
Abstract
Strigolactones (SLs) are natural products with promising applications as agrochemicals to prevent infestation with parasitic weeds due to their ability to trigger seed germination. However, their use is still limited because of the low yields in which they are isolated from natural sources. As such, numerous studies have led to strategies for obtaining them, and various structural analogues, by organic synthesis. These analogues have focused attention on the study of SLs, as some of them are easier to synthesize and possess enhanced properties, such as the level of bioactivity. This review provides an overview of the synthesis of SLs, subsequently focusing on the production of analogues with the canonical structure. The germinating activity of the compounds is also described herein, with positive effects on different species of the problematic genera Striga, Orobanche and Phelipanche having been found. The highly active analogue GR24 is currently the most widely studied in the literature, and relevant structural-activity relationships have been proposed as a result of the study of derivatives functionalized in different positions. Analogues based on other natural SLs such as strigol and orobanchol have also been developed, as have some novel canonical SLs derived from eudesmanolide or guaianolide sesquiterpene lactones. This review aims to provide useful information for the development of bioactive compounds applicable in new generation herbicides, in an attempt to employ similar compounds to those produced naturally in ecosystems that provoke effective herbicide effects at low concentrations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Strigolactones (SLs) are a family of natural products synthesized by some plant species from carotenoids (López-Ráez et al. 2008). They have been studied since the second half of the twentieth century as activities of agronomical and pharmacological (Carrillo et al. 2019) interest have been discovered. Based on the structure of the molecule, SLs can be classified into two types.
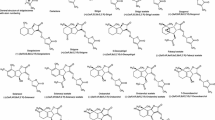
The first type is canonical SLs. These molecules have a fused tricyclic scaffold (ABC-rings) with a fourth ring (D-ring) bonded to the C-ring, via an enol ether bond (highlighted in red in Fig. 1). The C-ring must be a lactone and the D-ring a methyl furanone, whereas the AB-rings vary. Thus, this latter system can contain different functional groups and double bonds, and, in the case of the A-ring, different numbers of carbon atoms. To the best of our knowledge, a total of 23 canonical SLs have been identified and characterized to date, being 7β-hydroxy-5-deoxystrigol the most recent one (Yoneyama et al. 2018). As examples of the most representative, Fig. 1 shows the structures of strigol and orobanchol, together with medicaol and the synthetic GR24. One isomeric feature, namely the configuration of both 3a and 8b, which results in an upwards or backwards orientation of the C-ring, must be considered for canonical SLs. Indeed, some authors commonly classify SLs as being of the strigol or orobanchol type.

Examples of canonical (top) and non-canonical (bottom) SLs
The second type comprises non-canonical SLs. The structure of these compounds is simpler than for their canonical counterparts, with carlactone being the most representative (Fig. 1). In comparison to canonical SLs, this second type lacks one or two rings of the ABC-system, or the fusion between them (see avenaol). The presence of a methyl furanone as the D-ring is mandatory.
Despite the structural differences between the two types, bioactivities of particular interest have been observed for both. This has been attributed to the maintenance of the D-ring in a conjugated system. One of the most relevant activities is stimulation of the germination of seeds of parasitic weeds, with SLs being considered, in terms of activity, as suitable phytochemicals for the control of several damaging species as part of the preventive strategy of suicidal germination. In addition, other agronomic applications are being studied for SLs, highlighting their use as biofertilizers to increase the colonization of crop roots with symbiotic arbuscular mycorrhizal fungi (AMF) (López-Ráez et al. 2017). The activity shown by the different structures of natural SLs on this kind of fungi, as well as on parasitic weeds, were recently reviewed by our research group (Soto-Cruz et al. 2021). Moreover, studies such as the conducted by Yoshimura et al. (2020) have proved certain benefits that the application of SLs could generate to crop plants. It particularly proved the effectiveness of some non-canonical SLs to accelerate the germination of corn seeds.
Nevertheless, the study and use of natural SLs in herbicides is limited to a large extent by the low yields in which they are isolated from plant materials. Thus, some research groups have focused on their synthesis, providing routes involving many steps due to the complexity of their structures. One of the main handicaps is the selectivity of the reactions concerned as different chiral centers must be generated and activity is highly dependent on stereochemistry.
In order to simplify the production of canonical SLs, the design of more efficient routes that lead to simpler bioactive compounds have been published. This review will present an updated overview of the state of the art as regards the synthesis of canonical and non-canonical SLs, subsequently focusing on the synthesis of canonical SL analogues, which represent the most efficient strategy for obtaining canonical SLs on a multigram scale. The results from parasitic weed bioassays will also be presented from a structure–activity relationship perspective. All these points have been considered relevant to provide practical information for the commercial use of SLs that are the same as, or similar to, those produced naturally.
Synthesis of natural canonical SLs
Several synthetic routes for natural strigolactones have been reported in the last few decades, with the access to single enantiomers and the implementation of scalable processes that allow gram-scale synthesis being a particular challenge for organic chemists. This is due to the high stereospecificity of the biological activity shown by SLs and the multiple chiral centers present in their structures. Almost all such strategies involve three key steps: synthesis of the ABC scaffold, functionalization of the A/B-ring, and attachment of the D-ring (Zwanenburg et al. 2016). The first syntheses of strigolactones required the separation of racemic mixtures by formation of the corresponding diastereomers and their purification by exhaustive chromatography. This process was improved with the development of chiral columns. However, in the last few years, the publication of enantioselective syntheses has increased in an effort to improve the maximum resolution yield of 50% obtained in this process. This progress can be exemplified with the case of strigol. The first synthetic approach to ( +)-strigol was reported in 1976 and involved the chiral auxiliary resolution shown in Scheme 1 (Heather et al. 1976).

Heather's chiral auxiliary resolution strategy to isolate ( +)-strigol
Subsequently, Samson et al. (1991) and Reizelman et al. (2000) employed chiral HPLC columns to isolate the single enantiomer of strigol. Hirayama and Mori (1999)reported the application of a chemoenzymatic method in the only gram-scale synthesis of canonical strigolactone reported to date. Finally, Takahashi et al. (2016) published the only enantioselective synthesis of the tricyclic core of ( +)-strigol reported to date (Scheme 2). Compound 1 was generated using a catalytic Corey–Bakshi–Shibata (CBS) reduction with excellent levels of enantiomeric excess. After a few steps, compound 2 was isolated in a reasonable yield, but with only moderate diastereoselectivity. The mixture of diastereomers was saponified, brominated and cyclized. Then, the bromine atom was eliminated to obtain 3 after a simple column chromatographic separation. As only the desired diastereomer was able to participate in the base-catalyzed elimination, compound 3 was obtained as a single stereoisomer. Removal of the protecting group gave the tricyclic core of ( +)-strigol.

