane nucleus as compound 46 but with a d-glucose attached by α-glycosidic bond to the phenol. Another new skeleton was compound 87 which is a symmetrical dimer of two units of compound 76 connected through a double bond across C-8′ and C-8‴. It was also noted that phenylspirodrimane analogues with an 8β-CH3 orientation, i.e. compounds 88–91 were reported for the first time as natural products. Several of the isolated compounds displayed potent antihyperlipidemic activities in a HepG2 hepatocarcinoma cell model at a dose of 10 μM. Compounds 86 and 90 selectively reduced intracellular triglyceride (TG) levels, compounds 79, 87 and 89 reduced total cholesterol (TC) levels while compounds 80, 81 and N-(2-benzenepropanoic acid) stachybotrylactam (92) reduced both TG and TC levels (Li et al. 2014b). Another investigation of S. chartarum WGC-25C-6 also provided prenylated phthalimidines, chartarutines A-H (93–100) (Fig. 6). For compounds 95 and 96, this was the first report of a sulphate group in the TPP skeleton. However, compound 96 has an isoindolinimine skeleton rather than an isoindolinone. 2D-NMR fully proved the established structures. In anti-HIV bioassays, compounds 94, 99 and 100 exhibited significant inhibitory effects against HIV replication in one-cycle infection assays with IC50 values of 4.90, 5.57 and 5.58 μM, respectively. Although such activity is not comparable to the positive control (efavirenz, IC50 = 0.65 μM), their lower cell cytotoxicity (CC50 < 100 μM) warrants further investigation as anti-HIV drug candidates (Li et al. 2014a).

Chartarlactams and chartarutines isolated from the marine strain Stachybotrys chartarum WGC-25C-6
A marine driftwood-derived fungus Stachybotrys sp. strain MF347 has been reported to produce several phenylspirodrimanes, e.g. compounds 45–48, 57, 58, 86 and 90 along with stachyin A (101) and B (102). Compound 102 was the first example of a spirocyclic drimane coupled with a spirodihydrobenzofuranlactam unit and a spirodihydroisobenzofuran unit with the connecting position being N–C instead of an N–N. Intriguingly, only the spirocyclic drimanes with two sesquiterpene-spirobenzofuran structural units (i.e. compounds 57, 58 and 102) showed antibacterial activity against the gram-positive strains B. subtilis, Staphylococcus epidermidis and MRSA. This implied that a structural feature of two sesquiterpene-spirobenzofuran units with either a N–C or a N–N linkage of spirocyclic drimanes is essential for antibiotic activity. Furthermore, only compound 102 showed weak cytotoxic activity for the mouse fibroblasts cell line NIH-3T3 and for the carcinoma cell line HepG2 with IC50 values of 13.01 and 14.27 μM, respectively (Wu et al. 2014).
Stachybotrin H (103) and its analogues were separated from the deep-sea derived fungus Stachybotrys sp. MCCC 3A00409 (Ma et al. 2019). The same fungus and another marine-derived Stachybotrys sp. MF347 are also reported to produce spirobenzofuranlactam derivatives featuring either an N-leucine (104) or N-phenylalanine moiety (105). These compounds and other derivatives were earlier isolated from S. chartarum ST002348, DSM 14952, and were patented by Sanofi-Aventis Deutschland GmbH (Eder et al. 2007). The endophytic fungus S. chartarum further produces stachartins C-E (106–108), differing in the N-linked side chain, in addition to the ubiquitous stachybotrylactam (46). Although no activity was reported for the isolated metabolites (Chunyu et al. 2016), but their unique structural motifs can be of interest for organic and medicinal chemists. Since the fungus Stachybotrys sp. MCCC 3A00409, obtained from deep sea, also produced compound 108; this indicates that Stachybotrys sp. obtained from different sources sometimes produce the same metabolites, which could be used as a chemotaxonomic marker for these strains.
The Shionogi Research laboratories in Japan reported the isolation of anti-influenza A metabolites, stachyflin (109) and acetylstachyflin (110), from solid cultures of Stachybotrys sp. RF-7260. Compound 109 showed potent in-vitro activity against influenza A virus (H1N1) with an IC50 of 0.003 μM, which is exceptionally superior to that of the known antivirals amantadine (IC50 = 5.3 μM) and zanamivir (IC50 = 0.75 μM) (Minagawa et al. 2002a, 2002c). Its novel pentacyclic skeleton including a cis-fused decalin and a pyranoisoindolinone ring system makes it not structurally related to other known anti-influenza virus agents, which is accountable for its unique mode of action as a potent hemagglutinin inhibitor. Hence, its unique chemistry and novel antiviral mechanism of action makes it an interesting synthetic target to develop new anti-influenza agents (Minagawa et al. 2002b; Sakurai et al. 2011; Taishi et al. 1998; Watanabe et al. 2010; Wildermuth et al. 2017).
Chemical investigation of a commercial strain of S. chartarum CGMCC 3.5365 afforded four farnesylated phthalimidine derivatives, stachybotrysams A–D (111–114), and one farnesyl-cyclised analogue, stachybotrysam E (115), along with the congener 94. Compounds 112–114 showed an attached sulphate moiety, which was the second report of a 4-O-sulphation in the prenylated isoindolinone scaffold (i.e. compounds 95 and 96). Intriguingly, compound 114 has an α-d-glucopyranose attached as an N-substituent never encountered before in this skeleton. Compound 115 is a phenylspirodrimane analogue of compound 89 with the less common 8β-CH3 orientation, but with a butyric acid unit as an N-linked side chain. In a similar anti-HIV bioassay used for chartarutines (93–100), compounds 111–113 exhibited significant inhibitory effects against HIV-1 virus with IC50 values of 9.3, 1.0, and 9.6 μM, respectively (Zhao et al. 2017a). Compound 112 with its lower cytotoxicity and significant inhibitory activity is worthy for further investigation as an anti-HIV lead compound. Later, the re-investigation of S. chartarum CGMCC 3.5365 afforded another analogue with the same phenylspirodrimane skeleton, except for a methyl group attached on the nitrogen, and was given the name stachybotrin E (116), besides the fungus also produces compound 46 (Feng et al. 2019). Both compounds 46 and 116 were evaluated for inhibitory effect on potassium channel Kv1.3 but were found to be inactive. It should be noted that the name ‘stachybotrin E’ was also given earlier to a methylated congener of K-76 (70) isolated from the sponge-derived fungus S. chartarum MXH-X73 (Ma et al. 2013). This is not the first incident of compounds with different chemical structures being assigned the same name by different authors (e.g. 32 and 49, 74 and 75), especially with the growing number of compounds isolated and reported daily. Another strain of the endophytic fungus S. chartarum PT2-12 separated from the rhizomes of Pinellia ternate in China was found to produce compounds 45 and 46, and a 10′-OH acetylated derivative of 45. To fit with other metabolites isolated in the communication, the new stachybotramide derivative was named stachybochartin F (117) and was devoid of cytotoxic activity (Zhang et al. 2019).
Based on gene sequence data, the genus Memnoniella is synonymous to Stachybotrys and likewise was found to produce similar metabolites (Wang et al. 2015a, 2015e). From the rice culture of Memnoniella echinata, and with the help of solvent extraction and radial silica chromatography, memnobotrins A (118) and B (119) were identified (Fig. 7) (Hinkley et al. 1999). These are pyrano-fused phenyldrimanes, structurally similar to the aforementioned stachyflins (109 and 110). However, the pyran ring in the memnobotrins is fused to the C-8 and C-9 of the drimane ring rather than C-9 and C-10 seen in the stachyflins (109 and 110). Although the relative stereochemistry for the acetate (C-3) is retained, that for the drimane ring juncture and the C-12 methyl is different. Compounds 118 and 119 were assayed against 3 cancer cell lines and although 118 was less cytotoxic compared to 119, it showed selective cytotoxicity against the MCF7 (breast) cell line (Intaraudom et al. 2019). Researchers in Aventis Pharma Deutschland isolated terpene peptide, memnopeptide A (120) from culture of the fungus M. echinata FH2272 (Fig. 7). Compound 120 consists of the known phenylspirodrimane subunit linked to the proline-rich decapeptide Met-His-Gln-Pro-His-Gln-Pro-Leu-Pro-Pro. Partial structure of 120 was characterized by NMR while the amino acid sequence was determined from the mass spectrometric fragmentation pattern and was found homologous to the subsequence of β-casein. This suggested that 120 is not a bona fide SM, but rather a metabolic product arising from reaction with the constituents of the nutrient medium. However, its formation raises questions regarding nitrogen sequestration by the fungus as a mechanism of storage of amines for safeguarding supplies of vital nitrogen building blocks. Although initial chemical investigation targeted screening for inhibitors of glucose-6-phosphate translocase (G6PT), 120 was found as a weak inhibitor of G6TP, with an IC50 of 78 μM. It was also weakly antibacterial against gram-positive bacteria. However, it effected half-maximal activation of sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA2) at a concentration of 12.5 μM, where SERCA2 activators are expected to have a beneficial effect on heart failure (Vertesy et al. 2001). Consequently, Aventis Pharma Deutschland GmbH filed a patent for the production of a pharmaceutical for the treatment of cardiac insufficiency and/or the treatment of bacterial infection from memnopeptides (Vertesy et al. 2003). All other phthalimidines reported from various Stachybotrys spp. are outlined in Fig. 7.

More phthalimidines isolated from various Stachybotrys spp
Phthalimidines from fungal species of the order Xylariales, phylum Ascomycota
The Xylariales form one of the biggest orders of the class Sordariomycetes, and genera like Xylaria, Daldinia, Hypoxylon and Entonaema form fruiting-body like structures (ascostromata). Xylactams, a class of fungal phthalimidines, were reported from a number of Xylaria strains. The first one xylactam (121) was separated from the cultures of the fruiting bodies of a rot-wood-inhabiting Chinese Xylaria sp. (Wang et al. 2005). A decade later an analogue, assigned as xylactam B (122), with a saturated and shorter ketone side chain, was extracted from an endophytic Xylaria sp. NP-1A (Piyasena et al. 2015). The discovery of these compounds from fungal fruiting bodies continued with the isolation of xylactams C (123) and D (124) (Xylaria polymorpha) (Brown et al. 2018). Compounds 123 and 124 are (E,E)-conjugated dienone analogues of compound 121, while 124 has an additional lactam-N-isopentyl sub-unit. No bioactivity was reported for compounds 121, 123 and 124, while compound 122 showed anticrustacean activity against brine shrimp nymphs (Artemia salina) (Piyasena et al. 2015).
Another class of phthalimidines with 3-aryl moieties were described from the fruiting body of an Asian fungus that was identified as Daldinia concentrica (Kamauchi et al. 2018; Lee et al. 2012). According to the mycologists, the geographical distribution of this species has been confined to mild temperate and subtropical climates of western and southern Europe and reports of D. concentrica from temperate regions of Asia needs to be revised (Stadler et al. 2014). This fungus produced daldinans A-C (125–127) all featuring a 2-hydroxy-3-methoxy-5-methylbenzene moiety connected to C-3 of the phthalimidine nucleus, whose stereochemistry was not determined, but differing only in the N-substitution (Kamauchi et al. 2018). Compound 125 displayed antioxidant effect in a free radical scavenging and reducing power assays (Lee et al. 2012), while 126 and 127 were inactive in anti-angiogenesis assays (Kamauchi et al. 2018). Structurally similar to daldinans, but with a methoxy group at C-2′ of the aryl substituent, is childinin C (128), characterized from Daldinia childiae (Zhao et al. 2017b). The racemates of 128 were separated by chiral phase HPLC and the enantiomers absolute configurations determined with ECD. Although, compound 128 was inactive in antibacterial, cytotoxicity and anti-inflammatory assays, it may have other activities if investigated further. Other analogues of Daldinia phthalimidines were isolated from an Australian rainforest fungus Entonaema sp. and were identified as entonalactams A-C (129–131) (Choomuenwai et al. 2015). Interestingly 131, affords a hemiaminal functionality due to a hydroxy group attached to C-8. Such a moiety was previously found in fumschleicherine and fumadensine alkaloids isolated from Fumaria spp. (Kiryakov et al. 1980; Zarga et al. 1987). The three compounds were isolated as a racemic mixture and none of them showed promising antimalarial activity when tested in-vitro for growth inhibitory activity against Plasmodium falciparum 3D7 malaria parasites, thus, chiral separation was not undertaken.
The fendlerinines B-G (132–137) were found as unique drimane/isoindolinone derivatives in the static culture of a wood-inhabiting fungus Hypoxylon fendleri BCC32408 (Intaraudom et al. 2017, 2019). It is stated that 137 was obtained from cultivation on rotary shakers at 200 rpm, with the aim to stimulate metabolite production (Intaraudom et al. 2019). Compounds 133 and 136 are carbonyl regioisomers with a glutamate unit. Although no activity has been reported for these metabolites, their chemistry can be interesting for medicinal chemists to design and study its synthetic analogues for bioactivities. Compounds 121–137 isolated from fungal species belonging to the order Xylariales are shown in Fig. 8.