Takahashi’s enantioselective synthesis of the tricyclic core of ( +)-strigol (4)
The last step in strigolactone synthesis is commonly attachment of the D-ring. In the majority of cases this follows the same sequence, namely formylation of the ABC scaffold with (m)ethyl formate in the presence of an appropriate base and treatment of the enolate thus obtained with either chloro or bromo butenolide. A racemic D-ring is used and a mixture of two epimers is obtained in equal amounts. However, all strigolactones possess the R-configuration on this ring. To obtain the desired stereochemistry in the D-ring, Sugimoto et al. (1998) and Thuring et al. (1997a) employed a homochiral D-ring approach, although the subsequent retro-Diels–Alder reaction requires forcing conditions which may not be compatible with all target molecules. In addition, use of Trost's asymmetric allylic alkylation (Pd catalyzed) methodology to install the D-ring in a stereoselective manner has been described for the synthesis of strigolactones analogues (Bromhead et al. 2014) but, surprisingly, not for natural strigolactones.
The syntheses of natural strigolactones published prior to 2017 were reviewed by Bromhead and McErlean (2017) in that year. Subsequently, and to the best of our knowledge, only one enantioselective synthesis of (–)-solanacol has been published (Bromhead et al. 2018). In that article, authors synthesized (–)-solanacol using a dynamic kinetic resolution (DKR) in the stereo-defining step (Scheme 3). This synthesis starts with an intermolecular Stetter reaction between 2,3-dimethylbenzaldehyde and methyl acrylate to obtain the keto ester 5. Aldol condensation of 5 with formalin and in situ saponification then leads to the formation of enone 6 in good overall yield. The next step is a Nazarov cyclization with sulfuric acid in a one-pot process to obtain the indanone 7. The Nazarov reaction is used in different synthetic routes to form the cyclopentenone B or C ring of the target strigolactones (as will be seen throughout this review), so it is worth citing at this point the recent study by Nejrotti et al. (2020), in which this reaction was optimized through the use of natural deep eutectic solvents to provide alternative methods closer to the green chemistry principles. The tricyclic core of (–)-solanacol (8) is obtained by performing a Noyori asymmetric transfer hydrogenation with a DKR (Bromhead et al. 2018), thus providing excellent enantioselectivity (> 99% ee).

Bromhead’s enantioselective synthesis of the tricyclic core of (−)-solanacol (8)
Synthesis of non-canonical SL and analogues
The synthesis of a non-canonical strigolactone as a single enantiomer has not been reported to date. Carlactone and its analogues carlactonoic acid, methyl carlactonoate and 19-hydroxycarlactone (Fig. 1) were the first non-canonical SLs synthesized as racemates. As these SLs lack the B- and C-rings, steps to form the bicyclic and tricyclic scaffold are not necessary.
Carlactone is the most representative and, after it was first isolated in 2012 (Alder et al. 2012), Seto et al. (2014) published its synthesis using 2,6-dimethylcyclohexanone as starting material. This route can be divided into three parts (Scheme 4): functionalization of the ring (also including a conjugated side chain in substitution of the carbonyl group) to obtain β-ionone, the formation of an aldehyde at the end of the chain (product 9), and introduction of the furanone ring. The two geometric isomers (Z/E) at C-9 of carlactone were obtained, but with a low yield for the last step (Scheme 5).

Synthesis of carlactone

Synthesis of heliolactone (non-selective) and isoheliolactone
Mori et al. (2016) used the previous strategy to synthesize carlactonoic acid, methyl carlactonoate and 19-hydroxycarlactone (all functionalized at C-19). However, in the case of the former two, a later enantiomerically pure synthesis was published (four steps in 36% overall yield and three steps in 50% overall yield, respectively; Dieckmann et al. 2018). These syntheses are based on a Stille cross-coupling of a stannane of the A-ring precursor compound with 10 (this strategy will be detailed below for the synthesis of heliolactone; Scheme 6).

Strategies for the synthesis of heliolactone
Two more hydroxylated analogues of carlactone were synthesized by Mori et al. (2016) by adapting the strategy shown in Scheme 4: at C-4, using α-ionone as starting material; and at C-18 using methyl 3,3-dimethyl-2-oxocyclohexane-1-carboxylate as starting material. The yields for the furanone addition step were notoriously low, as mentioned previously for carlactone. These results highlight that the optimization or design of an alternative reaction would be a great opportunity to improve the synthesis of these products. Furthermore, the same study presented a set of new analogues synthesized in two steps (employing formyl Meldrum’s acid as starting material), all of them with a distinctive ester at C-7. These analogies are therefore more closely related to canonical SL as the D-ring is connected via an enol ether bond. Different types of six-membered ring were used as the A-ring.
In the case of heliolactone, the recent synthetic procedure published by Yamamoto et al. (2020), which employs dimedone as starting material (Scheme 5), should be noted. The first intermediate (11) was obtained by the procedure of Yoshino et al. (2006), which is then allylated by reaction with LDA and allyl bromide. To introduce the butenolide ring, the new double bond must be oxidized to an aldehyde (12), which is then transformed into a diester by reaction with dimethyl malonate. These reactions require prior protection of the oxygenated function located at the ring. After butenolide addition, the final steps are deprotection and isomerization using a Lewis acid, with the best yield being obtained with TMSOTf. The product is a racemic and diastereomeric mixture of heliolactone (13), obtained in 2.6% overall yield. This yield could be significantly improved if the butenolide addition step was optimized, as its yield is much lower (22%) than those for the other steps (60–91%). If LiBF4 is employed in the last step, the product obtained is the isomer isoheliolactone (1% overall yield) as migration of the double bond in the six-membered ring does not occur.
Alternative routes were recently reported by Yoshimura et al. (2019) (Scheme 6). The first steps are aimed at synthesizing a stannane (14) from 4-ketoisophorone, and the key step (route A in Scheme 6) is the subsequent Stille cross-coupling reaction with 10 and a palladium catalyst, which leads to selective formation of the C-11R epimers of isoheliolactone (15) and heliolactone (16) if 10 is used enantiomerically pure. This strategy was improved to selectively synthesize heliolactone (route B in Scheme 6) after the prior transformation of 14 into 17, and subsequent HPLC purification using a chiral column. In the next steps, formation of the stannane of 17, and subsequent Stille cross-coupling with 10 (both reactions including a Pd catalyst), leads to heliolactone. In addition, reaction of the pure (S) enantiomer of 14 with 10 also provided pure isoheliolactone. This study also afforded the C-12 methylated analogues of heliolactone and isoheliolactone. The stability of this methylated heliolactone was considered by the authors to be improved (in comparison with heliolactone) in acidic and mildly basic aqueous solutions, while the biological activities were retained. An alternative synthesis of 17 (and pure heliolactone) was published by Woo and McErlean (2019), which requires fewer steps and used ( ±)18 as starting material. The first four steps allow addition of the iodine atom (oxidation of the carbonyl group to carboxylic acid, bonding to a phthalimide moiety, borylation and iodination), while the fifth step involves allylic oxidation to introduce the carbonyl group at the ring, thus resulting in 17.
In a subsequent study, Yoshimura et al. (2020) treated the corresponding stannane (20, synthesized from 19 in good yield) with 10 to also obtain the non-canonical zealactone with selectivity at C-2′, although as a racemate at C-6 (Scheme 7). It should be noted that zealactone, also named methyl zealactonoate (Xie et al. 2017), is a natural product isolated from corn roots as a mixture of epimers at C-6, proving not to epimerize under the isolation conditions (Yoshimura et al. 2020). The overall yield of the route shown in Scheme 7 is 21%. Analogues methylated at C-2′ were also synthesized in this study.

Synthesis of zealactone as a mixture of epimers at C-6
With regard to the non-canonical SL avenaol (Fig. 1), a total synthesis of this compound has also been published (Scheme 8; Yasui et al. 2017). This synthesis is characterized by the complexity of forming the AB-ring system (11 steps from 2,2-dimethylpent-4-enal, with good and high yields for each step, always over 68%). This strategy paid particular attention to formation of the appropriate cyclopropane B-ring, which was achieved using the rhodium catalyst Rh2(OAc)4.

Total synthesis of avenaol (C-2′R)
In addition to the natural non-canonical SLs, it is worth highlighting at this point other synthetic derivatives that simplify the canonical structures. That is, they lack their A- or B-rings, maintaining the moiety formed by the CD-rings and the enol ether bond. This moiety corresponds with the bioactiphore, the part that is primarily responsible for the bioactivity of the molecule (Zwanenburg et al. 2016). Some examples of bioactive compounds belonging to this type of derivatives (proven to be active as parasitic germination or branching arbuscular mycorrhizal fungi stimulators) are shown in Fig. 2 (Hýlová et al. 2019; Kondo et al. 2007; Lumbroso et al. 2016; Zwanenburg et al. 2016, 2009).