Phthalimidines isolated from fungal species of the order Xylariales
Miscellaneous phthalimidines from other Ascomycetes
Pestalotiopsis fungal sp. are endophytes and are an important source of bioactive compounds (Xu et al. 2010, 2014; Yang et al. 2012). For example, pestalachloride A (138) was the first fungal 3-aryl substituted phthalimidine, reported as inseparable diastereomer from Pestalotiopsis adusta. This compound features a 2,4-dichloro-5-methoxy-3-methylphenol moiety and a prenyl group attached to the basic phthalimidine skeleton. 2D-NMR and X-ray crystallography indicated the diasteromeric mixture due to restricted rotation about the C8-C9 bond. Biologically, 138 displayed potent antifungal activity against the plant pathogen Fusarium culmorum, with an IC50 value of 0.89 μM (Li et al. 2008). After chemical synthesis, its antifungal potency against various plant pathogens was retested. In addition to its moderate activity against F. culmorum (ED50 = 72.8 μM), it was also found active against Pyrenophora teres (ED50 = 75.5 μM) (Augner et al. 2013). Bioassay guided isolation from antifungal crude extract of the endophytic fungus Pestalotiopsis photiniae yielded zinnimidine (28), which exhibited antifungal activity (MIC of 24.1 μM) against three plant pathogens Botrytis cinerea, Fusarium graminearum, and Phytophthora nicotianae. However, a previously isolated compound from Alternaria sp. was found inactive against tested fungi (Yang et al. 2011).
Meyeroguilline A (139) where isolated from the mangrove endophytic yeast-like fungus Meyerozyma guilliermondii (Saccharomycetes). Compound 139 is an N-substituted phthalimidine with a valeric acid moiety, which exhibited strong α-glucosidase inhibitory activity with an IC50 value of 50.3 ± 1.3 μM (Chen et al. 2015). A further addition to this family were meyeroguillines C (140) and D (141) reported from another mangrove endophyte, Diaporthe phaseolorum SKS019. Similar to 139, re-isolated from the same fungus, compounds 140 and 141 are N-substituted phthalimidines. Compound 140 is analogous to 139 but with an additional methoxy group in the attached valeric acid moiety, while 141 has a hydroxyethyl moiety attached to the N atom. In bioactivity assays on several human cell lines, none of the meyeroguillines showed any cytotoxic activity (Cui et al. 2017b).
Successive phytochemical investigation of another endophyte Diaporthe sp. SYSU-HQ3, isolated from the mangrove plant Excoecaria agallocha yielded the diaporisoindoles A − E (142–146) (Cui et al. 2017a, 2018a). They are considered as the first example of isoprenylisoindole alkaloids with a rare 1,4-benzodioxan moiety, widely distributed in plant lignans (Bolchi et al. 2020). Compounds 142 and 143 are epimers, while 145 and 146 are C-8-methoxy derivatives of 142 and 143 respectively. Since only 142 showed significant inhibitory activity with an IC50 value of 4.2 μM (compared to the positive control oleanolic acid, 22.1 μM) against Mycobacterium tuberculosis protein tyrosine phosphatase B (MptpB), it explains that absence of methoxy group and stereochemistry at C-8 are important for the anti-TB activity of these compounds. Overall this compound can be a strong candidate to develop anti-TB drug (Cui et al. 2017a). Furthermore, compounds 142 and 143 exhibited potent inhibitory activity against nitric oxide (NO) production in RAW 264.7 cells with IC50 values less than 10 μM, while 145 and 146 displayed moderate activity with IC50 values of 22.7 and 18.2 μM respectively, and are proposed to drive new anti-inflammatory drug discovery (Cui et al. 2018a).
A 50-L culture of the marine fungus Phomopsis lithocarpus FS508 (synonym Diaporthe lithocarpi) afforded a new isoindolinone lactam lithocarlactam A (147) along with compounds 142 and 143. Compound 147, isolated as a minor metabolite, lacks the dioxane scaffold and the isoprenyl at C-6, and could be envisioned as an intermediate in the biosynthesis of the diaporisoindoles (142–143). Compound 147 did not display cytotoxic activity against four tumor cell lines (SF- 268, MCF-7, HepG-2, and A549) (Hu et al. 2022).
Another class of phthalimidine derivatives with interesting structural features are marilines A-C (148–150), which were purified from culture extract of a Stachylidium sp., isolated from the sponge Callyspongia cf. C. flammea. Compounds 148 and 149 contain an O-geranyl side chain but differing in the N-substitution. Compound 148 has a tri-substituted benzene ring while 149 shows a hydroxyethyl moiety connected to the phthalimidine nucleus. On the other hand compound 150 lacked all the substituents observed in 148 and 149, i.e. geranyl side chain and N-substituent. The marilines were all isolated as C-8 racemic mixture. Compound 148 was resolved through chiral HPLC into its enantiomers (mariline A1 and A2), and the absolute configuration was determined by combination of Electron Capture Detector (ECD) spectroscopy and quantum-chemical CD calculations. The marilines were subjected to a myriad of bioactivity testing such as antiproliferative and antiplasmodial activity, and effect towards several receptors including cannabinoid receptor CB2, histamine receptor H2, dopamine receptor DAT and β3-adrenergic receptor. Mariline A1/A2 (148) exhibited activity towards the serine protease human leukocyte elastase (HLE), with an IC50 value of 0.86 μM (Almeida et al. 2012). Inhibitors of HLE are medicinally relevant in the development of drugs for the treatment of inflammatory diseases including adult respiratory distress syndrome, chronic obstructive pulmonary disease (COPD) and cystic fibrosis. Thus, compound 148 could assist in the development of small molecular weight inhibitors of HLE, and was verified in subsequent efforts for their biomimetic synthesis (Augner and Schmalz 2015).
Diastereomeric spirocollequins A (151) and B (152), with a spirotetrahydrofuran moiety through ether bond between C-9 and C-12, were isolated from the plant endophyte Colletotrichum boninense AM-12–2 (Ariefta et al. 2021). Although spirocyclic compounds are ubiquitous in nature, still spiroisoindolinones are less reported (Chupakhin et al. 2019). In anti-plasmodial assays against a chloroquine-sensitive strain of Plasmodium falciparum (3D7), compounds 151 and 152 displayed IC50 values of 9.72 and 4.71 μM, respectively (Ariefta et al. 2021). Recently, total synthesis of these compounds was undertaken which in addition to confirming their structures, would allow a better understanding of their mode of action and structural modifications as anti-plasmodial drug candidates (Ichikawa et al. 2022).
Applying the OSMAC approach on the deep-sea-derived fungus Phialocephala sp. FL30r, afforded sorbicillasins A (153) and B (154) as isomers (Zhang et al. 2018a). Although classified under the family of sorbicillinoids, isolated from several fungal strains, the sorbicillasins are structurally unique with a novel hexahydropyrimido-[2,1-α]-isoindole moiety (Harned and Volp 2011; Meng et al. 2016). These isomers were fully characterized through NMR, ECD analysis and using the time-dependent density functional theory (TD-DFT). These metabolites were tested against K562 and MGC-803 cell lines and for antioxidant activity in DPPH (2,2-diphenyl-1-picrylhydrazyl) radical scavenging assays, but have been reported as inactive (Zhang et al. 2018a).
Another strategy applied to induce or upregulate the biosynthesis of natural products, is the use of small-molecule epigenetic modifiers such as; DNA methyltransferase inhibitors and HDACi (Williams et al. 2008). Furthermore, fermentation media amended with osmolites (e.g. glycerol or salt), applies a selective pressure on the SM profile of a fungus (Overy et al. 2017). Researchers using this strategy have isolated several novel metabolites including phthalimidines. For example sea-foam derived fungus Asteromyces cruciatus produced primarolides A and B (155) through “cooperative induction” strategy using the HDACi, suberoylanilide hydroxamic acid (SAHA), and sodium chloride (Igboeli et al. 2019). It could be postulated that compound 155 was only produced secondary to primarolide A, where the aniline moiety of SAHA serves as a source of the N-phenyl γ-lactam in 155. This was also encountered in various fungal species utilizing HDACi to yield aniline-containing SMs (Adpressa et al. 2017; Zhang et al. 2018b). Only the planar structure of the primarolides was attained and this compound was found inactive in antimicrobial assay (Igboeli et al. 2019).
Chaetosisoindolinone (156), named after its producer Chaetosphaeronema achilleae an endophyte isolated from English yew (Taxus baccata), was recently reported. The investigators reported 156 to be cytotoxic against cervix carcinoma cell line KB-3–1 and mouse fibroblast cell line L929, with IC50 values 105 and 110 μM, respectively (Narmani et al. 2019). A congener identified as (3R)-5,7-dihydroxy-3-methylisoindolin-1-one (157), was earlier reported from an unidentified mangrove endophyte, and the endolichenic fungus Hypoxylon fuscum, but did not exhibit any cytotoxic activity (Basnet et al. 2019; El Amrani et al. 2012). It can be suggested that the nature of substitution on the phenyl ring could be important for cytotoxicity. Phthalimidines isolated from various Ascomycetes are illustrated in Fig. 9.

Phthalimidines isolated from other Ascomycetes
Phthalimidines from Hericium species (phylum Basidiomycota)
Hericium spp. are a rich source of phthalimidine-derived metabolites, which are mostly isolated from the fruiting bodies or cultures of Hericium erinaceus, Hericium coralloides, and Hericium caput-medusae. Among these H. erinaceus, commonly known as Lion’s Mane Mushroom or Hedgehog Mushroom, has a long history of usage in Traditional Chinese Medicine (TCM), due to its health promoting value, e.g. anticarcinogenic, antidiabetic, antioxidant, neuronal disease protecting activities (Jiang et al. 2014; Khan et al. 2013; Thongbai et al. 2015). Many researchers investigated this fungus for its SMs and isolated a variety of phthalimidines, for example in 1990, Kawagishi et al. isolated hericenone B (158) from its fruiting bodies, along with its phthalide analogue hericenone A (Kawagishi et al. 1990). Cytotoxicity assays on HeLa cells revealed that compound 158 is strongly cytotoxic with a MIC of 14.5 μM as compared to the phthalide analogue hericenone A, which had a MIC of 333 μM. This potent cytotoxicity could be attributed to the β-lactam functionality and its phenyl ethyl N-substituent. A year later, from the same source, hericerin (159), a 5′-deoxo analogue of compound 158, was also isolated, which exerted inhibitory activity against pine pollen germination and tea pollen growth (Kimura et al. 1991). Almost two decades later, total synthesis by Kobayashi et al. led to the revision of the structures of both 158 and 159 as the carbonyl regioisomer of the reported structures (Kobayashi et al. 2012, 2014). Miyazawa and co-workers identified further analogues isohericerin (160) and N-De-phenylethyl-isohericerin (161) from this fungus (Miyazawa et al. 2012). However, based on comparison of spectral data for compounds 159 and 160, it was speculated that both independently isolated metabolites are actually the same. Similarly, isohericenone isolated by Kim et al. is in fact compound 159 (Kim et al. 2012). Compound 161 was also isolated as geranylated aromatic compound from H. erinaceus by Yaoita et al. in the same year (Yaoita et al. 2012). Li et al. isolated another analogue, hericerin A (162) with an ethanolic group attached to the nitrogen atom (Li et al. 2015). Interestingly, they also obtained the carbonyl regioisomers 159 and 160, illustrating the prospect of the existence of both regioisomers (Li et al. 2017). For clarity, and based on chemical synthesis, compound 159 could be characterized as (E)-5-(3,7-Dimethylocta-2,6-dienyl)-4-hydroxy-6-methoxy-2-phenethylisoindolin-1-one, and compound 160 as (E)-6-(3,7-Dimethylocta-2,6-dienyl)-7-hydroxy-5-methoxy-2-phenethylisoindolin-1-one (Kobayashi et al. 2012). A non-geranylated analogue of compound 159, named isohericerinol A (163) was recently reported from the same organism (Ryu et al. 2021).
Biologically, compound 158 exhibited potent cytotoxicity against A549, SK-OV-3, SK-MEL-2 and HCT-15 cell lines (IC50: 2.6, 3.1, 1.9 and 2.9 μM, respectively) (Kim et al. 2012). Compounds 160 and 162 inhibited the growth of HL-60 leukaemia cells with IC50 values of 3.06 and 5.47 μM, respectively. Although, compounds 159 and 161 were found less cytotoxic on HL-60 leukaemia cells, with IC50 values of 59.74 and 62.24 μM, respectively (Li et al. 2015), they exhibited promising α-glucosidase inhibitory activity with an IC50 value of 12.3 μM (Miyazawa et al. 2012). Furthermore, compounds 159–161, showed moderate antioxidant and anti-osteoporotic activities (Li et al. 2017). In neurotrophic assays, compounds 159 and 163 significantly increased nerve growth factor (NGF) production in C6 glioma cells culture compared to control, whereas 161 showed little effect on NGF production (Ryu et al. 2021). Mediated by NGF synthesis, compounds 159 and 163 increased the neurite outgrowth up to 116.82 ± 1.97% and 122.82 ± 2.02%, respectively. This is explained through the upregulated expressions of NGF protein by both compounds, in addition, 163 increased the expression of brain-derived neurotrophic factor (BDNF) protein. Neurotrophins such as NGF and BDNF play a critical role in regulating neuronal function (Weissmiller and Wu 2012), thus compounds 159 and 163 maybe beneficial in neurodegenerative disorders such as Alzheimer’s disease (Ma et al. 2010). The story of getting phthalimidines from fruiting bodies of H. erinaceus continued with the isolation of erinacerin A (164), which was identified as a cyclized product of compound 158, in fact acid-catalysed cyclisation of 158 using camphorsulfonic acid (CSA) generated 164 in quantitative yields (Kobayashi et al. 2014). Compound 164 was isolated as a racemic mixture which was resolved by chiral HPLC (Yaoita et al. 2005). In a later study, it was found to possess moderate antiproliferative activity on four human cancer cell lines (Kim et al. 2012). Since the solid-state fermentation (SSF) is a useful method to stimulate the biosynthesis of SMs in edible and medicinal mushrooms (Robinson et al. 2001), therefore, in another study, this technique was applied to the fungus H. erinaceus to get erinacerins C-L (165–174) and erinacerins Q-T (175–178). In contrast to the first reported erinacerin A (164), the new erinacerins (165–178) lack the dihydropyran ring, instead have a geranyl moiety attached to the phthalimidine skeleton. Compounds 169 and 170, 171 and 172, and 173 and 174 are pairs of double bond positional isomers, while compounds 177 and 178 are phthalimides with an isoindoline-1,3-dione skeleton. Erinacerins (165–178) exhibited α-glucosidase inhibitory activity with IC50 values ranging from 5.3 to 145.1 μM (Wang et al. 2015c, 2015d). In addition, compounds 175–178 showed inhibitory activity against protein tyrosine phosphatase-1B (PTP1B) another potential therapeutic target for treatment of diabetes mellitus (DM) type 2 (Wang et al. 2015c). A further derivative, erinacerin U (179) was obtained from the submerged fermentation of H. erinaceus mycelia (Herbst et al. 2018). It is a derivative of compound 165 but with a [5-(hydroxymethyl)furan-2-yl]methyl group attached to the nitrogen atom of the isoindolinone ring. Compound 179 was inactive in anti-inflammatory assay on NO production from LPS-activated microglia cells (Lin et al. 2018). Erinacerin M (180) and N (181) are structurally-related derivatives to compound 164 obtained from a 50% ethanolic extract of the fruiting bodies of H. erinaceus. Compounds 180 and 181 have a methyl and butyric acid moiety respectively, as an N-substituent. Like compound 164, they show the characteristic asymmetric C-3′, however their CD spectra exhibited a negative Cotton effect at 209 nm [Δε −1.30] for compound 180 and at 213 nm [Δε −2.0] for compound 181. Unlike 164, compounds 180 and 181 lacked any cytotoxic activity (Ashour et al. 2019).
Further investigation of the fruiting bodies of H. erinaceus led to the isolation of the erinaceolactams A-E (182–186) (Fig. 10). These compounds are closely related to another H. erinaceus phthalimidines. For example, compound 182 is a N-De-phenylethyl-derivative of compound 158 and compound 184 is a N-De-phenylethyl-derivative of compound 164. At the same time, compounds 184 and 185 are the cyclised form of compounds 182 and 183, respectively. This cyclisation between the olefinic carbon at C-3′ and the hydroxyl group at C-4 creates the dihydropyran ring and the stereocentre at C-3′ for compounds 184–186, with a zero specific optical rotation indicating a racemate mixture for each. These were separated by chiral HPLC into two equi-proportional peaks suggesting a non-stereoselective cyclisation. In cytotoxicity assays against human liver cancer cell lines SMMC-7221 and MHCC-97H, erinaceolactams (182–186) exhibited significant cell growth inhibition comparable to or more potent than the known anticancer 5-fluorouracil (Wang et al. 2016). Interestingly, compounds 164, 182 and 184–186, inhibited the growth of SMMC-7221, and compounds 161 (re-isolated from this fungus) and 186 inhibited the growth of MHCC-97H in a dose-dependent manner.

Phthalimidines reported from Hericium erinaceus
The metabolites 159 and 161 were also isolated from fruiting bodies of Hericium coralloides (synonym H. clathroides), along with other compounds corallocins B (187) and C (188) (Fig. 11). Compound 159 has a phenylethyl N-substituent, whereas compounds 187 and 188 feature a p-hydroxyphenylethyl and indolylethyl moiety, respectively. Compound 187 showed cytotoxic activity against HUVEC (IC50 2.1 μM), MCF-7 (IC50 9.2 μM), and KB-3–1 (IC50 11.5 μM) human cell lines (Wittstein et al. 2016). Furthermore, the corallocins (187 and 188) induced different patterns of neurotrophin expression in human astrocytes on both NGF and/or BDNF expression. It could be speculated that for the neurotrophic activity observed, a bulky N-substituent rather than a gernyl side chain would be essential as outlined for several Hericium metabolites tested so far. These low molecular weight compounds can easily cross the blood brain barrier, mimicking/potentiating the effect of neurotrophins and could be seen as an alternative drug candidate for treatment of brain disease where neurotrophin-based therapy has proven to be challenging (Ma et al. 2010; Phan et al. 2014).

Phthalimidines isolated from Hericium coralloides and Hericium caput-medusae
Chromatographic purification of the ethyl acetate extract of fruiting bodies of H. caput-medusae yielded caputmedusins A-K (189–199) (Fig. 11). Compounds 189 and 190 are analogous meroterpene dimers, with an additional methyl group in compound 190 linking the bicyclic isoindolinone skeleton. Compounds 191 and 192 are closely related to compound 173 with slight modifications in the geranyl side chain. Thus, the absolute configuration at C-1′ was proposed as S by comparison of specific optical rotation values with that of compound 173. Detailed spectroscopic analysis of compounds 198 and 199 established as diastereomers with a C-6, C-9-ether bridge featuring a five-membered ring and a γ-lactone, with a 3′-carboxypropyl group linked to the nitrogen. These metabolites were evaluated for their hypoglycemic activity in an in-vitro α-glucosidase inhibition assay, where only compounds 189–191 displayed moderate inhibitory activity, with IC50 values of 39.2, 36.2, 40.8 μM, respectively (Chen et al. 2017b).
Miscellaneous phthalimidines from other Basidiomycetes
The SSF culture of Stereum mushroom from which a group of phthalimidines designated as sterenins were characterized. The sterenins A-D (200–203) are isoprenyl-isoindolinone analogues with an O-orsellinic acid attached via an ester bond to C-6 and only varying in the N-linked side chain (Fig. 12). Intriguingly, the sterenins are regarded as the first natural metabolites that showed potent and selective 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitory activity. Notably, compounds 200 and 202 displayed significant inhibitory activity with IC50 values of 0.24 and 0.23 μM, respectively (Ito-Kobayashi et al. 2008). Since 11β-HSD1 plays an important role in type 2 DM and metabolic syndrome, keeping in view this medicinal importance, the investigators synthesized compounds 200, 202 and 203 for further studies, and hypothesized that a further investigation may allow biological evaluation in the search for therapeutics for metabolic diseases (Shinozuka et al. 2008).

Miscellaneous phthalimidines isolated from other Basidiomycetes
The sterenin class has recently seen an expansion with the addition of three new members, namely sterenin K-M (204–206). In addition to sterenin A-C, these compounds were isolated from S. hirsutum cultivated on potato dextrose agar slants. The six sterenins demonstrated inhibitory effects on α-glucosidase, an enzyme responsible for catalyzing carbohydrate digestion. They also exhibited notable 11β-HSD1 inhibitory activity, consistent with observations for sterenin A-D. These findings suggest the potential for developing these compounds into novel hypoglycemic drugs (Wang et al. 2014). Sterenin D was additionally identified in a liquid mycelial fermentation process of S. hirsutum from Chile. It exhibited antifungal activity, inhibiting both the sporogenesis and mycelial growth of Botrytis cinerea. This suggests its potential use as a biofungicide (Aqueveque et al. 2017).
Aldose reductase (AR) is another key enzyme involved in the complications of DM; thus inhibitors of this enzyme represent a potential pharmacological avenue in the treatment of side effects of this metabolic disorder (Tang et al. 2012). The salfredins are a family of AR inhibitors produced by Crucibulum sp. and are divided into A-, B- and C-type, based on structural features. Of relevance to this review are the A-type salfredins, A3 (207), A4 (208) and A7 (209) with a furo-phthalimidine skeleton and an attached propionic acid residue (Fig. 12). The N atom in compounds 207–209 originates from glutamic acid, glycine and alanine, respectively, and addition of 1.0% glycine to the fermentation media markedly enhanced the production of compound 208 verifying this assumption. The relative stereostructure of compound 208 was confirmed by X-ray crystallographic analysis and implied for compounds 207 and 209. In AR inhibitory assays on rat lens, compound 208 was found to be the most active (Matsumoto et al. 1995). Another salfredin type A metabolite (210) was isolated from cultures of Crucibulum laeve DSM 1653 and DSM 8519 (Fig. 12). Compound 210 afforded an N-ethylphenyl substituent (Neumann et al. 1999).
The natural antioxidant clitocybin A (211) was isolated from the culture broth of the mushroom Hygrophoropsis aurantiaca (Fig. 12) (Kim et al. 2008). It is a 4,6-dihydroxy-isoindolinone with a p-hyrdroxybenzene moiety attached to the N-atom. Its antioxidant activity was evaluated by measuring the free radical scavenging effects in three different assays. It exhibited potent superoxide radical scavenging activity and significant ABTS [2,2’-azino-bis-(3-ethylbenzothiazoline-6-sulfonic acid)] radical cation scavenging effects with an IC50 value of 10.3 and 6.4 μM, respectively, but displayed no scavenging activity against DPPH radical. Furthermore, it had a protective effect against cellular DNA damage induced by oxidative stress (Süssmuth and Mainz 2017). Synthetic modifications in compound 211 provided further derivatives, clitocybin B (212) and C (213) (Fig. 12) (Moon et al. 2009). Compounds 211–213 inhibited intracellular reactive oxygen species (ROS) and H2O2-induced cell death mediated by the reduction of caspase 3 and 9 activation, cytochrome c release from mitochondria and NF-KB activation. Recently the anti-wrinkle capacity of compound 211, supported through elastase inhibitory activity, stimulation of procollagen synthesis and inhibition of matrix metalloproteinase-1 (MM-1), all suggests its use in anti-wrinkle cosmetic products (Lee et al. 2017). In fact, the extract of H. aurantiaca and clitocybin derivatives have been patented as an active ingredient in anti-aging preparations (Yoo et al. 2010). However, clinical trials are crucially required before its commercial usage. Extended studies on bioactivities of compounds 211 and 212 revealed that the natural analogue 211 was found to block the platelet-derived growth factor (PDGF)-BB-induced proliferation through G1 phase arrest by regulating the phosphatidylinositol-3-kinase (PI3K)/Akt pathway in vascular smooth muscle cells (VSMCs) (Yoo et al. 2012a). However, compound 211 did not change the expression levels of extracellular signal-related kinase (ERK) 1/2, phospholipase C-γ1 (PLC-γ1), and PDGF-Rβ phosphorylation. While the synthetic derivative 212 significantly inhibited the phosphorylation of Akt, ERK 1/2, PLC-γ1, and PDGF-Rβ phosphorylation in the PDGF-BB signaling pathway (Yoo et al. 2012b). Altogether, these anti-proliferative activities on VSMCs make them a potential therapeutic agent for preventing or treating vascular disease such as atherosclerosis, hypertension and restenosis.
The story of compound 211 continued with its re-isolation along with its non-nitrogen substituted analogue, 4,6-dihydroxy-2,3-dihydro-1H-isoindol-1-one (214) from Langermannia fenzlii, a fungus used in TCM as a remedy for bleeding (Fig. 12). Both compounds were devoid of cytotoxic activity when tested against A549, PC-3, U87, and HeLa tumor cells in the concentration range of 12.5–100 μM (Lü et al. 2013).
Phthalimidines from Basidiomycete and Ascomycete co-culture
From a co-culture of the two endophytic fungi, Irpex lacteus and Phaeosphaeria oryzae of the fungal phyla Basidiomycota and Ascomycota, respectively, irpexine (215) was isolated, which was not identified in monocultures (Fig. 13). This expanded the array of the so far known 3-aryl substituted phthalimidines, and due to hindered rotation around the C8–C9 bond also seen in pestalachloride A (138); compound 215 was also isolated as a mixture of atropisomers. However, this mixture was resolved on chiral-phase column and the absolute configuration determined using ECD of a simplified model with a methyl group replacing the prenyl group. At a concentration of 50 μM, compound 215 was inactive in cytotoxicity and antimicrobial assays (Sadahiro et al. 2020).

Phthalimidines reported from Basidiomycete and Ascomycete co-culture
Clinical development of fungal phthalimidines
SMTP-7 (42) is a novel thrombolytic agent that demonstrates effective improvement in ischemic stroke in rodent models (Ito et al. 2014). To enhance the translation of animal findings to humans and ensure the safety and efficacy for drug development, studies in higher-order species are crucial. In evaluating the therapeutic potential of 42, a monkey occlusion model with thrombotic middle cerebral artery (MCA) was employed. The findings demonstrated its effectiveness in treating thrombotic stroke in monkeys, with the added relevance of their cerebral vascular anatomy closely resembling that of humans. This observation aligns with earlier results obtained in rodent models (Sawada et al. 2014). In addition, 42 was identified as having a protective effect against renal damage in a mouse model of acute kidney injury (AKI) (Shibata et al. 2021). As a member of the SMTP family of compounds, TMS-007 was subsequently progressed into clinical studies. In a phase 1 randomized, double-blind, placebo-controlled, dose-escalation study, registered as JapicCTI-142 654 at the Japan Pharmaceutical Information Center Clinical Trials Information, involving healthy 40 male volunteers in Japan, TMS-007 exhibited favorable tolerability. Administered intravenously at doses up to 6 mg/kg, the compound showed no indication of serious adverse reactions, including any signs or symptoms of bleeding (Moritoyo et al. 2023). Subsequently, a phase 2a study involving 90 acute ischemic stroke (AIS) patients in Japan was conducted. This study, registered as JapicCTI-183 842 at the Japan Pharmaceutical Information Center Clinical Trials Information, involved the administration of a single intravenous infusion approximately 9 h after the last known normal (LKN) (Nishimura et al. 2022). Biogen Inc., a leading biotechnology company focused on neuroscience, acquired TMS-007 (renamed BIIB131) as an investigational drug for AIS 6, building upon encouraging results from phase 2 study. The current gold standard treatment for AIS is tissue-type plasminogen activator (t-PA) therapy, which activates the thrombolytic enzyme plasmin by cleaving plasminogen. Nevertheless, the restricted time frame for administering t-PA within 3–4.5 h limits its applicability. Moreover, the use of t-PA benefits only a limited percentage (3%–9%) of AIS patients, primarily because of the elevated risk of fatal hemorrhagic transformation (HT) (dela Peña et al. 2017). The distinctive mode of action of TMS-007, which encompasses profibrinolytic, antioxidative, and anti-inflammatory activities, holds the potential to extend the time window for AIS treatment. These characteristic underscores the importance of developing TMS-007 into the next generation of thrombolytic agents. Such advancements are urgently required to address the critical need for reducing mortality and morbidity in stroke patients. Additionally, there is a need to clinically investigate the use of TMS-007 in other thromboembolic and thrombotic diseases, such as pulmonary embolism and myocardial infarction.
The initial assessment of physicochemical properties through absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiling is essential. This preliminary assessment assists in forecasting the drug-like properties of potential drug candidates, thereby mitigating the risk of expensive failures in the later stages of drug development. Stachyflin (109), while exhibiting promising in-vitro anti-viral activity, displayed diminished effectiveness when orally administered to mice infected with the influenza virus. The lipophilic nature and high water insolubility of 109 explain its limited gastrointestinal bioavailability. Various vehicles, including soybean oil, polyethylene glycol (PEG), and surfactants, were experimented with to enhance solubility and oral absorption. Positive in-vivo activity was observed when the substance was administered as an aqueous solution of a phosphate ester prodrug or as a solution in PEG4000 (Yagi et al. 1999; Yoshimoto et al. 2000). While the intraperitoneal injection of 109 in mice did not induce toxicity, its antiviral activity in mice was comparable to clinically used antiviral neuraminidase inhibitors, such as oseltamivir. Notably, this effect was achieved at a significantly higher dose of 100 mg/kg/day, as opposed to the 20 mg/kg/day used for oseltamivir (Motohashi et al. 2013). In a recent computational screening study aimed at discovering new inhibitors for Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV), 109 exhibited a positive ADMET profile. Further investigations through molecular dynamics (MD) simulations and molecular docking demonstrated a strong binding affinity between 109 and the amino acid residues of the Mpro active site, a critical protease involved in viral replication. However, the MD simulation of the Mpro–Stachyflin docking complex suggested a less stable interaction (Bharadwaj et al. 2021). Currently, the prospect of advancing 109 into clinical development does not appear promising. However, in a separate in-silico investigation utilizing docking and MD simulations, sterenin M (206) emerged as a potential SARS-CoV-2-Mpro inhibitor within the Medicinal Fungi Secondary Metabolites and Therapeutics (MeFSAT) database. All ADME parameters analyzed fell within recommended ranges, indicating that sterenin M holds promise as a ligand against SARS-CoV-2. Nevertheless, further in vitro and in vivo studies are warranted for comprehensive evaluation (Prajapati et al. 2021).
Culinary medicinal mushrooms, exemplified by H. erinaceus, offer a rich source of bioactive phthalimidine compounds, presenting potential for future advancements. Despite indications of their neuro-health benefits, derived from the combined and synergistic effects of numerous bioactive compounds in the fruiting bodies or cultured mycelium, the availability of clinical trials is currently limited (Brandalise et al. 2023). The primary reason for the limited availability of clinical trials is that naturally occurring compounds in their original state may lack patentability, making them less appealing for pharmaceutical companies to invest in.
Cultivating medicinal mushrooms submerged in bioreactors and fine-tuning fermentation conditions is essential for achieving maximum biomass production. This approach helps overcome challenges related to unpredictable metabolite profiles caused by seasonal variations. In order to generate substantial quantities of TMS-007 for clinical studies, a large-scale fermentation strategy involving precursor amine feeding was implemented. Although an alternative method involving the total synthesis of an SMTP congener was devised, it consisted of a 10-step process with relatively modest production yields (Hasumi and Suzuki 2021). Likewise, there was a need for optimizing the fermentation process on a large scale to produce clitocybin A (211) from C. aurantiaca for mass production. This was pursued with the goal of utilizing clitocybin A as a novel anti-wrinkle agent in cosmetics (Kim et al. 2014).
In summary, fungal phthalimidine metabolites boast diverse scaffolds and intricate structural complexity. Despite these advantages, few pharmaceutical industries are actively exploring drug leads from natural sources. On the other hand, several synthetic small molecule ligands, featuring a phthalimidine core, such as iberdomide, JM-1232, and CC-885—show promise as clinical candidates for treating multiple myeloma (Charliński et al. 2021). Conversely, obstacles in supplying adequate quantities for drug development, particularly in cases where chemical synthesis is impractical, significantly impede the progress of drug leads derived from natural sources. Another prevalent challenge lies in the limited dissolution/solubility and permeation of hydrophobic natural compounds. An integrated approach, which merges in-silico ADMET analysis with MD simulation studies, and is complemented by in-vivo studies, is crucial for propelling fungal phthalimidines into clinical development or validating them as leads for drug targets.
Table 1 below summarizes the fungal phthalimidines and phthalimidine-containing metabolites outlined in this review, including their origin and biological activity.
Biosynthesis of fungal phthalimidines
Fungal phthalimidines have shown diversity in their structural features and bioactivities, therefore, we have discussed here biosynthetic routes of these metabolites established or proposed in different fungi. Fungal phthalimidines belong to the class of fungal aromatic polyketides, synthesized by the non-reducing group of iterative polyketide synthases (NR-PKSs), which utilise simple carboxylic acid precursors to assemble the polyketide backbone (Crawford and Townsend 2010). Canonical PKS domains include a ketosynthase domain (KS) for decarboxylative Claisen-like condensation of the extender unit, an acyltransferase domain (AT) for extender unit selection and transfer and an acyl-carrier protein (ACP) for extender unit loading. Fungal NR-PKS feature two additional types of dedicated domains, a starter unit ACP transacylase (SAT) domain for starter unit selection and a product template (PT) domain involved in chain-length determination and cyclisation (Crawford et al. 2008; Herbst et al. 2018). For product release from the PKS, a thioesterase (TE) domain, a thioesterase/Claisen cyclase (TE/CLC) domain or a reductase (R) domain is tethered to the C-terminus, although some NR-PKS have an enzymatic domain that acts in trans to release the nascent polyketide chain, or may not require any releasing domain enzyme (Chiang et al. 2010).
Biosynthetic labelling studies in Stachylidium sp.
Biosynthetic examples of phthalimidines and phthalides have been reported from fungal secondary metabolism, and several fungal strains simultaneously produce both types of metabolites. For example, the marine-derived Stachylidium strain produces both marilones (phthalides) and marilines (phthalimidines) (Almeida et al. 2012, 2011a). Feeding experiments using 13C-labeled acetate, propionate and methionine established the tetraketide nature of the phthalide/phthalimidine core skeleton and the mevalonate origin of the attached geranyl side chain in both marilone A (216) and mariline B (149) (Fig. 14) (El Maddah et al. 2019). It is well-established that fungi exclusively use the mevalonate pathway (MVA) for the synthesis of their terpenoid units (Lombard and Moreira 2011). In addition to confirming the assumption of an analogous biosynthetic origin for phthalide/phthalimidine frameworks, an important conclusion from these feeding studies was the involvement of a methylated acetate starter unit for initiation of the biosynthesis, which is considered unique for fungal metabolites (El Maddah et al. 2019).