Examples of synthetic bioactive compounds based on simplified structures of SLs

GR24 analogues methylated at the A-ring
With special emphasis, compounds MP1, MP3, MP13 and MP26 have shown promising results in the germination of the problematic parasitic weed Striga hermonthica, including in greenhouse and field trials through suicidal germination strategy (Jamil et al. 2019; Kountche et al. 2019). MP3 can be synthetized from methyl phenylacetate, and MP1 by nitrification of MP3 (Jamil et al. 2018), whereas MP13 could be obtained from 3-dimethylamino-2-methyl-2-propenal, and MP26 by reaction of MP13 with methyl (triphenylphosphoranylidene) acetate (Scheme 9; Jamil et al. 2019).

Synthesis of bioactive compounds of the MP family
Analogues of canonical strigolactones
The synthesis of SL analogues and mimics represents the most efficient method for obtaining compounds related to canonical SLs on a multigram scale. Among these synthetic compounds, those with optimal bioactivity, stability and solubility could be the best candidates for use as agrochemicals to prevent parasitic weeds. The following sections will present a brief summary of the synthesis of canonical SL analogues, focusing on the main characteristics, complexity and yield of the routes. In addition, the activity of compounds in germination bioassays (if carried out) will be discussed from a structure–activity perspective.
From a structural point of view, SL analogues are molecules which, in comparison to natural canonical SLs, present a lactone group as the C-ring and a methyl furanone as the D-ring, both linked via an enol ether bond. Analogues differ in terms of the presence of new structural features like alternative A- or B-rings, or functional groups.
Analogues are typically obtained by total or partial synthesis, using accessible molecules as starting material (usually commercially obtained, but also isolated from natural sources). In comparison to total synthesis strategies, partial synthesis allows the process to start with molecules related to the structure of the target compounds, thereby significantly reducing the number of steps. This point is highly important, as the majority of synthetic routes are focused on construction of the tricyclic scaffold (ABC-rings) of SLs. As intermediate compounds in the synthesis usually present different chiral or reaction centers, analogue synthesis is usually non-selective. This can be interpreted in two ways: positive, as analogues with alternative isomeric features can be obtained; or negative, as lower yields are obtained for the synthesis of a single analogue. C-2′ is a common chiral center in all SLs and analogues, and it must be noted that the study of its configuration requires experiments other than NMR spectroscopy, namely circular dichroism and computational calculations.
GR24 family
The most widely studied SL analogue, known as GR24, was first synthesized and evaluated in a bioassay by Johnson et al. (1981). Since then, this phenolic analogue has been the subject of numerous studies due to its bioactivities in the fields of medicine (for example, GR24 was recently proposed as promising drug to control Alzheimer’s disease; Kurt et al. 2020) or agriculture (as a herbicide). Eliciting the germination of parasitic weed seeds is one of the most widely studied applications, thus making it a promising herbicide for the preventive strategy of suicidal germination. In fact, GR24 is commonly used, as a C-2′ racemate, in germination bioassays as standard positive control, due to its ability to germinate different species of the problematic genera Striga, Orobanche and Phelipanche.
Malik et al. (2010) reported that all synthetic routes published until 2010 employed the commercial byciclic compound indan-1-one as starting material. The synthetic route to this compound was optimized to four steps, with an overall yield of 44% (route A in Scheme 10), in which the first three steps concern enantioselective formation of the tricyclic scaffold (21) (Wigchert et al. 1999; Malik et al. 2010). The first step introduces a carboxylic acid group into an exocyclic position of the indanone, with the double bond being reduced in the second step to obtain 22. The third step involves the use of NaBH4 to reduce the carbonyl group to an alcohol, followed by treatment of the crude product with acid to form a new lactone group, thus giving the tricyclic molecule 21. Efforts to optimize the synthesis of GR24 in the literature have focused on the efficient construction of this intermediate. Indeed, synthesis of this lactone accounts for most of the steps in the synthetic routes developed to obtain any analogue. One recent example is the attempt by Xiong et al. 2019 to simplify the last step to obtain 21. This compound was synthesized in 90% yield by treating 22 with a Ru(II) catalyst (Scheme 10) in the presence of the azeotropic mixture HCOOH/Et3N as hydrogen source and solvent. The main benefit arising from use of this catalyst is its possible application with different substrates, as subsequently confirmed by authors by the high yield synthesis of the tricyclic scaffold using different derivatives of intermediate 22 (characterized by a halogen atom in the A-ring, or containing a cyclohexane or cycloheptane as the B-ring, amongst others). Once 21 has been obtained, the common final step is an initial formylation under basic conditions to form an enolate, which subsequently acts as a nucleophile in a nucleophilic substitution reaction with compound 23 (a brominated or chlorinated butenolide ring) to form the strigolactone-type product GR24. Pospíšil (2021) recently published some procedures for the attachment of 23 to phenols or carboxylic acids, which could be useful for the development of new SL analogues or mimics.

Synthesis of GR24 (C-2′R) from indan-1-one (route A) and indene (route B)
Reagent 23 is unstable and must be synthesized immediately prior to use. It can be easily obtained in a radical-substitution reaction catalyzed by AIBN, although 23 can also be synthesized via more complex routes requiring up to six steps (Mangnus et al. 1992).
An alternative route to synthesize 21 was published in 2014 (route B in Scheme 10; Bromhead et al. 2014). This synthesis is based on opening of the cyclopentane ring in the indene (starting material) to form an aldehyde, which forms the carbonyl group after subsequent cyclization. The selective formation of 21 (> 99% after recrystallization) was accomplished using Noyori’s (S,S)-RuTsDPEN catalyst in the next step. Similarly, if the (R,R)- RuTsDPEN catalyst is used, (–)-21 is generated, thus allowing isolation of the corresponding (–)-GR-24 isomers after addition of the D-ring (23).
Another relevant route to synthesize 21 (Malik et al. 2010), which differs in the use of benzaldehyde as starting material, is shown in Scheme 11. This preparation starts with a Stobbe condensation of benzaldehyde with dimethyl succinate to obtain an ester intermediate, which forms the bicyclic product 24 after hydrogenation and a Friedel–Crafts transposition with oxalyl choride. Finally, the tricyclic scaffold is obtained by saponification of 24 with LiOH and subsequent reduction and acid-catalyzed lactonization reactions.

Synthesis of the GR24 scaffold (21) from benzaldehyde
The overall yield is 30%, which is lower than that achieved following the routes shown in Scheme 10 (55% for route A, and 48% for route B) due to the 45% yield of the Stobbe condensation step. Despite this decrease in overall yield, one advantage of the route depicted in Scheme 11 is the possibility to obtain new GR24 analogues differing in the A-ring (Fig. 2) when methylated benzaldehyde derivatives are used as starting material (Malik et al. 2010). Epimeric compounds at C-2′ were also obtained, and the evaluation of all these compounds in bioassays allowed the influence of the C-2′ configuration on the activity to be evaluated. Thus, all analogues germinated S. hermonthica, with maximum germination percentages of around 60% at the highest concentration tested (3.3·10–5 M). It could therefore be concluded that the configuration C-2′R, similar to that of natural SL, is better than C-2′S as regards obtaining higher levels of activity. This preference has been observed in different studies. With regard to substituents at the A-ring, the presence of H atoms or methyl groups did not have a marked influence on the activity.
The synthesis of GR24 derivatives methylated at the A-ring is also feasible using the strategy of Chen et al. (2013) (Scheme 12), which also allows different substituents (OH, F or OAcyl) to be introduced at C-4. Moreover, enantiomerically pure analogues (including GR24) can be obtained as a kinetic resolution is applied. The starting material is commercial 2-bromostyrene, or 3,4-dimethylphenol for methylated analogues. Both compounds form the corresponding bicyclic intermediate 25 via a two- or nine-step route that concludes with a ring-closing metathesis (RCM) using the Grubbs ruthenium catalyst I. After enzymatic kinetic resolution (using Candida antarctica lipase), the tricylic scaffold is obtained in three steps: RCM, trichloroacetylation and atom-transfer radical cyclization with copper(I) coordinated to dHbipy. Prior to butenolide ring addition, the hydroxyl group is incorporated at C-4 by nucleophilic substitution (an additional Mitsunobu reaction is required to invert the configuration of C-4S products), and dechlorination of the C-ring with Zn is then carried out.