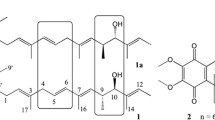
Observed labeling pattern in marilone A (216) and mariline B (149) after feeding [1-13C]sodium acetate, [2-13C]sodium acetate, and L-[Me-13C]methionine. Smaller bullets (■) indicates indirect incorporation
The mariline analogue (148) has a unique O-prenylated tri-substituted benzene ring attached to the nitrogen of the phthalimidine backbone putatively tyrosine-derived. Interestingly, similar motifs; the stachylines were independently isolated from the same fungal strain (Almeida et al. 2011b) and it could be speculated that 148 is a product resulting from crosstalk between the phthalimidine and the stachyline pathways. However, this needs further exploration for determination of the BGC for each building block and the enzyme(s) catalyzing the coupling, and several dimeric fungal natural products provide excellent examples for such biosynthetic crosstalks, e.g. aspercryptin (6) (Dai et al. 2022). As for the N-substituent in 149 this is likely provided from amino acid metabolism, e.g. ethanolamine from serine.
Biosynthetic studies in Aspergillus nidulans
As an established molecular genetic system, A. nidulans has successfully served as a model fungus for heterologous expression of fungal SMs (Chiang et al. 2013; Yaegashi et al. 2014). The group of Clay Wang has developed a targeted gene deletion strategy employing the genome of A. nidulans. By directly replacing the promoters of the NR-PKS genes as well as other genes required for compound production or release, they were successful in determining NR-PKS genes required for the synthesis of the simple phthalimidine cichorine (2), prenylated phthalimidine aspernidine A (10) and the hybrid metabolite aspercryptin (6). This strategy helped to increase the titre of the target metabolite of the biosynthetic gene cluster (BGC) (when the promoters of all genes in the cluster are replaced) as well as pathway intermediates (when some of the genes in the cluster are replaced), which allowed them to propose a biosynthetic pathway for the above metabolites (Ahuja et al. 2012).
For cichorine (2), the BGC in A. nidulans includes a PKS gene AN6448 (cicF), a transcriptional activator (regulatory) gene (cicD), a transporter gene (cicA), and four tailoring genes (cicB, cicC, cicE and cicH) (Fig. 15A). The ΔcicB and the ΔcicC knockout strains produced the biosynthetic intermediates 3-methylorsellinic acid (217) and nidulol (218), respectively. The cichorine analogs 3–5 are also products of the cicF, which provides the basic phthalimidine backbone (Liao et al. 2019). However, 3 and 4 feature an additional methoxy group at C-8, probably introduced at a later stage mediated by tailoring enzymes, e.g. hydroxylase, methyltransferase. As for the N-carboxybutyl substituent in 5, this is postulated to be derived from amino acid metabolism, e.g. 5-aminovaleric acid from proline, and an amino acid transporter gene (AN11921) has been annotated upstream of cichorine BGC. A proposed biosynthetic scheme for cichorine (2) is outlined in Fig. 15B (Sanchez et al. 2012).

A Organization of genes surrounding the PKS cicF involved in cichorine (2) biosynthesis. B Proposed biosynthesis of cichorine (2) and related analogues (3–5) in Aspergillus nidulans (metabolites in boxes have been analytically identified). Abbreviations: SAT, starter unit ACP transacylase; KS, ketosynthase; AT, acyltransferase; PT, product template; ACP, acyl carrier protein; C-MeT, C-methyltransferase; TE; thioesterase; SAM, S-adenosyl-L-methionine
Manipulating the expression of kinases, which play a role in aspects of regulation and signal transduction, is a novel approach for activating cryptic gene clusters (De Souza et al. 2013). This approach was employed for elucidating the genetic basis for the biosynthesis of aspernidine A (10). Through screening a genome-wide kinase knockout (KO) library of A. nidulans (a total of 98 kinase KO strains), one strain with a mitogen-activated protein kinase gene knockout (ΔmpkA) was detected. Sequential targeted gene deletions in this strain allowed the characterization of aspernidine A (10) BGC and further related metabolites (aspernidine C-E) (219–221) probably involved in the biosynthesis. In silico analysis using the Aspergillus Genome Database (AspGD, http://www.aspgd.org/) correlated well with results of targeted gene deletion predicting the aspernidine BGC to contain 6 genes pkfA-pkfF (Fig. 16A), although other gene involved in the biosynthesis cannot be excluded. For example, no gene accounting for the methylation was annotated in the aspernidine BGC. Based on gene deletion data and isolated intermediates a biosynthetic scheme for aspernidine A (10) biosynthesis was proposed starting with the production of orsellinaldehyde (222) by the PKS PkfA (Fig. 16B) (Yaegashi et al. 2013).

A Organization of genes surrounding the PKS pkfA involved in aspernidine A (10) biosynthesis. B Proposed biosynthesis of aspernidine A (10) in Aspergillus nidulans (metabolites in boxes have been analytically identified). Abbreviations: SAT, starter unit ACP transacylase; KS, ketosynthase; AT, acyltransferase; PT, product template; ACP, acyl carrier protein; C-MeT, C-methyltransferase; R, reductase
For the production of cichorine (2) and aspernidine A (10) in A. nidulans, the CicF and PkfA, respectively, provide the phthalimidine core structure. Both CicF and PkfA belong to the NR-PKS in subclade III, distinguished by the C-methyl transferase (C-MeT) domain, that adds one or more S-adenosylmethionine(SAM)-derived methyl groups at the α-position of the polyketide chain (Herbst et al. 2018). Additionally, C-MeT acts as a gatekeeper, where α-methyl groups function as check-point tags for KS, thus controlling chain elongation cycles and polyketide formation by iterative PKS (Storm et al. 2017). Subclade III is further sub-divided into group VI with the domain architecture SAT-KS-AT-PT-ACP-(ACP)-CMeT-TE and group VII with the domain architecture SAT-KS-AT-PT-ACP-(CMeT)-R. Phylogenetic analysis revealed that CicF belongs to group VI while PkfA belongs to group VII (Ahuja et al. 2012). Both groups VI and VII NR-PKS produce monocyclic aromatics with a C2-C7 regioselective cyclisation under the control of the PT domain. However, group VI terminates with a TE that cleaves the thioester bond and releases the acid product, e.g. 3-methylorsellinic acid (217), and group VII has a NADPH-dependent R domain to release the product typically as an aldehyde, e.g. orsellinaldehyde (222).
Aspercryptin (6) is the product of two distinct BGCs physically separated in the genome of A. nidulans strain LO8030. The NR-PKS containing AN6448 (cicF) cluster and a sole non-ribosomal peptide synthetase (NRPS), AN7884 (atnA). Twelve additional genes (atnB-atnM) are involved in the biosynthesis of aspercryptin (6) (Fig. 17) (Chiang et al. 2016). Analogous to PKS, NRPS are multifunctional mega-enzymes organised structurally into modules, in which each module fulfils a cycle of peptide elongation. A minimal NRPS module consists of 3 core catalytic domains; an adenylation (A) domain for amino acid selection and activation, which is then tethered onto a thiolation (T)/peptidyl carrier protein (PCP) domain through the phosphopantetheine arm, which is then shuttled to the condensation (C) domain for peptide bond formation (Süssmuth and Mainz 2017; Vassaux et al. 2019). The final peptide is then released with the help of a TE domain, a R domain, a terminal C domain or with no obvious release domain (Du and Lou 2010).

Organization of genes surrounding the NRPS atnA involved in aspercryptin (6) biosynthesis in Aspergillus nidulans
The 6 module NRPS AtnA is a type A linear NRPS, with each module responsible for the incorporation of a single amino acid residue (Chiang et al. 2016). The second module harbors an epimerise (E) domain which is responsible for generating the d-allo-threonine residue, while modules 4 and 6 incorporate the unusual amino acids 2-aminocaprylic acid and 2-aminododecanoic acid, respectively. These are believed to be the products of the fatty acid synthases (atnF and atnM), dehydrogenase (atnD), cytochrome P450 (atnE) and the amino acid transferases (atnJ and atnH) genes. Similar enzymes have earlier been proposed for the introduction of non-canonical amino acids in cyclosporine (Bushley et al. 2013), apicidin (Niehaus et al. 2014) and HC-toxin (Cheng et al. 1999) and d-amino acids and non-canonical amino acids are common in fungal NRP (Eisfeld 2009). Similar to aspernidine A (10), a C-terminal R domain in module 6 of the the NRPS would be involved in the reductive release of the aldehyde hexapeptide (Chiang et al. 2016). A proposed biosynthetic pathway for aspercryptin (6) is depicted in Fig. 18 using cichorine-serine as a precursor.

Proposed biosynthetic pathway for aspercryptin (6) in Aspergillus nidulans. Abbreviations: A, adenylation domain; T, thiolation domain; C, condensation domain; E, epimerization domain; R, reductase
The proposed biosynthetic pathways (Figs. 15B, 16B and 18) for the A. nidulans phthalimidines cichorine (2), aspernidine A (10) and aspercryptin (6) were based on the analysis of deletion mutants and isolated metabolites, and further work would be essential to confirm those pathways especially that other genes expected to be involved in the biosynthesis where so far not found within the cluster. For example, genes required for the conversion of the lactone nidulol (218) to a lactam in cichorine (2) and genes involved in the biosynthesis of the terpenoid moiety of apsernidine A (10). The O-methyltransferase cicE is believed to play a role in adding the methyl group to cichorine (10). However, a gene with a similar methyl functionality was not identified in the aspernidine (2) BGC. It is proposed that methylation could be carried out by a separate, standalone methyltransferase located outside of the gene cluster (Chen et al. 2019a). It would also be relevant to search for homologous cichorine (2) biosynthetic genes in A. cichorii the first source from which cichorine (2) was isolated; however, whole genome sequencing of A. cichorii was so far not undertaken. The predicted core genes involved in cichorine (2), aspernidine A (10) and aspercryptin (6) biosynthesis are summarized in Table 2.
Biosynthetic studies in Stachybotrys spp.
Genome mining of the sequenced Stachybotrys strains, S. chartarum and Stachybotrys chlorohalonata genome was undertaken by Zhang et al. to identify the BGC involved in the synthesis of the structurally related Stachybotrys phthalimidines. A cluster containing 10 genes (idlA-I, R) was identified in both genomes (Fig. 19A) (Yin et al. 2017). Genes homologous to the stb cluster, earlier reported in Stachybotrys bisbyi PYH05-7 genome, were identified (Table 3). Heterologous expression of the stb genes in Aspergillus oryzae probed the functionality of the biosynthetic enzymes, revealing that the stb cluster has a NR-PKS (StbA), an NRPS-like enzyme (StbB), and a UbiA-like prenyltransferase (PTase) (StbC) involved in the biosynthesis of ilicicolin B (LL-Z1272β) (223) (Li et al. 2016). Ilicicolin B (223) is a prenylated aryl-aldehyde, considered to be a common precursor in the biosynthesis of Stachybotrys phthalimidines. The NR-PKS (StbA) provides the orsellinic acid starter unit (224), which is then farnesylated by the StbC using farnesyl pyrophosphate (FPP) as a prenyl donor to give grifolic acid (225). Here, the aldehyde moiety in ilicicolin B (223) is not generated by the R domain covalently appended at the end of the NR-PKS as seen in aspernidine A (10) biosynthesis, but is instead catalysed by the subtype II NRPS-like enzyme (StbB), considered as the first NRPS-like enzyme which activates a prenylated acid (225) (Fig. 19B) (Tao and Abe 2021).

A Predicted BGC for phthalimidine derivatives in Stachybotrys chlorohalonata IBT 40285. B Proposed biosynthetic pathway of the putative intermediate ilicicolin B (223) (metabolites in boxes have been analytically identified). Abbreviations: SAT, starter unit ACP transacylase; KS, ketosynthase; AT, acyltransferase; PT, product template; ACP, acyl carrier protein; C-MeT, C-methyltransferase; TE; thioesterase; R, reductase
Accordingly, a biosynthetic scheme was proposed for Stachybotrys phthalimidines, starting with the common precursor 223. They suggested two modes of cyclisation of the farnesyl side chain, initiated by an acid-catalysed epoxidation on different double bonds of the farnesyl group to provide different classes of Stachybotrys phthalimidine derivatives (Fig. 20) (Yin et al. 2017). Stereospecific olefin epoxidation, usually catalysed by a flavin-dependent monooxygenase (FMO) or P450 monooxygenase, precedes cyclisation by a terpene cyclase (TC) (Matsuda and Abe 2016; Quan et al. 2019). However, the detailed mechanism and enzymes involved in this catalysis still remain unclear. These scaffolds are then modified by tailoring enzymes in versatile manners to provide the huge structural diversity observed for Stachybotrys phthalimidines.

Proposed biosynthetic pathway for phthalimidine derivatives in Stachybotrys sp. starting with ilicicolin B (223)
Additionally, homologous genes of a NR-PKS, an NRPS-like enzyme and two putative short-chain dehydrogenases seen in cichorine BGC in A. nidulans, and homologous PTase genes seen in the austinols and terretonin BGC in A. nidulans and A. terreus, respectively, were found in the Stachybotrys genome. The authors postulated that the nitrogen in the phthalimidine core skeleton is probably introduced non-enzymatically by the reaction of the phthalic aldehyde precursor (226 and 227) and ammonium ions or amino compounds (Yin et al. 2017). This was already proposed for the biosynthesis of the pyridine ring in rubrolone tropolone alkaloids, where absence of candidate genes in the rub cluster suggested the capture of free amines from solution by a reactive 1,5-dione moiety (Yan et al. 2016). In an earlier study, focusing on the production of SMTP congeners by feeding amine precursors to S. microspora cultures, significant amounts of 223 and 226 were found to accumulate during the ‘starvation phase’ before the addition of amines to the culture, and were rapidly consumed following the addition of amines, which strongly supports their involvement in the biosynthesis of SMTPs (Nishimura et al. 2012). They also suggested that the nitrate reductase (NR) gene in the Stachybotrys cluster is responsible for the balance of the ammonium ion concentration, where NR is the rate limiting enzyme for nitrate reduction prior to the formation of ammonium ions (Yin et al. 2017).
Biosynthetic studies in Hericium sp.
In contrast to Ascomycetes, genes and enzymes involved in the biosynthesis of phthalimidines in Basdiomycetes remain obscure. Initial work focused on the characterisation of two NR-PKSs and two PTases in the genome of a stereaceous Basidiomycete BY1. Both PKSs were found to produce orsellinic acid, a key intermediate involved in the biosynthesis of fungal phthalimidines (Braesel et al. 2017). Furthermore, the genome of Hericium sp. (H. erinaceus and H. coralloides) have been recently sequenced and published (Chen et al. 2017a; Gong et al. 2020; Zhang et al. 2017). Gene cluster prediction for SMs biosynthesis using the fungal anti-SMASH platform revealed that the genome sequence harbors a large number of terpenoid gene clusters and a single type I PKS, these could be expected to be involved in the biosynthesis of Hericium phthalimidines. However, this is not conclusive as the anti-SMASH platform is not highly efficient in annotating fungal genomes especially for those belonging to the Basidiomycota and future biochemical and genetic studies are essential. Through sequence similarity networks (SNNs) analysis, they identified and heterologously expressed an orsellinic acid synthase gene herA in the genome of H. erinaceus. Although, the biosynthesis of Hericium phthalimidines still remains to be decoded, the ability of the heterologous host used, i.e. a filamentous fungi A. oryzae, to express basidiomycetes genes, opens new possibilities for the production of mushroom polyketide compounds, in addition to promoting research on basidiomycetes phthalimidines, which has been lagging behind (Han et al. 2023).
Conclusion and future prospects
This review article summarizes the discovery, chemodiversity and bioactive potential of more than 200 phthalimidines from various fungal species. Literature search revealed that the fungi Stachybotrys is the major producer of these metabolites, since about 90 phthalimidines are reported so far from these fungi. The next in line are Hericium (~ 53 compounds), Aspergillus (~ 27 compounds) and Xylaria species (~ 17 phthalimidines). Only few compounds showed no activity in some bioassay, whereas, most of them afford different bioactivities including phytotoxicity, cytotoxicity, anti-tumor, antioxidant, antidiabetic, anti-inflammatory, neuroprotective and antimicrobial (Table 1).
Recently there is a growing interest among organic chemists to synthesize substituted, chiral and enantiopure phthalimidines (Di Mola et al. 2014, 2015; Thapa et al. 2018), largely due to their specific binding affinities to a wide range of drug targets, which could be exploited in the drug design process (Chen et al. 2019b; Papeo et al. 2019). As outlined, several fungal phthalimidines have proven to be important ligands to many pharmacologically relevant receptors and could be perceived as strong contributors to the drug discovery pipeline, e.g. sterenin M (206). Interestingly, several fungal phthalimidines showed comparable or superior in-vitro bioactivity as to the known standards, e.g. stachyflin (109) (Minagawa et al. 2002c) and erinaceolactams (182–186) (Wang et al. 2016), and many have already been patented for their unique chemistry and promising therapeutic activity (e.g. SMTP-7 (orniplabin) (42) (Itoh et al. 2022), spirobenzofuranlactams (60 and 61) (Eder et al. 2007), stachyflin (109) (Tang et al. 2021), memnopeptide A (120) (Vertesy et al. 2003) and clitocybin A (208) (Yoo et al. 2010). A potent SMTP-congener, TMS-007, is currently in clinical trials as a next-generation thrombolytic for acute ischemic stroke (Hasumi and Suzuki 2021). Furthermore, endeavors of synthetic chemists to develop specific and interesting methodological approaches for the total synthesis of fungal phthalimidines has helped confirm and sometimes revise the structure of many of the isolated compounds e.g. porritoxin (30) and hericenone B (158) (Cornella and Kelly 2004; Kobayashi et al. 2014). This was particularly important for the unambiguous attribution of the relative and absolute configuration of phthalimidines’ stereogenic centers for stachybotrin C (34) (Jacolot et al. 2013; Kuroda et al. 2018). In addition, biomimetic synthetic approaches probing proposed biosynthetic pathways in-vitro e.g., the conversion of pestalone in the presence of ammonia to rac-pestalachloride A (138) (Slavov et al. 2010) and the condensation of keto-aldehyde intermediates with primary amines under mild conditions to give racemic marilines (148–150) (Augner and Schmalz 2015; Min et al. 2017), strongly supported a non-stereoselective, non-enzymatic formation of the γ-lactam ring. Phthalimidines with an 8-substituted core skeleton were isolated as racemates, due to the stereogenic C-8, as evident from specific optical rotation measurements and ECD spectra, frequently pertinent to further separation using chiral HPLC. Synthetic chemists proposed that reaction of 2-formylarylketone (228) with a primary amine forms initially a cyclic hemiaminal (229), which then undergoes tautomerisation via an isoindole intermediate (230) to give the isoindolinone racemate (231) (Fig. 21) (Augner and Schmalz 2015; Min et al. 2017; Slavov et al. 2010).

A potential biosynthetic pathway to phthalimidines core skeleton; SAM, S-adenosyl-L-methionine
Thus far, a common biosynthetic precursor for fungal phthalimidine metabolites appears to be a tetraketide orsellinate derivative (232), a product of iterative NR-PKSs and a known intermediate in the biosynthesis of several fungal metabolites (Geris and Simpson 2009; Matsuda and Abe 2016). This was also the case for their oxygenated analogues (phthalides), e.g. mycophenolic acid, silvaticol and nidulol, which confirms the close yet divergent biosynthetic relationship between phthalide and phthalimidine metabolites employing similar enzymes and mechanisms and explains their frequent co-isolation (Ayer and Racok 1990; Regueira et al. 2011). For example cicH, encoding a P450 monooxygenase, in the cichorine (2) BGC, is homologous to mpaD in the mycophenolic acid BGC, and cytochrome P450 genes pkfB and atnE were also annotated in both the aspernidine (10) and aspercryptin (6) BGC, respectively (Sanchez et al. 2012). The P450 monoxygenase (mpaD) was predicted to catalyse ring closure and lactonisation of mycophenolic acid and could be postulated to have a similar function in fungal phathalimidines ring cyclisation (Regueira et al. 2011).
The nitrogen atom in the lactam ring of phthalimidines is postulated to be derived from amino acid metabolism (e.g. SMTPs containing serine, leucine, phenylalanine or tryptophan partial structures), and feeding fungal cultures with amine precursors was found to increase their production, e.g. salfredins (Matsumoto et al. 1995), and was used to generate novel derivatives, e.g. SMTPs (Hasegawa et al. 2010; Koide et al. 2012). Although, non-enzymatic amination of intermediate precursors, not involving amino acids, could also be envisioned. For example, the formation of SMTPs and phthalimidine-type phenylspirodrimanes from 226 and 227, respectively (Hasumi and Suzuki 2021).
Continued advances in fungal genome sequencing and modern molecular genetic techniques will ensure a better understanding of phthalimidine BGCs and could be extended for genome mining of structurally-related compounds, i.e. phthalides. The so far identified BGCs could be used as query to search for homology genes in newly sequenced fungal genomes helping link clusters to phthalimidine metabolites. And as more fungal phthalimidines biosynthetic pathways are isolated through genome mining efforts, characterization of NR-PKSs producing the archetypal orsellinate-derivatives, forming their backbone structure, is highly intriguing as these have variable domain architecture and could be used as a starting point in the search for fungal phthalimidine BGCs. In-silico genome mining of the newly sequenced and assembled Stachylidium genome identified several biosynthetic genes, of interest is an iterative PKS with an orsellinic acid synthase domain architecture and a NRPS-like gene suggested to initiate marilines (148–150) biosynthesis. Phylogenetic analysis of these such orsellinate synthases could provide a basis for future biosynthetic classification of fungal phthalimidines among major fungal groups and related domains (bacteria and plants). Moreover, genes flanking the NR-PKSs would provide a better understanding of downstream reactions, including the introduction of the nitrogen atom for indole ring formation and ring cyclisation. It is still challenging trying to directly predict the product of an iterative NR-PKSs, based on their amino acid sequences and domain architecture due to lack of understanding of how tailoring domains in individual iterations are programmed and number of iterations performed. Besides the metabolite identified from the PKS rarely seems to be the final product in the biosynthesis, but usually undergoes further modifications by tailoring enzymes modifying the core structure, which could be localized outside the BGC in other parts of the fungal genome. Additionally, metabolic engineering efforts could help increase phthalimidine yields for future commercial production and simultaneously allow the generation of novel analogues. While, new chemical insights obtained in elucidating phthalimidine metabolic pathways would complement the ongoing efforts of synthetic chemists by helping design more efficient chemoenzymatic synthesis routes for complex enantiopure phthalimidines.
Abbreviations
- PLC-γ1:
-
1/2, Phospholipase C-γ1
- 11β-HSD1:
-
11β-Hydroxysteroid dehydrogenase type 1
- ADMET:
-
Absorption, distribution, metabolism, excretion, and toxicity
- ABTS:
-
2,2’-Azino-bis-(3-ethylbenzothiazoline-6-sulfonic acid)
- AIS:
-
Acute ischemic stroke
- AKI:
-
Acute kidney injury
- ACP:
-
Acyl-carrier protein
- AT:
-
Acyltransferase domain
- A:
-
Adenylation
- AR:
-
Aldose reductase
- BGC:
-
Biosynthetic gene cluster
- BDNF:
-
Brain-derived neurotrophic factor
- COPD:
-
Chronic obstructive pulmonary disease
- C-MeT:
-
C-methyl transferase
- C:
-
Condensation
- COX:
-
Cyclooxygenase
- DM:
-
Diabetes mellitus
- DPPH:
-
2,2-Diphenyl-1-picrylhydrazyl
- ECD:
-
Electron Capture Detector
- CBL38:
-
Endophytic fungus Aspergillus variecolor CLB38
- ET:
-
Endothelin
- EGF:
-
Epidermal growth factor
- ERK:
-
Extracellular signal-related kinase
- FPP:
-
Farnesyl pyrophosphate
- FMO:
-
Flavin-dependent monooxygenase
- FGFC1:
-
Fungi fibrinolytic compound 1
- FGFC2:
-
Fungi fibrinolytic compound 2
- G6PT:
-
Glucose-6-phosphate translocase
- SERCA2:
-
Sarco(endo)plasmic reticulum Ca2 + ATPase
- HT:
-
Hemorrhagic transformation
- HPLC:
-
High-Performance Liquid Chromatography
- Tie2:
-
Homology domains
- HLE:
-
Human leukocyte elastase
- TG:
-
Intracellular triglyceride
- KS:
-
Ketosynthase domain
- LKN:
-
Last known normal
- HK-ZJ:
-
Mangrove endophytic Aspergillus sp.
- MM-1:
-
Matrix metalloproteinase-1
- MeFSAT:
-
Medicinal Fungi Secondary Metabolites and Therapeutics
- MRSA:
-
Methicillin-resistant Staphylococcus aureus
- MVA:
-
Mevalonate pathway
- MCA:
-
Middle cerebral artery
- MIC:
-
Minimal inhibitory concentration
- MD:
-
Molecular dynamics
- MptpB:
-
Mycobacterium tuberculosis protein tyrosine phosphatase B
- NGF:
-
Nerve growth factor
- NR:
-
Nitrate reductase
- NNRTI:
-
Non-nucleoside reverse transcriptase inhibitors
- NR-PKSs:
-
Non-reducing group of iterative polyketide synthases
- NRPS:
-
Non-ribosomal peptide synthetase
- NMR:
-
Nuclear Magnetic Resonance
- NO:
-
Nitric oxide
- OSMAC:
-
One-strain many compounds
- PCP:
-
Peptidyl carrier protein
- PI3K:
-
Phosphatidylinositol-3-kinase
- PDGF:
-
Platelet-derived growth factor s
- PEG:
-
Polyethylene glycol
- PKS:
-
Polyketide synthase
- PTase:
-
Prenyltransferase
- PT:
-
Product template
- PTP1B:
-
Protein tyrosine phosphatase-1B
- CD:
-
Quantum-chemical
- ROS:
-
Reactive oxygen species
- R:
-
Reductase
- RT:
-
Reverse transcriptase
- SAM:
-
S-Adenosylmethionine
- SM:
-
Secondary metabolite
- SMASH:
-
Secondary Metabolite Analysis Shell
- SARs:
-
Severe Acute Respiratory Syndrome
- SARS-CoV:
-
Severe Acute Respiratory Syndrome Coronavirus
- SSF:
-
Solid-state fermentation
- SMTPs:
-
Stachybotrys microspora Triprenyl phenols
- SAR:
-
Structure–activity relationship
- SAT:
-
Starter unit ACP transacylase
- SAHA:
-
Suberoylanilide hydroxamic acid
- TC:
-
Terpene cyclase
- TE:
-
Thioesterase
- CLC:
-
Claisen cyclase
- T:
-
Thiolation
- TD-DFT:
-
Time-dependent density functional theory
- t-PA:
-
Tissue-type plasminogen activator
- TC:
-
Total cholesterol
- TCM:
-
Traditional Chinese Medicine
- TPPs:
-
Triprenyl phenols
- VSMCs:
-
Vascular smooth muscle cells
References
Achenbach H, Mühlenfeld A, Kohl W, Brillinger G-U (1985) Stoffwechselprodukte von mikroorganismen, XXXI [1]. Duricaulinsäure, ein neuer naturstoff vom phthalimidin-Typ aus Aspergillus duricaulis/Metabolie products of microorganisms, XXXI [1]. Duricaulic acid, a new natural product of the phthalim idine type from Aspergillus duricaulis. Zeitschrift Für Naturforschung B 40:1219–1225
Adpressa DA, Stalheim KJ, Proteau PJ, Loesgen S (2017) Unexpected biotransformation of the HDAC inhibitor vorinostat yields aniline-containing fungal metabolites. ACS Chem Biol 12:1842–1847
Ageta H, Ageta T (1984) Ericaceous constituents: seventeen triterpenoids isolated from the buds of Rhododendron macrocepalum. Chem Pharm Bull 32:369–372
Ahuja M, Chiang Y-M, Chang S-L, Praseuth MB, Entwistle R, Sanchez JF, Lo H-C, Yeh H-H, Oakley BR, Wang CC (2012) Illuminating the diversity of aromatic polyketide synthases in Aspergillus nidulans. J Am Chem Soc 134:8212–8221
Almeida C, Kehraus S, Prudêncio M, König GM (2011a) Marilones A-C, phthalides from the sponge-derived fungus Stachylidium sp. Beilstein J Org Chem 7:1636–1642
Almeida C, Part N, Bouhired S, Kehraus S, König GM (2011b) Stachylines A−D from the sponge-derived fungus Stachylidium sp. J Nat Prod 74:21–25
Almeida C, Hemberger Y, Schmitt SM, Bouhired S, Natesan L, Kehraus S, Dimas K, Gütschow M, Bringmann G, König GM (2012) Marilines A-C: novel phthalimidines from the sponge-derived fungus stachylidium sp. Chem-Eur J 18:8827–8834
Aqueveque P, Céspedes CL, Becerra J, Aranda M, Sterner O (2017) Antifungal activities of secondary metabolites isolated from liquid fermentations of Stereum hirsutum (Sh134-11) against Botrytis cinerea (grey mould agent). Food Chem Toxicol 109:1048–1054
Ariefta NR, Koseki T, Nishikawa Y, Shiono Y (2021) Spirocollequins A and B, new alkaloids featuring a spirocyclic isoindolinone core, from Colletotrichum boninense AM-12-2. Tetrahedron Lett 64:152736
Ashour A, Amen Y, Allam AE, Kudo T, Nagata M, Ohnuki K, Shimizu K (2019) New isoindolinones from the fruiting bodies of the fungus Hericium erinaceus. Phytochem Lett 32:10–14
Augner D, Schmalz H-G (2015) Biomimetic synthesis of isoindolinones related to the marilines. Synlett 26:1395–1397
Augner D, Krut O, Slavov N, Gerbino DC, Sahl HG, Benting J, Schmalz HG (2013) On the antibiotic and antifungal activity of pestalone, pestalachloride A, and structurally related compounds. J Nat Products 76(8):1519–1522
Ayer WA, Miao S (1993) Secondary metabolites of the aspen fungus Stachybotrys cylindrospora. Can J Chem 71:487–493
Ayer WA, Racok JS (1990) The metabolites of Talaromy ces flavus: Part 2. Biological activity and biosynthetic studies. Can J Chem 68:2095–2101
Azumaya I, Kagechika H, Fujiwara Y, Itoh M, Yamaguchi K, Shudo K (1991) Twisted intramolecular charge-transfer fluorescence of aromatic amides: conformation of the amide bonds in excited states. J Am Chem Soc 113:2833–2838
Basnet BB, Chen B, Suleimen YM, Ma K, Guo S, Bao L, Huang Y, Liu H (2019) Cytotoxic secondary metabolites from the endolichenic fungus Hypoxylon fuscum. Planta Med 85:1088–1097
Bharadwaj KK, Sarkar T, Ghosh A, Baishya D, Rabha B, Panda MK, Nelson BR, John AB, Sheikh HI, Dash BP (2021) Macrolactin a as a novel inhibitory agent for SARS-CoV-2 M pro: bioinformatics approach. Appl Biochem Biotechnol 193:3371–3394
Bode HB, Bethe B, Höfs R, Zeeck A (2002) Big effects from small changes: possible ways to explore nature’s chemical diversity. ChemBioChem 3:619–627
Bolchi C, Bavo F, Appiani R, Roda G, Pallavicini M (2020) 1, 4-Benzodioxane, an evergreen, versatile scaffold in medicinal chemistry: a review of its recent applications in drug design. Eur J Med Chem 200:112419
Braesel J, Fricke J, Schwenk D, Hoffmeister D (2017) Biochemical and genetic basis of orsellinic acid biosynthesis and prenylation in a stereaceous basidiomycete. Fungal Genet Biol 98:12–19
Brandalise F, Roda E, Ratto D, Goppa L, Gargano ML, Cirlincione F, Priori EC, Venuti MT, Pastorelli E, Savino E (2023) Hericium erinaceus in neurodegenerative diseases: from bench to bedside and beyond, how far from the Shoreline? J Fungi 9:551
Brown CE, Liscombe DK, McNulty J (2018) Three new polyketides from fruiting bodies of the endophytic ascomycete Xylaria polymorpha. Nat Prod Res 32:2408–2417
Bushley KE, Raja R, Jaiswal P, Cumbie JS, Nonogaki M, Boyd AE, Owensby CA, Knaus BJ, Elser J, Miller D (2013) The genome of Tolypocladium inflatum: evolution, organization, and expression of the cyclosporin biosynthetic gene cluster. PLoS Genet 9:e1003496
Charliński G, Vesole DH, Jurczyszyn A (2021) Rapid progress in the use of immunomodulatory drugs and cereblon e3 ligase modulators in the treatment of multiple myeloma. Cancers 13:4666
Chaudhari K, Surana S, Jain P, Patel HM (2016) Mycobacterium Tuberculosis (MTB) GyrB inhibitors: An attractive approach for developing novel drugs against TB. Eur J Med Chem 124:160–185
Chen S, Liu Z, Liu Y, Lu Y, He L, She Z (2015) New depsidones and isoindolinones from the mangrove endophytic fungus Meyerozyma guilliermondii (HZ-Y2) isolated from the South China Sea. Beilstein J Org Chem 11:1187–1193
Chen J, Zeng X, Yang YL, Xing YM, Zhang Q, Li JM, Ma K, Liu HW, Guo SX (2017a) Genomic and transcriptomic analyses reveal differential regulation of diverse terpenoid and polyketides secondary metabolites in Hericium erinaceus. Sci Rep 7:10151