Synthesis of enantiomerically pure GR24 analogues (HFIP = hexafluoroisopropanol)
Xiao et al. (2017) published a strategy to obtain the GR24 scaffold (21) by nickel-promoted reductive cyclization of the acetal of o-iodophenyl allyl alcohol. This reaction was also used to obtain GR24 analogues chlorinated at the A-ring, as shown in Scheme 13. The vinyl alcohol reacts initially with ethyl vinyl ether and NIS (or NBS) to form the acetal, which is transformed into the tricyclic intermediate by nickel-promoted reductive cyclization and subsequent oxidation with m-CPBA. This strategy appears to be suitable for obtaining new analogues with new functionalities at the A-ring as synthetic derivatives of o-iodobenzaldehyde can be employed as starting material.

Synthesis of chlorinated GR24
Lachia et al. (2014b) published several strategies for synthesizing up to thirteen GR24 analogues with different substituents at the B-ring. These were then tested on O. cumana seeds, and those with an C-2′R configuration were found to be more active than their respective C-2′S epimers, especially at lower doses (a C-2′R analogue can show 79% of activity, with no effects for the C-2′S epimer). This structure–activity relationship coincides with that previously mentioned for S. hermonthica. Some of the analogues reported contain a methyl group at C-4 (and therefore more similar to natural SL) or C-8. The starting material for their synthesis is 2-iodophenyl acid, and intramolecular [2 + 2] cycloaddition is used to form the first tricyclic system (26), which is transformed into the scaffold related to 21 via a subsequent Baeyer–Villiger oxidation catalyzed by H2O2. As an example, Scheme 14 shows the synthesis of analogue 27 (overall yield of 4.2%).

Synthesis of a GR24 analogue methylated at C-4 (27)
In light of the bioactivity with respect to O. cumana, the most promising analogues reported by Lachia et al. (2014b) included oxygenated groups at C-4 (hydroxy, methoxy, acetate or -OTBS), with the presence thereof increasing the germinating activity of the molecules by up to 20% in comparison to analogues methylated at C-4 or with no such substituent (GR24). The most active ones, with activities of 76–79% at the lowest dose tested (0.001 mg·l−1), are shown in Scheme 15, together with the route for their synthesis from the tricyclic system. Given that this tricyclic intermediate can be synthesized in 55% yield following route A in Scheme 10, the global yield for synthesis of 28 is 4.4% (1.8% in the case of 29). In a subsequent study, 28 showed activity in the germination of S. hermonthica (but only limited activity for Striga gesnerioides), and was found to be more active than the GR24 analogue epimer at C-4 (Ueno et al. 2015). Similarly, the study by Malik et al. (2011) presents the synthesis of similar analogues hydroxylated or acetylated at C-4, as well as others containing a ketone group. These analogues showed that hydroxylated analogues present higher activity when the hydroxyl group has a trans orientation with the C-ring, as is the case for product 27. The high activity levels achieved by the analogues evaluated in this study for O. ramosa seeds, especially the acetyl and ketone derivatives with percentages of 60–100% in the range 0.033–3.3·10–5 µM, should be noted.

Synthesis of highly active oxygenated GR24 analogues
Since more active analogues than GR24 could be synthesized, Morris and McErlean (2016) focused on optimization of the synthesis of 4-hydroxy-GR24 (28) in an effort to reduce the number of steps and avoid the formation of undesired stereoisomers. This study led to the route depicted in Scheme 16. The first step is perhaps the most innovative improvement as the tricyclic scaffold is obtained from a single reaction between phthalaldehyde, 30 and 2,5-dimethyl-N-acryloylpyrrole. As the 38% overall yield is the main disadvantage of this reaction, the search for more efficient conditions would represent a great achievement. The next step is formation of the hydroxyl group at the B-ring by hydrogenation in the presence of Noyori’s (S,S)-RuTsDPEN catalyst. This step is also significant for resolving the racemate, as only the ( −)-ketone stereoisomer is transformed. This is the key to obtaining up to four analogues in the final step. The unreacted ( +)-ketone (31) is reduced to the alcohol via a Luche hydrogenation (using CeCl3 and NaBH4). Once the hydroxyl group is present in the target orientation (by Mitsunobu inversion with benzoic acid and subsequent hydrolysis with K2CO3), the butenolide is added as usual.

Improved synthesis of hydroxylated GR24 analogues
At this point it is interesting to mention some of the compounds in the so-called EGO family, the scaffold of which is related to that of GR24 and characterized by the presence of a tertiary amine at C-4 and no oxygen atom in the C-ring. Of these, EGO10 is the simplest as well as the most similar to GR24. This compound can be synthesized from commercial 3-(indol-3-yl)propanoic acid, in 26% overall yield, following a three-step strategy (Scheme 17; Prandi et al. 2011). Use of this molecule simplifies the route as it is a bicyclic compound with a carboxylic acid function, and therefore a precursor of the C-ring (formed by cyclization with polyphosphoric acid). In bioassays, EGO10 induced the germination of P. aegyptiaca with high germination percentages (always up to 85%) in the range of 10–0.1 µM (Sanchez et al. 2018). A comparison of activity levels with GR24 showed that similar maximum response values were obtained, although the EC50 is higher for EGO10 (Cohen et al. 2013). The study of Artuso et al. (2015) showed that the C-2′R configuration improves the activity of EGO10 for the germination of P. aegyptiaca to some extent, as occurs with the non-nitrogen-containing GR24 analogues.

Synthesis of EGO10
Bhattacharya et al. (2009) previously reported two routes to obtain EGO derivatives methylated at the C-ring, which belong to the so-called ST series. The route with the best yield is shown in Scheme 18. The starting material is commercial 1-methylindolin-2-one, the ketone group of which is first transformed into an enol triflate. A subsequent Suzuki coupling with a dienyl boronate (previously synthesized from an α,β-unsaturated diethyl acetal) leads to the intermediate compounds (32), which undergo a Nazarov cyclization (catalyzed by the Brønsted acid o-benzenedisulfonimide) to form the triclyclic scaffold (33). After butenolide ring addition, the two analogues PLN655 and PLN655a were obtained in 43% and 48% overall yield respectively. The former, together with its non-nitrogen-containing derivative PLC655, which was also synthesized, were tested on O. aegyptiaca as racemates and their activity levels found to be higher than those for GR24: both were highly active at 10–4 and 10–6 M, with percentages of around 80–90%, and PLN655 retained this level down to 10–8 M.