Chen L, Li Z-H, Yao J-N, Peng Y-L, Huang R, Feng T, Liu J-K (2017b) Isoindolinone-containing meroterpenoids with α-glucosidase inhibitory activity from mushroom Hericium caput-medusae. Fitoterapia 122:107–114
Chen M, Liu Q, Gao S-S, Young AE, Jacobsen SE, Tang Y (2019a) Genome mining and biosynthesis of a polyketide from a biofertilizer fungus that can facilitate reductive iron assimilation in plant. Proc Natl Acad Sci 116:5499–5504
Chen X, Zhao S, Li H, Wang X, Geng A, Cui H, Lu T, Chen Y, Zhu Y (2019b) Design, synthesis and biological evaluation of novel isoindolinone derivatives as potent histone deacetylase inhibitors. Eur J Med Chem 168:110–122
Cheng Y-Q, Ahn J-H, Walton JD (1999) A putative branched-chain-amino-acid transaminase gene required for HC-toxin biosynthesis and pathogenicity in Cochliobolus carbonum. Microbiology 145:3539–3546
Chi C-L, Xu L, Li J-J, Liu Y, Chen B-Q (2022) Synthesis, antiproliferative, and antimicrobial properties of novel phthalimide derivatives. Med Chem Res 31:120–131
Chiang Y-M, Oakley BR, Keller NP, Wang CC (2010) Unraveling polyketide synthesis in members of the genus Aspergillus. Appl Microbiol Biotechnol 86:1719–1736
Chiang Y-M, Oakley CE, Ahuja M, Entwistle R, Schultz A, Chang S-L, Sung CT, Wang CC, Oakley BR (2013) An efficient system for heterologous expression of secondary metabolite genes in Aspergillus nidulans. J Am Chem Soc 135:7720–7731
Chiang YM, Ahuja M, Oakley CE, Entwistle R, Asokan A, Zutz C, Wang CC, Oakley BR (2016) Development of genetic dereplication strains in Aspergillus nidulans results in the discovery of aspercryptin. Angew Chem Int Ed 55:1662–1665
Choomuenwai V, Beattie KD, Healy PC, Andrews KT, Fechner N, Davis RA (2015) Entonalactams A-C: isoindolinone derivatives from an Australian rainforest fungus belonging to the genus Entonaema. Phytochemistry 117:10–16
Chunyu WX, Ding ZG, Li MG, Zhao JY, Gu SJ, Gao Y, Wang F, Ding JH, Wen ML (2016) Stachartins A-E, phenylspirodrimanes from the tin mine tailings-associated fungus Stachybotrys chartarum. Helv Chim Acta 99:583–587
Chupakhin E, Babich O, Prosekov A, Asyakina L, Krasavin M (2019) Spirocyclic motifs in natural products. Molecules 24:4165
Cornella I, Kelly TR (2004) Synthesis of porritoxin. J Org Chem 69:2191–2193
Crawford JM, Townsend CA (2010) New insights into the formation of fungal aromatic polyketides. Nat Rev Microbiol 8:879–889
Crawford JM, Vagstad AL, Ehrlich KC, Townsend CA (2008) Starter unit specificity directs genome mining of polyketide synthase pathways in fungi. Bioorg Chem 36:16–22
Cui H, Lin Y, Luo M, Lu Y, Huang X, She Z (2017a) Diaporisoindoles A-C: three isoprenylisoindole alkaloid derivatives from the mangrove endophytic fungus Diaporthe sp. SYSU-HQ3. Org Lett 19:5621–5624
Cui H, Yu J, Chen S, Ding M, Huang X, Yuan J, She Z (2017b) Alkaloids from the mangrove endophytic fungus Diaporthe phaseolorum SKS019. Bioorg Med Chem Lett 27:803–807
Cui H, Liu Y, Li J, Huang X, Yan T, Cao W, Liu H, Long Y, She Z (2018a) Diaporindenes A-D: four unusual 2, 3-dihydro-1 H-indene analogues with anti-inflammatory activities from the mangrove endophytic fungus Diaporthe sp. SYSU-HQ3. J Org Chem 83:11804–11813
Cui H, Liu Y, Li T, Zhang Z, Ding M, Long Y, She Z (2018b) 3-Arylisoindolinone and sesquiterpene derivatives from the mangrove endophytic fungi Aspergillus versicolor SYSU-SKS025. Fitoterapia 124:177–181
Dai G, Shen Q, Zhang Y, Bian X (2022) Biosynthesis of fungal natural products involving two separate pathway crosstalk. J Fungi 8:320
De Souza CP, Hashmi SB, Osmani AH, Andrews P, Ringelberg CS, Dunlap JC, Osmani SA (2013) Functional analysis of the Aspergillus nidulans kinome. PLoS ONE 8:e58008
dela Peña, I., Borlongan, C., Shen, G., Davis, W., (2017) Strategies to extend thrombolytic time window for ischemic stroke treatment: an unmet clinical need. J Stroke 19:50
Deng W-P, Zhong M, Guo X-C, Kende AS (2003) Total synthesis and structure revision of Stachybotrys Spirolactams. J Org Chem 68:7422–7427
Di Mola A, Tiffner M, Scorzelli F, Palombi L, Filosa R, De Caprariis P, Waser M, Massa A (2015) Bifunctional phase-transfer catalysis in the asymmetric synthesis of biologically active isoindolinones. Beilstein J Org Chem 11:2591–2599
Du L, Lou L (2010) PKS and NRPS release mechanisms. Nat Prod Rep 27:255–278
Eder C, Kurz M, Toti L (2007) Spirobenzofuranlactam derivatives, methods for their preparation, and use thereof. European Patent EP1572697 21
Eisfeld K (2009) Non-ribosomal peptide synthetases of fungi. Physiol Genet Select Basic Appl Aspect pp 305–330
El Amrani M, Debbab A, Aly AH, Wray V, Dobretsov S, Müller WE, Lin W, Lai D, Proksch P (2012) Farinomalein derivatives from an unidentified endophytic fungus isolated from the mangrove plant Avicennia marina. Tetrahedron Lett 53:6721–6724
El Maddah F, Eguereva E, Kehraus S, König GM (2019) Biosynthetic studies of novel polyketides from the marine sponge-derived fungus Stachylidium sp. 293K04. Org Biomol Chem 17:2747–2752
Feng J-M, Li M, Zhao J-L, Jia X-N, Liu J-M, Zhang M, Chen R-D, Xie K-B, Chen D-W, Yu H-B (2019) Three new phenylspirodrimane derivatives with inhibitory effect towards potassium channel Kv1. 3 from the fungus Stachybotrys chartarum. J Asian Nat Products Res
Frisvad JC, Andersen B, Thrane U (2008) The use of secondary metabolite profiling in chemotaxonomy of filamentous fungi. Mycol Res 112:231–240
Geris R, Simpson TJ (2009) Meroterpenoids produced by fungi. Nat Prod Rep 26:1063–1094
Gong W, Wang Y, Xie C, Zhou Y, Zhu Z, Peng Y (2020) Whole genome sequence of an edible and medicinal mushroom, Hericium erinaceus (Basidiomycota, Fungi). Genomics 112:2393–2399
Guo R, Zhang Y, Duan D, Fu Q, Zhang X, Yu X, Wang S, Bao B, Wu W (2016) Fibrinolytic evaluation of compounds isolated from a marine fungus Stachybotrys longispora FG216. Chin J Chem 34:1194–1198
Han H, Yu C, Qi J, Wang P, Zhao P, Gong W, Xie C, Xia X, Liu C (2023) High-efficient production of mushroom polyketide compounds in a platform host Aspergillus oryzae. Microb Cell Fact 22:1–10
Harned AM, Volp KA (2011) The sorbicillinoid family of natural products: isolation, biosynthesis, and synthetic studies. Nat Prod Rep 28:1790–1810
Hasegawa K, Koide H, Hu W, Nishimura N, Narasaki R, Kitano Y, Hasumi K (2010) Structure–activity relationships of 11 new congeners of the SMTP plasminogen modulator. J Antibiot 63:589–593
Hasumi K, Suzuki E (2021) Impact of SMTP targeting plasminogen and soluble epoxide hydrolase on thrombolysis, inflammation, and ischemic stroke. Int J Mol Sci 22:954
Hasumi K, Ohyama S, Kohyama T, Ohsaki Y, Takayasu R, Endo A (1998) Isolation of SMTP-3, 4, 5 and-6, novel analogs of staplabin, and their effects on plasminogen activation and fibrinolysis. J Antibiot 51:1059–1068
Hasumi K, Hasegawa K, Kitano Y (2007) Isolation and absolute configuration of SMTP-0, a simplest congener of the SMTP family nonlysine-analog plasminogen modulators. J Antibiot 60:463–468
Helaly SE, Thongbai B, Stadler M (2018) Diversity of biologically active secondary metabolites from endophytic and saprotrophic fungi of the ascomycete order Xylariales. Nat Prod Rep 35:992–1014
Henke MT, Soukup AA, Goering AW, McClure RA, Thomson RJ, Keller NP, Kelleher NL (2016) New aspercryptins, lipopeptide natural products, revealed by HDAC inhibition in Aspergillus nidulans. ACS Chem Biol 11:2117–2123
Herbst DA, Townsend CA, Maier T (2018) The architectures of iterative type I PKS and FAS. Nat Prod Rep 35:1046–1069
Hinkley SF, Fettinger JC, Dudley K, Jarvis BB (1999) Memnobotrins and memnoconols: novel metabolites from Memnoniella echinata. J Antibiot 52:988–997
Horiuchi M, Maoka T, Iwase N, Ohnishi K (2002) Reinvestigation of structure of porritoxin, a phytotoxin of Alternaria porri. J Nat Prod 65:1204–1205
Horiuchi M, Ohnishi K, Iwase N, Nakajima Y, Tounai K, Yamashita M, Yamada Y (2003) A novel isoindoline, porritoxin sulfonic acid, from Alternaria porri and the structure-phytotoxicity correlation of its related compounds. Biosci Biotechnol Biochem 67:1580–1583
Horiuchi M, Tokuda H, Ohnishi K, Yamashita M, Nishino H, Maoka T (2006) Porritoxins, metabolites of Alternaria porri, as anti-tumor-promoting active compounds. Nat Prod Res 20:161–166
Hu W, Ohyama S, Hasumi K (2000) Activation of fibrinolysis by SMTP-7 and-8, novel staplabin analogs with a pseudosymmetric structure. J Antibiot 53:241–247
Hu J, Wang N, Liu H, Li S, Liu Z, Zhang W, Gao X (2022) Secondary metabolites from a deep-sea derived fungal strain of Phomopsis lithocarpus FS508. Natural Product Research, pp 1–8
Ichikawa K, Inuzuka T, Yoda H, Sengoku T (2022) Total synthesis and structural confirmation of (±)-spirocollequins A and B. Tetrahedron Lett 107:154109
Igboeli HA, Marchbank DH, Correa H, Overy D, Kerr RG (2019) Discovery of primarolides A and B from marine fungus Asteromyces cruciatus using osmotic stress and treatment with suberoylanilide hydroxamic acid. Mar Drugs 17:435
Intaraudom C, Bunbamrung N, Dramae A, Boonyuen N, Kongsaeree P, Srichomthong K, Supothina S, Pittayakhajonwut P (2017) Terphenyl derivatives and drimane–Phathalide/isoindolinones from Hypoxylon fendleri BCC32408. Phytochemistry 139:8–17
Intaraudom C, Punyain W, Bunbamrung N, Dramae A, Boonruangprapa T, Pittayakhajonwut P (2019) Antimicrobial drimane–phthalide derivatives from Hypoxylon fendleri BCC32408. Fitoterapia 138:104353
Iqbal A, Herren F, Wallquist O (2009) Isoindolinone pigments. High Performance Pigments, pp 243–259
Ito A, Niizuma K, Shimizu H, Fujimura M, Hasumi K, Tominaga T (2014) SMTP-7, a new thrombolytic agent, decreases hemorrhagic transformation after transient middle cerebral artery occlusion under warfarin anticoagulation in mice. Brain Res 1578:38–48
Itoh T, Nagata K, Kanemitsu T, Miyazaki M, Tomisawa Y, Misa M (2022) Chemical method of producing SMTP groups or SMTP-7 and intermediates used in the method. Google Patents
Ito-Kobayashi M, Aoyagi A, Tanaka I, Muramatsu Y, Umetani M, Takatsu T (2008) Sterenin A, B, C and D, novel 11β-hydroxysteroid dehydrogenase type 1 inhibitors from Stereum sp. SANK 21205. J Antibiot 61:128–135
Jacolot M, Jean M, Tumma N, Bondon A, Chandrasekhar S, van de Weghe P (2013) Synthesis of stachybotrin C and all of its stereoisomers: structure revision. J Org Chem 78:7169–7175
Jarvis BB, Salemme J, Morals A (1995) Stachybotrys toxins. 1. Nat Toxins 3:10–16
Jiang S, Wang S, Sun Y, Zhang Q (2014) Medicinal properties of Hericium erinaceus and its potential to formulate novel mushroom-based pharmaceuticals. Appl Microbiol Biotechnol 98:7661–7670
Kamalov L, Aripova S, Isaev M (1997) Low-molecular-mass metabolites of fungi I. Stachybotrin from Stachybotrys alternans. Chem Nat Compd 33:462–468
Kamalov L, Aripova S, Tashkhodzhaev B, Isaev M (1998) Low-molecular-weight metabolites of fungi. II. Refinement of the structure of stachybotrin. Chem Nat Compd 34:605–608
Kamalov L, Aripova S, Isaev M (1999) Low-molecular-mass metabolites of fungi IV. the structures of stachybotrin A and stachybotral. Chem Nat Compd 35:82–85
Kamauchi H, Shiraishi Y, Kojima A, Kawazoe N, Kinoshita K, Koyama K (2018) Isoindolinones, phthalides, and a naphthoquinone from the fruiting body of Daldinia concentrica. J Nat Prod 81:1290–1294
Kawagishi H, Ando M, Mizuno T (1990) Hericenone A and B as cytotoxic principles from the mushroom Hericium erinaceum. Tetrahedron Lett 31:373–376
Kawahara N, Nozawa K, Nakajima S, Udagawa S-I, Kawai K-I (1988) Studies on fungal products. XVI.: new metabolites related to 3-methylorsellinate from Aspergillus silvaticus. Chem Pharm Bull 36:398–400
Kende AS, Deng W-P, Zhong M, Guo X-C (2003) Enantioselective total synthesis and structure revision of spirodihydrobenzofuranlactam 1. total synthesis of stachybotrylactam. Org Lett 5:1785–1788
Kerru N, Gummidi L, Maddila S, Gangu KK, Jonnalagadda SB (2020) A review on recent advances in nitrogen-containing molecules and their biological applications. Molecules 25:1909
Khan MA, Tania M, Liu R, Rahman MM (2013) Hericium erinaceus: an edible mushroom with medicinal values. J Complement Integr Med 10:253–258
Kim Y-H, Cho S-M, Hyun J-W, Ryoo I-J, Choo S-J, Lee S, Seok S-J, Hwang JS, Sohn ED, Yun B-S (2008) A new antioxidant, clitocybin A, from the culture broth of Clitocybe aurantiaca. J Antibiot 61:573–576
Kim KH, Noh HJ, Choi SU, Lee KR (2012) Isohericenone, a new cytotoxic isoindolinone alkaloid from Hericium erinaceum. J Antibiot 65:575–577
Kim K-C, Lee H-W, Lee H-W, Choo S-J, Yoo I-D, Ha B-J (2014) Fermentation process for mass production of clitocybin A, a New anti-wrinkle agent from Clitocybe aurantiaca and evaluation of inhibitory activity on matrix metalloproteinase-1 expression. Microbiol Biotechnol Lett 42:194–201
Kimura Y, Nishibe M, Nakajima H, Hamasaki T, Shimada A, Tsuneda A, Shigematsu N (1991) Hericerin, a new pollen growth inhibitor from the mushroom Hericium erinaceum. Agric Biol Chem 55:2673–2674
Kiryakov H, Mardirossian Z, Hughes DW, MacLean DB (1980) Fumschleicherine, an alkaloid of Fumaria schleicheri. Phytochemistry 19:2507–2509
Kobayashi S, Inoue T, Ando A, Tamanoi H, Ryu I, Masuyama A (2012) Total synthesis and structural revision of hericerin. J Org Chem 77:5819–5822
Kobayashi S, Tamanoi H, Hasegawa Y, Segawa Y, Masuyama A (2014) Divergent synthesis of bioactive resorcinols isolated from the fruiting bodies of Hericium erinaceum: total syntheses of hericenones A, B, and I, hericenols B-D, and erinacerins A and B. J Org Chem 79:5227–5238
Kohyama T, Hasumi K, Hamanaka A, Endo A (1997) SMTP-1 and-2, novel analogs of staplabin produced by Stachybotrys microspora IFO30018. J Antibiot 50:172–174
Koide H, Hasegawa K, Nishimura N, Narasaki R, Hasumi K (2012) A new series of the SMTP plasminogen modulators with a phenylamine-based side chain. J Antibiot 65:361–367
Kuroda Y, Hasegawa K, Noguchi K, Chiba K, Hasumi K, Kitano Y (2018) Confirmation of the absolute configuration of Stachybotrin C using single-crystal X-ray diffraction analysis of its 4-bromobenzyl ether derivative. J Antibiot 71:584–591
Lee I-K, Kim S-E, Yeom J-H, Ki D-W, Lee M-S, Song J-G, Kim Y-S, Seok S-J, Yun B-S (2012) Daldinan A, a novel isoindolinone antioxidant from the ascomycete Daldinia concentrica. J Antibiot 65:95–97
Lee J-E, Lee I-S, Kim K-C, Yoo I-D, Yang H-M (2017) ROS scavenging and anti-wrinkle effects of clitocybin a isolated from the mycelium of the mushroom Clitocybe aurantiaca. J Microbiol Biotechnol 27:933–938
Li E, Jiang L, Guo L, Zhang H, Che Y (2008) Pestalachlorides A-C, antifungal metabolites from the plant endophytic fungus Pestalotiopsis adusta. Bioorg Med Chem 16:7894–7899
Li Y, Liu D, Cen S, Proksch P, Lin W (2014a) Isoindolinone-type alkaloids from the sponge-derived fungus Stachybotrys chartarum. Tetrahedron 70:7010–7015
Li Y, Wu C, Liu D, Proksch P, Guo P, Lin W (2014b) Chartarlactams A-P, phenylspirodrimanes from the sponge-associated fungus Stachybotrys chartarum with antihyperlipidemic activities. J Nat Prod 77:138–147
Li W, Zhou W, Kim E-J, Shim SH, Kang HK, Kim YH (2015) Isolation and identification of aromatic compounds in Lion’s Mane Mushroom and their anticancer activities. Food Chem 170:336–342
Li C, Matsuda Y, Gao H, Hu D, Yao XS, Abe I (2016) Biosynthesis of LL-Z1272β: discovery of a new member of NRPS-like enzymes for Aryl-aldehyde formation. ChemBioChem 17:904–907
Li W, Lee SH, Jang HD, Ma JY, Kim YH (2017) Antioxidant and anti-osteoporotic activities of aromatic compounds and sterols from Hericium erinaceum. Molecules 22:108
Li Q, Chen C, He Y, Wei M, Cheng L, Kang X, Wang J, Hao X, Zhu H, Zhang Y (2020) Prenylated quinolinone alkaloids and prenylated isoindolinone alkaloids from the fungus Aspergillus nidulans. Phytochemistry 169:112177
Liao L, Zhang X, Lou Y, Zhou C, Yuan Q, Gao J (2019) Discovery of three new phytotoxins from the fungus Aspergillus nidulans by pathway inactivation. Molecules 24:515
Lin C-F, Shiao Y-J, Chen C-C, Tzeng T-T, Chen C-C, Lee L-Y, Chen W-P, Shen C-C (2018) A xanthurenate and an isoindolinone from the mycelia of Hericium erinaceum. Phytochem Lett 26:218–221
Logrieco A, Moretti A, Solfrizzo M (2009) Alternaria toxins and plant diseases: an overview of origin, occurrence and risks. World Mycotoxin J 2:129–140
Lombard J, Moreira D (2011) Origins and early evolution of the mevalonate pathway of isoprenoid biosynthesis in the three domains of life. Mol Biol Evol 28:87–99
Lü WW, Gao YJ, Su MZ, Luo Z, Zhang W, Shi GB, Zhao QC (2013) Isoindolones from Lasiosphaera fenzlii Reich and their bioactivities. Helv Chim Acta 96:109–113
Ma B-J, Shen J-W, Yu H-Y, Ruan Y, Wu T-T, Zhao X (2010) Hericenones and erinacines: stimulators of nerve growth factor (NGF) biosynthesis in Hericium erinaceus. Mycology 1:92–98
Ma X, Li L, Zhu T, Ba M, Li G, Gu Q, Guo Y, Li D (2013) Phenylspirodrimanes with anti-HIV activity from the sponge-derived fungus Stachybotrys chartarum MXH-X73. J Nat Prod 76:2298–2306
Ma X, Wang H, Li F, Zhu T, Gu Q, Li D (2015) Stachybotrin G, a sulfate meroterpenoid from a sponge derived fungus Stachybotrys chartarum MXH-X73. Tetrahedron Lett 56:7053–7055
Ma X, Lv X, Zhang J (2018) Exploiting polypharmacology for improving therapeutic outcome of kinase inhibitors (KIs): an update of recent medicinal chemistry efforts. Eur J Med Chem 143:449–463
Ma X-H, Zheng W-M, Sun K-H, Gu X-F, Zeng X-M, Zhang H-T, Zhong T-H, Shao Z-Z, Zhang Y-H (2019) Two new phenylspirodrimanes from the deep-sea derived fungus Stachybotrys sp. MCCC 3A00409. Nat Prod Res 33:386–392
Matsuda Y, Abe I (2016) Biosynthesis of fungal meroterpenoids. Nat Prod Rep 33:26–53
Matsumoto K, Nagashima K, Kamigauchi T, Kawamura Y, Yasuda Y, Ishii K, Uotani N, Sato T, Nakai H, Terui Y (1995) Salfredins, new aldose reductase inhibitors produced by Crucibulum sp. RF-3817 I. Fermentation, isolation and structures of salfredins. J Antibiot 48:439–446
Matsumoto N, Suzuki E, Tsujihara K, Nishimura Y, Hasumi K (2015) Structure–activity relationships of the plasminogen modulator SMTP with respect to the inhibition of soluble epoxide hydrolase. J Antibiot 68:685–690
Meng J, Wang X, Xu D, Fu X, Zhang X, Lai D, Zhou L, Zhang G (2016) Sorbicillinoids from fungi and their bioactivities. Molecules 21:715
Min C, Lin Y, Seidel D (2017) Catalytic enantioselective synthesis of Mariline A and related isoindolinones through a biomimetic approach. Angew Chem Int Ed 56:15353–15357
Minagawa K, Kouzuki S, Kamigauchi T (2002a) Stachyflin and acetylstachyflin, novel anti-influenza a virus substances, produced by Stachybotrys sp. RF-7260 II. Synthesis and preliminary structure-activity relationships of Stachyflin derivatives. J Antibiot 55:165–171
Minagawa K, Kouzuki S, Tani H, Ishii K, Tanimoto T, Terui Y, Kamigauchi T (2002b) Novel stachyflin derivatives from stachybotrys sp. RF-7260 fermentation, isolation, structure elucidation and biological activities. J Antibiot 55:239–248
Minagawa K, Kouzuki S, Yoshimoto J, Kawamura Y, Tani H, Iwata T, Terui Y, Nakai H, Yagi S, Hattori N (2002c) Stachyflin and acetylstachyflin, novel anti-influenza A virus substances, produced by Stachybotrys sp. RF-7260 I. Isolation, structure elucidation and biological activities. J Antibiot 55:155–164
Miyazawa M, Takahashi T, Horibe I, Ishikawa R (2012) Two new aromatic compounds and a new D-arabinitol ester from the mushroom Hericium erinaceum. Tetrahedron 68:2007–2010
Di Mola A, Palombi L, Massa A (2014) An overview on asymmetric synthesis of 3-substituted isoindolinones. Targets in Heterocyclic Systems: Chemistry and Properties; Attanasi, OA, Ed, pp 113–140
Moon E-Y, Oh J-M, Kim Y-H, Ryoo I-J, Yoo I-D (2009) Clitocybins, novel isoindolinone free radical scavengers, from mushroom Clitocybe aurantiaca inhibit apoptotic cell death and cellular senescence. Biol Pharm Bull 32:1689–1694
Moritoyo T, Nishimura N, Hasegawa K, Ishii S, Kirihara K, Takata M, Svensson AK, Umeda-Kameyama Y, Kawarasaki S, Ihara R (2023) A first-in-human study of the anti-inflammatory profibrinolytic TMS-007, an SMTP family triprenyl phenol. Br J Clin Pharmacol 89:1809–1819
Motohashi Y, Igarashi M, Okamatsu M, Noshi T, Sakoda Y, Yamamoto N, Ito K, Yoshida R, Kida H (2013) Antiviral activity of stachyflin on influenza A viruses of different hemagglutinin subtypes. Virol J 10:1–10
Nakamura M, Ito Y, Ogawa K, Michisuji Y, Sato S-I, Takada M, Hayashi M, Yaginuma S, Yamamoto S (1995) Stachybocins, novel endothelin receptor antagonists, produced by Stachybotrys sp. M6222 I. taxonomy, fermentation, isolation and characterization. J Antibiot 48:1389–1395
Narmani A, Teponno RB, Helaly SE, Arzanlou M, Stadler M (2019) Cytotoxic, anti-biofilm and antimicrobial polyketides from the plant associated fungus Chaetosphaeronema achilleae. Fitoterapia 139:104390
Neumann T, Schlegel B, Hoffmann P, Heinze S, Gräfe U (1999) Isolation and structure elucidation of new salfredin-type metabolites from Crucibulum laeve DSM 1653 and DSM 8519. J Basic Microbiol Int J Biochem Physiol Genet Morphol Ecol Microorg 39:357–363
Niehaus E-M, Janevska S, von Bargen KW, Sieber CM, Harrer H, Humpf H-U, Tudzynski B (2014) Apicidin F: characterization and genetic manipulation of a new secondary metabolite gene cluster in the rice pathogen Fusarium fujikuroi. PLoS ONE 9:e103336
Nishimura Y, Suzuki E, Hasegawa K, Nishimura N, Kitano Y, Hasumi K (2012) Pre-SMTP, a key precursor for the biosynthesis of the SMTP plasminogen modulators. J Antibiot 65:483–485
Nishimura N, Niizuma K, Wald M, Hasegawa K, Tominaga T, Hasumi K (2022) Abstract WMP1: results from A Phase 2a study of TMS-007, an SMTP family anti-inflammatory prothrombolytic, on patients with acute ischemic stroke up To 12 hours after onset. Stroke 53, AWMP1-AWMP1
Nozawa Y, Ito M, Sugawara K, Hanada K, Mizoue K (1997a) Stachybotrin C and Parvisporin, novel neuritogenic compounds II. Struct Determ J Antibiot 50:641–645
Nozawa Y, Yamamoto K, Ito M, Sakai N, Mizoue K, Mizobe F, Hanada K (1997b) Stachybotrin C and parvisporin, novel neuritogenic compounds I. taxonomy, isolation, physico-chemical and biological properties. J Antibiot 50:635–640
Ogawa K, Nakamura M, Hayashi M, Yaginuma S, Yamamoto S, Furihata K, Shin-Ya K, Seto HS (1995) Novel endothelin receptor antagonists produced by Stachybotrys sp. M6222. J Antibiotics 48:1396–1400
Overy D, Correa H, Roullier C, Chi W-C, Pang K-L, Rateb M, Ebel R, Shang Z, Capon R, Bills G (2017) Does osmotic stress affect natural product expression in fungi? Mar Drugs 15:254
Papeo G, Orsini P, Avanzi NR, Borghi D, Casale E, Ciomei M, Cirla A, Desperati V, Donati D, Felder ER (2019) Discovery of stereospecific PARP-1 inhibitor isoindolinone NMS-P515. ACS Med Chem Lett 10:534–538
Phan C-W, Lee G-S, Hong S-L, Wong Y-T, Brkljača R, Urban S, Abd Malek SN, Sabaratnam V (2014) Hericium erinaceus (Bull.: Fr) Pers. cultivated under tropical conditions: isolation of hericenones and demonstration of NGF-mediated neurite outgrowth in PC12 cells via MEK/ERK and PI3K-Akt signaling pathways. Food Funct 5:3160–3169
Phuwapraisirisan P, Rangsan J, Siripong P, Tip-Pyang S (2009) New antitumour fungal metabolites from Alternaria porri. Nat Prod Res 23:1063–1071
Piyasena N, Schüffler A, Laatsch H (2015) Xylactam B, A new isobenzofuranone from an endophytic Xylaria sp. Nat Prod Commun 10:1715–1717
Prajapati J, Patel R, Goswami D, Saraf M, Rawal RM (2021) Sterenin M as a potential inhibitor of SARS-CoV-2 main protease identified from MeFSAT database using molecular docking, molecular dynamics simulation and binding free energy calculation. Comput Biol Med 135:104568
Quan Z, Awakawa T, Wang D, Hu Y, Abe I (2019) Multidomain P450 Epoxidase and a terpene cyclase from the ascochlorin biosynthetic pathway in Fusarium sp. Org Lett 21:2330–2334
Regueira TB, Kildegaard KR, Hansen BG, Mortensen UH, Hertweck C, Nielsen J (2011) Molecular basis for mycophenolic acid biosynthesis in Penicillium brevicompactum. Appl Environ Microbiol 77:3035–3043
Robinson T, Singh D, Nigam P (2001) Solid-state fermentation: a promising microbial technology for secondary metabolite production. Appl Microbiol Biotechnol 55:284–289
Roggo BE, Hug P, Moss S, Stämpfli A, Kriemler H-P, Peter HH (1996a) Novel spirodihydrobenzofuranlactams as antagonists of endothelin and as inhibitors of HIV-1 protease produced by Stachybotrys sp. II. Struct Determ J Antibiot 49:374–379
Roggo BE, Petersen F, SiLLS M, ROESEL, J. L., MOERKER, T., PETER, H. H., (1996b) Novel spirodihydrobenzofuranlactams as antagonists of endothelin and as inhibitors of HIV-1 protease produced by Stachybotrys sp. I. Fermentation, isolation and biological activity. J Antibiot 49:13–19
Ryu SH, Hong SM, Khan Z, Lee SK, Vishwanath M, Turk A, Yeon SW, Jo YH, Lee DH, Lee JK (2021) Neurotrophic isoindolinones from the fruiting bodies of Hericium erinaceus. Bioorg Med Chem Lett 31:127714
Sadahiro Y, Kato H, Williams RM, Tsukamoto S (2020) Irpexine, an isoindolinone alkaloid produced by coculture of endophytic fungi, Irpex lacteus and Phaeosphaeria oryzae. J Nat Prod 83:1368–1373
Sakai K, Watanabe K, Masuda K, Tsuji M, Hasumi K, Endo A (1995) Isolation, characterization and biological activities of novel triprenyl phenols as pancreatic cholesterol esterase inhibitors produced by Stachybotrys sp. F-1839. J Antibiot 48:447–456
Sakurai J, Kikuchi T, Takahashi O, Watanabe K, Katoh T (2011) Enantioselective Total Synthesis of (+)‐Stachyflin: a potential anti‐influenza a virus agent isolated from a microorganism. Wiley Online Library
Sanchez JF, Entwistle R, Corcoran D, Oakley BR, Wang CC (2012) Identification and molecular genetic analysis of the cichorine gene cluster in Aspergillus nidulans. MedChemComm 3:997–1002
Sawada H, Nishimura N, Suzuki E, Zhuang J, Hasegawa K, Takamatsu H, Honda K, Hasumi K (2014) SMTP-7, a novel small-molecule thrombolytic for ischemic stroke: a study in rodents and primates. J Cereb Blood Flow Metab 34:235–241