Synthesis of the ST analogues PLN655 and PLN655a
Ueno et al. (2015) synthesized the GR24 analogues monohydroxylated at each aromatic position of the A-ring (C-5, C-6, C-7 or C-8; Scheme 19). The starting materials in this case were four hydroxylated indan-1-one derivatives isolated from root exudates of hydroponically grown Sorghum bicolor. The synthesis of one of them is shown in Scheme 19 as an example. The synthesis starts with protection of the hydroxyl group by formation of an MOM ether. The yield for formation of the tricyclic scaffold (34) was much lower (13%) than for the other steps in this route (19%, 20% and 29% for the other starting materials), therefore optimization of these reactions could provide high-yielding synthetic routes. Analogues with similar and different isomeric features (C-ring geometry and C-2′ orientation) to GR24 were also obtained and evaluated in a bioassay. All analogues were active on S. hermonthica seeds, especially those with similar isomeric features to GR24, and analogues hydroxylated at C-7 and C-8 were significantly more active. With Striga gesnerioides seeds, only some analogues with a different C-ring geometry contrary to GR24 were active (below 20% at 10 or 0.1 µM), and the position of the hydroxyl group on the A-ring was not considered relevant.

Synthesis of GR24 analogues with a hydroxyl group on the A-ring
With regard to modifications on the D-ring, Mwakaboko and Zwanenburg (2016) focused on the introduction of methyl groups. The key to their success was the preparation of chlorobutenolides as precursors for the D-ring (instead of the brominated butenolide 23; Scheme 20). This was carried out by condensation of pyruvic acid with acetone or 2-butanone to synthesize hydroxylated butenolides (35), and subsequent chlorination with SOCl2. The final SL analogues (36 and 37) were tested on S. hermonthica and O. cernua seeds as racemates, with moderate activity being observed. Although these findings appeared to indicate a loss of activity when methyl groups are included in the D-ring, the authors state that firm conclusions can only be obtained after the evaluation of pure epimers.

Synthesis of chlorobutenolides and GR24 analogues with methylated D-rings
Lombardi et al. (2017) also synthesized new alternative D-rings for subsequent introduction into 1. The main aim of this study was to replace the butenolide with γ-lactams, thus forming GR24 derivatives containing this type of ring. The synthesis of brominated lactam rings (Scheme 21) starts with a condensation of allylamine or 2-methylallylamine (the latter to synthesize strigolactones containing a methylated D-ring) with methacryloyl chloride under basic conditions. The product (38) is an amide that must first be protected with Boc2O before subsequent cyclization catalyzed by the ruthenium catalyst catMETium-RF1. The resulting lactam (39) is then brominated with NBS.

Total synthesis of brominated lactam rings
Addition of the brominated lactams to 21 is carried out as usual, with very good yields (route A in Scheme 22). The final step corresponds to removal of the protecting group, with yields (21–25%) of around half those of the previous step.

Final steps in the synthesis of GR24 analogues containing a lactam as D-ring
Products with R = H were tested on Phelipanche aegyptiaca. All compounds were active at 10–5 M, with those with C-2′S exhibiting activities of more than 80% at this concentration. Similarly high percentages (around 75%) were obtained at 10–6 M, whereas the C-2′R epimers exhibited values of less than 35%. It must be noted that the C-2′S orientation of lactams is α for the butenolide ring, as in the SL with C-2′R, therefore the preference for the activity with this orientation also occurs in lactams. The high activity levels of C-2′S epimers were similar to those for GR24 at the same concentrations. In summary, the use of a lactam as the D-ring and the presence of a Boc group in it does not result in a loss of activity at the highest concentrations if the configuration of the D-ring is C-2′S.
Products 40 and 41, both lactams at the D-ring of EGO 10, were also synthesized by Lombardi et al. (2017) (route B in Scheme 22) and evaluated in a bioassay (Sanchez et al. 2018). These compounds showed poor activity for the germination of P. aegyptiaca, unlike the other strigolactams shown in Scheme 22, or EGO10 and PLN655.
With regard to the enol ether bond, different studies have shown that the modification thereof results in a loss of activity of the analogue for many parasitic weed species. Thus, Thuring et al. (1997b) synthesized the two geometric isomers (11Z for 43, and 11E for 44) of GR24 lacking the oxygen atom of the ether (route A in Scheme 23). The key was addition of the butenolide derivative 42 to the tricyclic GR24 scaffold. Different methods were employed to obtain each geometric isomer. Thus, 11Z was obtained by mesylation and subsequent elimination catalyzed by the base DBU, whereas 11E was obtained by reaction with 2-fluoro-1-methylpyridinium p-toluenesulfonate and Et3N. The final step is elimination of the thioether protecting group. In bioassays, both analogues (43 and 44) were found to be inactive for the germination of S. hermonthica and O. crenata. On the other hand, Kondo et al. (2007) synthesized the imino derivative of GR24 at the enol ether bond, in which a nitrogen atom is found at the exocyclic position of the double bond (route B in Scheme 23). This product (45) is obtained by reaction of the tricyclic GR24 scaffold with isoamyl nitrite to form an oxime, and subsequent addition of the furanone ring. Compound 45 was tested in a bioassay as a possible mixture of geometric isomers, and found to be moderately active for S. hermonthica (28.3% at 10 µM) but inactive at the highest concentration tested (10 µM) for S. gesnerioides, O. minor or O. crenata. These results show that some modifications to the enol ether bond do not provoke a total loss of activity for some parasitic species. This could be promising if this modification generated more stability and better solubility for the molecule, which would increase its suitability for use in herbicides.

Synthesis of GR24 analogues with modifications at the enol ether
The final GR24 derivatives presented herein are the promising strigolactams reported by Lachia et al. (2015), which contain an amide at the C-ring instead of the canonical ester. Two synthetic routes were reported to obtain the tricyclic scaffold (46; Scheme 24). The first (route A in Scheme 24) is related to the cycloaddition strategy previously shown in Scheme 14 to obtain 26. The key step is the subsequent Beckman rearrangement catalyzed by 2,4,6-trimethylbenzenesulfonamide to obtain 46. However, this reaction gave only a low yield (8%) of the desired product, therefore the second method (route B in Scheme 24) is clearly better in terms of overall yield (> 80%). Thus, starting from compound 24 (synthesized as shown in Scheme 11), a reductive amination is carried out to form an oxime (47), which is reduced to a lactam with Zn in the following step. Once 46 has been obtained, after protection of the amine with Boc2O, the enol intermediate 48 is formed by formylation with Bredereck reagent and subsequent hydrolysis and removal of the protecting group with trifluoroacetic acid (TFA). The butenolide is added as usual to obtain strigolactam 49 and its C-2′ epimer. Compound 50 is obtained by methylation of 49 with NaH and MeI.

Strategies to synthesize strigolactams at the C-ring
Recently, Ashida et al. (2020) reported the enantioselective synthesis of 49 using a nickel-catalyzed asymmetric cycloaddition. This study broadens the possibilities for the enantioselective synthesis of lactams and other compounds containing two contiguous stereogenic carbon centers.
Both 49 and 50 proved to be more active for the germination of O. cumana (82–93% activity in the range 10–1-10–3 mg·L−1) than GR24 (57–82% in the same range; Lachia et al. 2015). At lower concentrations, compound 49 stood out as 80% of activity was observed at 10–5 mg·L−1, a concentration at which 26 or GR24 exhibited low percentages (3% and 19% respectively). The study of Lumbroso et al. (2016) also focused on the synthesis of strigolactams, specifically on the obtaining of simpler analogues possessing a bicyclic scaffold that lack of the canonical A-ring. They were based on the structure of GR-28 (Fig. 2). Showing different activity profiles, some of these compounds proved to be potent germination stimulants of O. cumana, reaching a maximum activity of 96% (0.01 mg·L−1).
The structure–activity relationships mentioned in this section are in accordance with the general lines provided by Mwakaboko and Zwanenburg (2011a): “for the germination activity, A-ring modifications affect to a minor extent, stereochemistry is important, and the enol ether bond and D-ring are essential.”
All compounds discussed in this section are structurally based on GR24. This skeleton is the most common canonical SL analogue published in the literature, not only by studies on parasitic weeds, but also for its applicability to develop stimulators for the AMF growth and branching. As example of recent study, the results published by Borghi et al. (2021) showed the potential of GR24 and some analogues, including a strigolactam, for AMF branching promotion.
Strigol, orobanchol and sorgomol families
Strigol, the first strigolactone isolated and tested in a parasitic weed bioassay, has also been considered in different studies to synthesize structurally related analogues. The study by Reizelman et al. (2000) could be a good starting point in this regard since these authors reported the total synthesis of the eight stereoisomers of strigol (Scheme 25). The first steps correspond to the efficient three-steps methodology reported by Steiner and Willhalm (1952) to synthesize the intermediate 51, which have been included in Scheme 25 to provide a complete synthetic route. The corresponding carboxylic acid of 51 was then synthesized to obtain the first bicyclic intermediate (52) by reaction with vinyl silane and a subsequent Nazarov reaction (SnCl4-catalyzed cyclization). After insertion of a new carboxylic acid group, the tricyclic scaffold (53) was formed by a Luche reduction with CeCl3 and NaBH4. The next steps could be the most interesting for the design of new analogues since oxygenated functional groups are introduced at the A-ring using a strategy that also allows purification of the stereoisomers. To this end, a hydroxyl group is placed at C-5 by reaction with SeO2, and then overoxidized to a ketone in order to form products more suitable for purification by column chromatography. The ketone is then reduced via a Luche reaction to obtain the target hydroxyl group at C-5. The resulting molecules are then formylated and bonded to the butenolide group. The diastereoisomers were resolved by cellulose triacetate HPLC.

Total synthesis of strigol and its stereoisomers
5-Deoxystrigol is a natural SL similar to strigol but lacking the characteristic hydroxyl group. It was considered a SL analogue before its first isolation (Akiyama et al. 2005). Frischmuth et al. (1991) reported the first synthesis of 5-deoxystrigol together with other analogues based on functionalization at C-5 with hydroxyl or acetate groups. Alternative synthetic strategies were published later (Reizelman et al. 2000; Shoji et al. 2009), including the synthesis of 5-deoxystrigol together with its C-2′ epimer, and their C-ring stereoisomers (the latter, corresponding with analogues of orobanchol, namely 4-deoxyorobanchol), reported by Lachia et al. (2014a) (route A in Scheme 26). The first steps result in the acid 54, which then forms an amide with (S,S)- or (R,R)-2,5-bis(methoxymethyl)pyrrolidine. The tricyclic system was prepared via an asymmetric intramolecular [2 + 2] cycloaddition of the ketene-iminium salts of the amides, catalyzed by Tf2O in the presence of 2-fluoropyridine as base. The butenolide ring is added as usual. We would like to recommend the review by De Mesmaeker et al. (2019) to those who intend to obtain a more detailed background on this synthesis of 5-deoxystrigol and its stereoisomers.

Strategies for the synthesis of 5-deoxystrigol and its stereoisomers
It is also worth highlighting the study recently published by Shiotani et al. (2021), which achieved the synthesis of 5-deoxystrigol as a racemate (therefore also obtaining 4-deoxyorobanchol) from a non-canonical SL structurally related to carlactone, by acid-mediated cascade cyclization (route B in Scheme 26). In a different study, Ueno et al. (2018)proved that 5-deoxystrigol acts like starting material for the natural synthesis in plants of the monohydroxylated SLs strigol and sorgomol. In the same way, some plants species synthetize orobanchol by bioconversion of 4-deoxyorobanchol, although other species would synthetize it from another compound.
Tanaka et al. (2013) reported a synthetic strategy to obtain the analogue 7-oxo-5-deoxystrigol in 11 steps from 2,2-dimethylcyclohexane-1,3-dione (Scheme 27). This route starts with protection of one of the ketone groups, which is then transformed into a dienone by way of Grignard and oxidation reactions. The bicyclic intermediate 55 is then obtained via a Nazarov cyclization catalyzed by H3PO4. After protection of the ketone group of the A-ring by formation of an MOM ether, the tricyclic scaffold (56) is formed. This requires the prior introduction of an acid group by reaction with the base LDA and ICH2CO2Et (formation of an ester) and subsequent hydrolysis. The keto-acid group generated allows cyclization by way of a Luche reduction. The final steps correspond to introduction of the butenolide, removal of the MOM group by treatment with LiBF4 and oxidation of the resulting hydroxyl group with Dess-Martin periodinane to form the ketone group at the A-ring. Although the overall yield for this synthesis is not available as the yield for the first step was not reported, it is noteworthy that the overall yield of the following steps is 19%.

Synthesis of 7-oxo-5-deoxystrigol
With regard to orobanchol, it should be emphasized that until the correction of its structure in 2011, the total syntheses previously reported did not lead to orobanchol. The total synthesis of this originally assigned orobanchol molecule was for example published by (Zwanenburg et al. 2016), together with its 2′ epimer (Scheme 28), both as racemates. Mesityl oxide is used as starting material, and the ABC scaffold is obtained in 12 steps in 25% overall yield as a racemate. The following step is the formation of a ketone group at the B-ring to obtain 57 via allylic oxidation. This reaction was found to be low yielding (14%) as the major product (40%) is an undesired ketone at A-ring (C-5). On the other hand, this procedure employs reagents that reduce environmental impact, compared to the previously published methodology. A subsequent Luche reduction of 57 led to the cis hydroxylated intermediate as major product, and a subsequent Mitsunobu inversion was then carried out to invert the configuration at C-4 by way of esterification of the hydroxyl group by treatment with diethyl azodicarboxylate (DEAD), and subsequent hydrolysis with K2CO3 (Matsui et al. 1999). These steps are similar to those previously described for the synthesis of hydroxylated derivatives of GR24 (see Scheme 16). Orobanchol and its epimer at C-2′ were obtained in similar yields after introduction of the butenolide ring.

Total synthesis of racemates of originally assigned orobanchol and its 2' epimer
Nomura et al. (2013) synthesized and purified all orobanchol stereoisomers (configuration at C-4 and C-2′; and enantiomers at the C-ring) from compound 57 by protecting the hydroxyl group with 2-trimethylsilylethoxymethyl (SEM) and subsequent deprotection using the strong acid TFA. In the bioassay, almost all orobanchol stereoisomers induced the germination of S. hermonthica, with orobanchol and its C-4’ epimer being the most active (around 70% of activity at 1 µM). For S. gesnerioides seeds, only two of the stereoisomers were active (medium activity at 10 and 1 µM), namely the two C-2′ epimers of the C-4R and C-ring enantiomers of orobanchol.
With regard to the strigolactone sorgomol, Kitahara et al. (2011) reported its synthesis from ethyl 2-oxocyclohexanecarboxylate (Scheme 29). The bicyclic intermediate is obtained first via a Nazarov cyclization using BrMgC≡CCH2OTHP, then the tricyclic scaffold is formed in two steps. The second step is a reduction with diisobutylaluminium hydride (DIBAL), the low yield of which (12%) is the main disadvantage of this strategy. After oxidation of the hydroxyl group on the C-ring using Ag2CO3, the D-ring was added as usual. The overall yield for each 2′ epimer is 1%. Nomura et al. (2013) used this strategy to obtain the eight stereoisomers of sorgomol (configuration of C-8 and C-2′; and enantiomers at C-ring), which were isolated and evaluated in a bioassay in a similar manner to the orobanchol stereoisomers discussed in the previous paragraph. All were active for S. hermonthica seeds, although sorgomol was the most active. For S. gesnerioides seeds, only the C-ring enantiomer of sorgomol was significantly active, with around 35% activity at 1 µM.