Scherlach K, Schuemann J, Dahse H-M, Hertweck C (2010) Aspernidine A and B, prenylated isoindolinone alkaloids from the model fungus Aspergillus nidulans. J Antibiot 63:375–377
Shibata K, Hashimoto T, Hasumi K, Nobe K (2021) Potent efficacy of Stachybotrys microspora triprenyl phenol-7, a small molecule having anti-inflammatory and antioxidant activities, in a mouse model of acute kidney injury. Eur J Pharmacol 910:174496
Shinohara C, Hasumi K, Hatsumi W, Endo A (1996) Staplabin, a novel fungal triprenyl phenol which stimulates the binding of plasminogen to fibrin and U937 cells. J Antibiot 49:961–966
Shinozuka T, Yamamoto Y, Hasegawa T, Saito K, Naito S (2008) First total synthesis of sterenins A, C and D. Tetrahedron Lett 49:1619–1622
Slavov N, Cvengroš J, Neudörfl JM, Schmalz HG (2010) Total synthesis of the marine antibiotic pestalone and its surprisingly facile conversion into pestalalactone and pestalachloride A. Angew Chem Int Ed 49:7588–7591
Stadler M, Læssøe T, Fournier J, Decock C, Schmieschek B, Tichy H-V, Peršoh D (2014) A polyphasic taxonomy of Daldinia (Xylariaceae). Stud Mycol 77:1–143
Stierle A, Hershenhorn J, Strobel G (1993) Zinniol-related phytotoxins from Alternaria cichorii. Phytochemistry 32:1145–1149
Storm PA, Herbst DA, Maier T, Townsend CA (2017) Functional and structural analysis of programmed C-methylation in the biosynthesis of the fungal polyketide citrinin. Cell Chem Biol 24:316–325
Suemitsu R, Ohnishi K, Horiuchi M, Kitaguchi A, Odamura K (1992) Porritoxin, a phytotoxin of Alternaria porri. Phytochemistry 31:2325–2326
Suemitsu R, Ohnishi K, Morikawa Y, Nagatomo S (1995) Zinnimidine and 5-(3′, 3′-dimethylallyloxy)-7-methoxy-6-methylphthalide from Alternaria porri. Phytochemistry 38:495–497
Süssmuth RD, Mainz A (2017) Nonribosomal peptide synthesis—principles and prospects. Angew Chem Int Ed 56:3770–3821
Suzuki E, Nishimura N, Yoshikawa T, Kunikiyo Y, Hasegawa K, Hasumi K (2018) Efficacy of SMTP-7, a small-molecule anti-inflammatory thrombolytic, in embolic stroke in monkeys. Pharmacol Res Perspect 6:e00448
Taishi T, Takechi S, Mori S (1998) First total synthesis of (±)-stachyflin. Tetrahedron Lett 39:4347–4350
Tang WH, Martin KA, Hwa J (2012) Aldose reductase, oxidative stress, and diabetic mellitus. Front Pharmacol 3:87
Tang C, Ren Q, Luo H, Yin J, Yi K, Lei Y, Wang Y, Zhang Y (2021) Influenza virus replication inhibitor and use thereof. Published online 2021
Tao H, Abe I (2021) Enzymology and biosynthesis of the orsellinic acid derived medicinal meroterpenoids. Curr Opin Biotechnol 69:52–59
Thapa P, Corral E, Sardar S, Pierce BS, Foss FW Jr (2018) Isoindolinone synthesis: selective dioxane-mediated aerobic oxidation of isoindolines. J Org Chem 84:1025–1034
Thongbai B, Rapior S, Hyde KD, Wittstein K, Stadler M (2015) Hericium erinaceus, an amazing medicinal mushroom. Mycol Prog 14:1–23
Tok F, Yang X, Tabanca N, Koçyiğit-Kaymakçıoğlu B (2023) Synthesis of Phthalimide derivatives and their insecticidal activity against caribbean fruit fly, Anastrepha suspensa (Loew). Biomolecules 13:361
Tsuruta Y, Inoue H (1998) 4-(5, 6-dimethoxy-2-phthalimidinyl)-2-methoxyphenylsulfonyl chloride as a fluorescent labeling reagent for determination of amino acids in high-performance liquid chromatography and its application for determination of urinary free hydroxyproline. Anal Biochem 265:15–21
Vassaux A, Meunier L, Vandenbol M, Baurain D, Fickers P, Jacques P, Leclère V (2019) Nonribosomal peptides in fungal cell factories: from genome mining to optimized heterologous production. Biotechnol Adv 37:107449
Vázquez MAJ, Vega A, Rivera-Sagredo A, Jiménez-Alfaro MAD, Díez E, Hueso-Rodríguez JA (2004) Novel sesquiterpenoids as tyrosine kinase inhibitors produced by Stachybotrys chortarum. Tetrahedron 60:2379–2385
Vertesy L, Kogler H, Markus A, Schiell M, Vogel M, Wink J (2001) Memnopeptide A, a novel terpene peptide from Memnoniella with an activating effect on SERCA2. J Antibiot 54:771–782
Vertesy L, Kogler H, Markus A, Schiell M (2003) Memno peptides, process for their preparation and use thereof. Google Patents
Wang X-N, Tan R-X, Liu J-K (2005) Xylactam, a new nitrogen-containing compound from the fruiting bodies of ascomycete Xylaria euglossa. J Antibiot 58:268–270
Wang BT, Qi QY, Ma K, Pei YF, Han JJ, Xu W, Li EW, Liu HW (2014) Depside α-glucosidase inhibitors from a culture of the mushroom Stereum hirsutum. Planta Med 80:918–924
Wang A, Xu Y, Gao Y, Huang Q, Luo X, An H, Dong J (2015a) Chemical and bioactive diversities of the genera Stachybotrys and Memnoniella secondary metabolites. Phytochem Rev 14:623–655
Wang G, Wu W, Zhu Q, Fu S, Wang X, Hong S, Guo R, Bao B (2015b) Identification and fibrinolytic evaluation of an isoindolone derivative isolated from a rare marine fungus Stachybotrys longispora FG216. Chin J Chem 33:1089–1095
Wang K, Bao L, Ma K, Liu N, Huang Y, Ren J, Wang W, Liu H (2015c) Eight new alkaloids with PTP1B and α-glucosidase inhibitory activities from the medicinal mushroom Hericium erinaceus. Tetrahedron 71:9557–9563
Wang K, Bao L, Qi Q, Zhao F, Ma K, Pei Y, Liu H (2015d) Erinacerins C-L, isoindolin-1-ones with α-glucosidase inhibitory activity from cultures of the medicinal mushroom Hericium erinaceus. J Nat Prod 78:146–154
Wang Y, Hyde KD, McKenzie EH, Jiang Y-L, Li D-W, Zhao D-G (2015e) Overview of Stachybotrys (Memnoniella) and current species status. Fungal Divers 71:17–83
Wang X-L, Xu K-P, Long H-P, Zou H, Cao X-Z, Zhang K, Hu J-Z, He S-J, Zhu G-Z, He X-A (2016) New isoindolinones from the fruiting bodies of Hericium erinaceum. Fitoterapia 111:58–65
Watanabe K, Sakurai J, Abe H, Katoh T (2010) Total synthesis of (+)-stachyflin: a potential anti-influenza A virus agent. Chem Commun 46:4055–4057
Weissmiller AM, Wu C (2012) Current advances in using neurotrophic factors to treat neurodegenerative disorders. Trans Neurodegener 1:1–9
Wildermuth R, Speck K, Haut F-L, Mayer P, Karge B, Brönstrup M, Magauer T (2017) A modular synthesis of tetracyclic meroterpenoid antibiotics. Nat Commun 8:2083
Williams RB, Henrikson JC, Hoover AR, Lee AE, Cichewicz RH (2008) Epigenetic remodeling of the fungal secondary metabolome. Org Biomol Chem 6:1895–1897
Wittstein K, Rascher M, Rupcic Z, Löwen E, Winter B, Köster RW, Stadler M (2016) Corallocins A-C, nerve growth and brain-derived neurotrophic factor inducing metabolites from the mushroom Hericium coralloides. J Nat Prod 79:2264–2269
Wu B, Oesker V, Wiese J, Malien S, Schmaljohann R, Imhoff JF (2014) Spirocyclic drimanes from the marine fungus Stachybotrys sp. strain MF347. Mar Drugs 12:1924–1938
Xu X, De Guzman FS, Gloer JB, Shearer CA (1992) Stachybotrins A and B: novel bioactive metabolites from a brackish water isolate of the fungus Stachybotrys sp. J Org Chem 57:6700–6703
Xu J, Ebada SS, Proksch P (2010) Pestalotiopsis a highly creative genus: chemistry and bioactivity of secondary metabolites. Fungal Divers 44:15–31
Xu J, Yang X, Lin Q (2014) Chemistry and biology of Pestalotiopsis-derived natural products. Fungal Divers 66:37–68
Xu D, Xue M, Shen Z, Jia X, Hou X, Lai D, Zhou L (2021) Phytotoxic secondary metabolites from fungi. Toxins 13:261
Yaegashi J, Praseuth MB, Tyan S-W, Sanchez JF, Entwistle R, Chiang Y-M, Oakley BR, Wang CC (2013) Molecular genetic characterization of the biosynthesis cluster of a prenylated isoindolinone alkaloid aspernidine A in Aspergillus nidulans. Org Lett 15:2862–2865
Yaegashi J, Oakley BR, Wang CC (2014) Recent advances in genome mining of secondary metabolite biosynthetic gene clusters and the development of heterologous expression systems in Aspergillus nidulans. J Ind Microbiol Biotechnol 41:433–442
Yagi S, Ono J, Yoshimoto J, Sugita K-I, Hattori N, Fujioka T, Fujiwara T, Sugimoto H, Hirano K, Hashimoto N (1999) Development of anti-influenza virus drugs I: improvement of oral absorption and in vivo anti-influenza activity of stachyflin and its derivatives. Pharm Res 16:1041–1046
Yan Y, Yang J, Yu Z, Yu M, Ma Y-T, Wang L, Su C, Luo J, Horsman GP, Huang S-X (2016) Non-enzymatic pyridine ring formation in the biosynthesis of the rubrolone tropolone alkaloids. Nat Commun 7:13083
Yang X-L, Zhang S, Hu Q-B, Luo D-Q, Zhang Y (2011) Phthalide derivatives with antifungal activities against the plant pathogens isolated from the liquid culture of Pestalotiopsis photiniae. J Antibiot 64:723–727
Yang X-L, Zhang J-Z, Luo D-Q (2012) The taxonomy, biology and chemistry of the fungal Pestalotiopsis genus. Nat Prod Rep 29:622–641
Yaoita Y, Danbara K, Kikuchi M (2005) Two New Aromatic Compounds from Hericium erinaceum (B ULL.: F R.) P ERS. Chem Pharm Bull 53:1202–1203
Yaoita Y, Yonezawa S, Kikuchi M, Machida K (2012) A new geranylated aromatic compound from the mushroom Hericium erinaceum. Nat Product Commun, 7, 1934578X1200700427
Yashavantha Rao H, Rakshith D, Harini BP, Gurudatt DM, Satish S (2017) Chemogenomics driven discovery of endogenous polyketide anti-infective compounds from endosymbiotic Emericella variecolor CLB38 and their RNA secondary structure analysis. PLoS ONE 12:e0172848
Yin Y, Fu Q, Wu W, Cai M, Zhou X, Zhang Y (2017) Producing novel fibrinolytic isoindolinone derivatives in marine fungus Stachybotrys longispora FG216 by the rational supply of amino compounds according to its biosynthesis pathway. Mar Drugs 15:214
Yoo K-D, Park E-S, Lim Y, Kang S-I, Yoo S-H, Won H-H, Kim Y-H, Yoo I-D, Yoo H-S, Hong JT (2012a) Clitocybin A, a novel isoindolinone, from the mushroom Clitocybe aurantiaca, inhibits cell proliferation through G1 phase arrest by regulating the PI3K/Akt cascade in vascular smooth muscle cells. J Pharmacol Sci 118:171–177
Yoo K-D, Park E-S, Lim Y, Kang S-I, Yoo S-H, Won H-H, Lee H-P, Kim Y-H, Yoo I-D, Yoo H-S (2012b) Clitocybin B inhibits rat aortic smooth muscle cell proliferation through suppressing PDGF-Rβ phosphorylation. Vascul Pharmacol 56:91–97
Yoo I, Ryoo I, Kim Y, Choo S, Lee S (2010) Novel clitocybin derivates, preparation method thereof and composition containing the same for prevention of aging as an active ingredient. Korea Patent
Yoshimoto J, Yagi S, Ono J, Sugita K, Hattori N, Fujioka T, Fujiwara T, Sugimoto H, Hashimoto N (2000) Development of anti-influenza drugs: II. Improvement of oral and intranasal absorption and the anti-influenza activity of stachyflin derivatives. J Pharm Pharmacol 52:1247–1255
Zarga MHA, Sabri SS, Firdous S, Shamma M (1987) Fumadensine a phthalideisoquinoline from Fumaria densiflora. Phytochemistry 26:1233–1234
Zhang G, Sun S, Zhu T, Lin Z, Gu J, Li D, Gu Q (2011) Antiviral isoindolone derivatives from an endophytic fungus Emericella sp. associated with Aegiceras corniculatum. Phytochemistry 72:1436–1442
Zhang C, Li C, Ye W, Yang M (2017) The complete mitochondrial genome of Hericium coralloides (Hericiaceae, Basidiomycota). Mitochondrial DNA Part B 2:385–386
Zhang Z, He X, Che Q, Zhang G, Zhu T, Gu Q, Li D (2018a) Sorbicillasins A-B and scirpyrone K from a deep-sea-derived fungus, Phialocephala sp. FL30r. Mar Drugs 16:245
Zhang Z, He X, Wu G, Liu C, Lu C, Gu Q, Che Q, Zhu T, Zhang G, Li D (2018b) Aniline-tetramic acids from the deep-sea-derived fungus Cladosporium sphaerospermum L3P3 cultured with the HDAC inhibitor SAHA. J Nat Prod 81:1651–1657
Zhang H, Yang M-H, Zhuo F-F, Gao N, Cheng X-B, Wang X-B, Pei Y-H, Kong L-Y (2019) Seven new cytotoxic phenylspirodrimane derivatives from the endophytic fungus Stachybotrys chartarum. RSC Adv 9:3520–3531
Zhao J, Liu J, Shen Y, Tan Z, Zhang M, Chen R, Zhao J, Zhang D, Yu L, Dai J (2017a) Stachybotrysams A-E, prenylated isoindolinone derivatives with anti-HIV activity from the fungus Stachybotrys chartarum. Phytochem Lett 20:289–294
Zhao Z-Z, Chen H-P, Huang Y, Zhang S-B, Li Z-H, Feng T, Liu J-K (2017b) Bioactive polyketides and 8, 14-seco-ergosterol from fruiting bodies of the ascomycete Daldinia childiae. Phytochemistry 142:68–75
Zhou D, Li L-J, Qi H, Pan J-J, Zhang H, Wang J-D, Xiang W-S (2015) A new stachybotrin congener from a soil fungus Stachybotrys parvispora strain HS-FG-843. J Antibiot 68:339–341
Zhou H, Sun X, Li N, Che Q, Zhu T, Gu Q, Li D (2016) Isoindolone-containing meroperpenoids from the endophytic fungus Emericella nidulans HDN12-249. Org Lett 18:4670–4673
Acknowledgements
We thank Dr. Matthew Jenner, Department of Chemistry, University of Warwick, for diligent proof-reading of the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
It is disclosed that there are no financial or non-financial interests that are directly or indirectly related to the work submitted for publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
El Maddah, F., Nazir, M., Ahmad, R. et al. Fungal phthalimidines-chemodiversity, bioactivity and biosynthesis of a unique class of natural products. Phytochem Rev (2024). https://doi.org/10.1007/s11101-024-09923-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11101-024-09923-1