Synthesis of sorgomol and its 2' epimer
To end this section, we suggest the design of synthetic routes to achieve sorgomol derivatives at C-4 via the introduction of oxygenated groups like hydroxyl or acetate. The resulting analogues could be more active for S. hermonthica germination as this structural improvement has been observed to be beneficial for GR24 and its hydroxylated analogue 5 described previously in this review.
SL analogues based on sesquiterpene lactones
Sesquiterpene lactones are a large group of metabolites synthesized by plants (mostly the Astaraceae family). They have been studied for their medicinal or herbicidal properties, and some have been shown to be highly active at low concentrations. According to their structure, they can be classified into five subclasses (Moujir et al. 2020). All these compounds are bicyclic or tricyclic, and one of the rings is a lactone that is directly implicated in the bioactivities of these compounds.
The guaianolide and eudesmanolide subclasses will be highlighted herein as their structures have been used to synthesize canonical SL analogues. The synthetic routes involved are simpler, as the use of these tricyclic compounds, which already contain a lactone ring, as starting material avoids the large number of steps required to obtain the tricyclic scaffold of the analogues (like 21). Thus, analogues can be synthesized via possible strategies that allow introduction of the butenolide ring via an enol ether bond in the lactone ring. From a practical point of view, the high-yielding synthesis is advantageous as some sesquiterpene lactones can be isolated on a multigram scale from natural sources. This is the case, for example, for dehydrocostulactone, a guaianolide-type lactone isolated from root extracts of Saussurea costus. Macías et al. (2009) reported a three-step synthesis of new analogues, known as guaianestrigolactones (GELs), derived from dehydrocostulactone (Scheme 30). The key step in this synthesis was the introduction of hydroxyl groups at the methylene of the lactone ring via a Michael addition with K2CO3, and the use of hexamethylphosphoramide as polar aprotic solvent. Monohydroxylated products were obtained in 51% yield (58), being then oxidized to the aldehyde by treatment with Dess-Martin periodinane. Finally, the butenolide ring was added to give an overall yield of 12%. This final step was also applied to 58 and 60 to synthesize GELs lacking an enol ether bond (11,13-dihydroGELs 59 and 61, respectively).

First synthesis reported for the synthesis of GEL and 11,13-dihydroGELs
The first step was improved by Cala et al. (2019), who reported a two-step procedure (Scheme 31) that reduces the reaction times significantly (from 7 to 2 days and 6 h), while also avoiding use of the toxic solvent HMPA and the formation of the dihydroxylated byproduct 60. The first target intermediate was an ether at C-13, which was obtained by a Michael addition of the alkoxide of 4-methoxybenzyl alcohol (formed using the base 1,8-diazabicyclo[5.4.0]-undec-7-ene). The resulting ether (selective to C-11R configuration) is then oxidized using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone to obtain the hydroxylated derivative 58. The overall yield was 54%.

Improved synthesis of the hydroxylated intermediate for the synthesis of GELs
In the bioassay, the C-2′R GEL was found to be highly active for O. cumana (nearly 80% at 10 µM) and Phelipanche ramosa (60% at 10 and 1 µM, and 46% at 0.1 µM), whereas its C-2′S counterpart was somewhat less active (differences of 5–25%). Both epimers of 59, which lack the enol ether, were also significantly active for P. ramosa (50–60% at 10 µM). With regard to O. cumana, the C-11S epimer stands out as an activity of 80% was observed at 10 µM and more than 60% was retained at the lowest dose tested (0.01 µM). Thus, the presence of the enol ether does not result in a loss of activity for the parasitic species evaluated, therefore the orientation of both C-2′ and C-11 must also be considered.
Recently, Zorrilla et al. (2020) selectively synthesized the C-11R epimers of 59 and isolated the epimers at C-2′. Both were tested in a bioassay, and significant activity was observed at 100 µM for P. ramosa (mean 83%) and O. cumana (53%). This activity was well retained at 10 µM, but only the C-2′R epimer was significantly active for P. ramosa (EC50 = 5.27 × 10−2 µM) at 1 µM or lower, thus highlighting the possibilities of this compound.
This same study also optimized the synthetic route shown in Schemes 30 and 31 for the synthesis of analogues based on eudesmanolides and thus obtain a new series of SL canonical analogues based on natural products, known as eudesmanestrigolactones (EDSLs). Three accessible eudesmanolides (α- and β-cyclocostunolide, and 3-deoxybrachylaenolide) were used as starting material. An important point of this strategy (Scheme 32) is the obtaining of SL analogues with both geometries of the enol ether bond (11E like natural SL, and 11Z) and configurations of C-2′ (R like natural SL, and S). Thus, up to four SL analogues could be obtained from each eudesmanolide, as well as 11,13-dihydroEDSLs like 62. This synthetic procedure is relevant in the context of the synthesis of analogues, specifically analogues based on other sesquiterpene lactones, due to its potential applicability to each compound that contains a methylene-lactone group (C-ring).

Synthesis of canonical SL analogues based on eudesmanolides
All EDSLs were tested in parasitic seed bioassays, showing high activity profiles for the germination of P. ramosa and O. cumana even at the lowest concentrations tested, and medium activity for O. crenata. This activity was similar to, or even better than, that observed for GR24. EDSLs with the same isomeric features as natural SL (11E and C-2′R) were found to be remarkably active, and some EDSLs with alternative features achieved better results for some species. One of the main conclusions of the SAR study is that the double-bond system of the A-ring and the geometry of the enol ether have a marked influence on activity levels. Indeed, compound 62 (lacking the enol ether) was found to have no activity as regards O. crenata germination.
Since other sesquiterpene lactones, or sesquiterpenes of the guaiane- and eudesmane-type, can be isolated from natural sources (Cárdenas et al. 2020; Macías et al. 2013), the application or adaptation of the synthetic strategies shown in this review could provide new active analogues or mimics of strigolactones based on natural products.
Other scaffolds for SL analogues
This final section presents the example of two products related to canonical analogues of SL that have not been as widely applied for the synthesis of analogues as the families mentioned previously.
The first example is the total synthesis of 4-deoxymedicaol (analogue of the natural SL medicaol) and its C-2′ epimer, reported by Tokunaga et al. (2015) (Scheme 33). Overall yields of 0.08 and 0.06% were obtained, respectively. These low yields are reasonable as overall yields for the five-step synthesis of the bicyclic intermediate 63 from ethyl 2-oxo-cyclohexanecarboxylate, and addition of the butenolide ring in the final step, are between 4–9%. The high-yielding step involving expansion of the A-ring in 63 to a seven-membered ring (64) should be noted as similar reactions are not common in the synthesis of analogues. The tricyclic scaffold is then synthesized by forming a keto acid at the B-ring, followed by treatment with DIBAL and acidification. 4-Deoxymedicaol was tested for the induction of hyphal branching of germinating spores of the arbuscular mycorrhizal fungus Gigaspora margarita rather than in a parasitic seed bioassay. Positive results were observed for both this compound and for medicaol.

Synthesis of the analogue 4-deoxymedicaol
The second example is provided by the study of Mwakaboko and Zwanenburg (2011b), in which different SL analogues of progressive structural complexity were synthesized. One of them is structurally comparable to canonical SL, although its structure is actually based on the nobornene system (Scheme 34), an unusual scaffold in the literature for the synthesis of analogues and mimics. A further distinctive feature is that the C- and D-rings are linked via an ether placed on a double bond on the C-ring rather than via an enol ether bond. In bioassays, this product germinated S. hermonthica (over 50% germination at 0.1 and 0.01 mg·L−1) and O. cernua seeds (over 80% at 1 mg·L−1 and around 55% at 0.1 mg·L−1). As such, it may be of interest to develop related compounds containing nobornene scaffolds to perform more in-depth structure–activity studies.

Synthesis of an SL analogue based on a nobornene scaffold
Conclusions
Strigolactone analogues are an alternative to the use of SL as agrochemicals given their improved activity levels and the more efficient methods available to synthesize them. Different synthetic strategies have been described, with most efforts focusing on construction of the main tricyclic scaffold in the final molecule. Although many of the steps in these routes have high yields, the use of at least one low-yielding step hinders the development of high-yielding routes, which is a particular disadvantage for routes involving a large number of steps. Thus, it is worth highlighting those routes that minimize the number of steps, principally by using a readily accessible bicyclic or tricyclic compound as starting material. In this regard, the synthesis of GR24 and some of the related analogues described in this review, as well as guaianestrigolactones (GELs) and eudesmanestrigolactones (EDSLs), are good examples.
With regard to the germination activity, recent advances in chromatography and the development of selective synthetic routes have led to the ability to evaluate pure SLs, thus demonstrating that the isomeric characteristics of these compounds could affect their activity levels. In the case of the GR24 family, the main relevance resides in the enol ether bridge and D-ring. However, these conclusions cannot be extrapolated to analogues containing other scaffolds, therefore more in-depth structure–activity relationship studies are required.
References
Akiyama K, Matsuzaki KI, Hayashi H (2005) Plant sesquiterpenes induce hyphal branching in arbuscular mycorrhizal fungi. Nature 435:824–827. https://doi.org/10.1038/nature03608
Alder A, Jamil M, Marzorati M, Bruno M, Vermathen M, Bigler P, Ghisla S, Bouwmeester H, Beyer P, Al-Babili S (2012) The path from β-carotene to carlactone, a strigolactone-like plant hormone. Science (80-). 335:1348–1351. https://doi.org/10.1126/science.1218094
Artuso E, Ghibaudi E, Lace B, Marabello D, Vinciguerra D, Lombardi C, Koltai H, Kapulnik Y, Novero M, Occhiato EG, Scarpi D, Parisotto S, Deagostino A, Venturello P, Mayzlish-Gati E, Bier A, Prandi C (2015) Stereochemical assignment of strigolactone analogues confirms their selective biological activity. J Nat Prod 78:2624–2633. https://doi.org/10.1021/acs.jnatprod.5b00557
Ashida K, Hoshimoto Y, Tohnai N, Scott DE, Ohashi M, Imaizumi H, Tsuchiya Y, Ogoshi S (2020) Enantioselective synthesis of polycyclic γ-lactams with multiple chiral carbon centers via Ni(0)-catalyzed asymmetric carbonylative cycloadditions without stirring. J Am Chem Soc 142:1594–1602. https://doi.org/10.1021/jacs.9b12493
Bhattacharya C, Bonfante P, Deagostino A, Kapulnik Y, Larini P, Occhiato EG, Prandi C, Venturello P (2009) A new class of conjugated strigolactone analogues with fluorescent properties: synthesis and biological activity. Org Biomol Chem 7:3413–3420. https://doi.org/10.1039/b907026e
Borghi L, Screpanti C, Lumbroso A, Lachia M, Gübeli C, De Mesmaeker A (2021) Efficiency and bioavailability of new synthetic strigolactone mimics with potential for sustainable agronomical applications. Plant Soil 465:109–123. https://doi.org/10.1007/s11104-021-04943-8
Bromhead LJ, McErlean CSP (2017) Accessing single enantiomer strigolactones: progress and opportunities. European J Org Chem 2017:5712–5723. https://doi.org/10.1002/ejoc.201700865
Bromhead LJ, Visser J, McErlean CSP (2014) Enantioselective synthesis of the strigolactone mimic (+)-GR24. J Org Chem 79:1516–1520. https://doi.org/10.1021/jo402722p
Bromhead LJ, Norman AR, Snowden KC, Janssen BJ, McErlean CSP (2018) Enantioselective total synthesis and biological evaluation of (-)-solanacol. Org Biomol Chem 16:5500–5507. https://doi.org/10.1039/c8ob01287c
Cala A, Zorrilla JG, Rial C, Molinillo JMG, Varela RM, Macías FA (2019) Easy access to alkoxy, amino, carbamoyl, hydroxy, and thiol derivatives of sesquiterpene lactones and evaluation of their bioactivity on parasitic weeds. J Agric Food Chem 67:10764–10773. https://doi.org/10.1021/acs.jafc.9b03098
Cárdenas DM, Cala A, Mejías FJR, Zorrilla JG, Macías FA, 2020. Sesquiterpenes in cereals and spices. In: Xiao J, Sarker SD, Asakawa Y (eds), Handbook of dietary phytochemicals. Springer, Singapore, pp 1–63. https://doi.org/10.1007/978-981-13-1745-3_16-1
Carrillo P, Martínez-Poveda B, Medina MÁ, Quesada AR (2019) The strigolactone analog GR-24 inhibits angiogenesis in vivo and in vitro by a mechanism involving cytoskeletal reorganization and VEGFR2 signalling. Biochem Pharmacol 168:366–383. https://doi.org/10.1016/j.bcp.2019.07.019
Chen VX, Boyer FD, Rameau C, Pillot JP, Vors JP, Beau JM (2013) New synthesis of A-ring aromatic strigolactone analogues and their evaluation as plant hormones in pea (Pisum sativum). Chem - A Eur J 19:4849–4857. https://doi.org/10.1002/chem.201203585
Cohen M, Prandi C, Occhiato EG, Tabasso S, Wininger S, Resnick N, Steinberger Y, Koltai H, Kapulnik Y (2013) Structure-function relations of strigolactone analogs: activity as plant hormones and plant interactions. Mol Plant 6:141–152. https://doi.org/10.1093/mp/sss134
De Mesmaeker A, Screpanti C, Fonné-Pfister R, Lachia M, Lumbroso A, Bouwmeester H (2019) Design, synthesis and biological evaluation of strigolactone and strigolactam derivatives for potential crop enhancement applications in modern agriculture. Chimia (aarau) 73:549–560. https://doi.org/10.2533/chimia.2019.549
Dieckmann MC, Dakas PY, De Mesmaeker A (2018) Synthetic access to noncanonical strigolactones: syntheses of carlactonic acid and methyl carlactonoate. J Org Chem 83:125–135. https://doi.org/10.1021/acs.joc.7b02465
Frischmuth K, Samson E, Kranz A, Welzel P, Meuer H, Sheldrick WS (1991) Routes to derivatives of strigol (the witchweed germiantion factor) modified in the 5-position. Tetrahedron 47:9793–9806
Heather JB, Mittal RSD, Sih CJ (1976) Synthesis of the witchweed seed germination stimulant (+)-strigol. J Am Chem Soc 98:3661–3669. https://doi.org/10.1021/ja00428a046
Hirayama K, Mori K (1999) Synthesis of (+)-strigol and (+)-orobanchol, the germination stimulants, and their stereoisomers by employing lipase-catalyzed asymmetric acetylationas the key step. Eur J Org Chem 1999:2211–2217. https://doi.org/10.1002/(SICI)1099-0690(199909)1999:9%3c2211::AID-EJOC2211%3e3.0.CO;2-O
Hýlová A, Pospíšil T, Spíchal L, Mateman JJ, Blanco-Ania D, Zwanenburg B (2019) New hybrid type strigolactone mimics derived from plant growth regulator auxin. N Biotechnol 48:76–82. https://doi.org/10.1016/j.nbt.2018.08.001
Jamil M, Kountche BA, Haider I, Guo X, Ntui VO, Jia K-P, Ali S, Hameed US, Nakamura H, Lyu Y, Jiang K, Hirabayashi K, Tanokura M, Arold ST, Asami T, Al-Babili S (2018) Methyl phenlactonoates are efficient strigolactone analogs with simple structure. J Exp Bot 69:2319–2331. https://doi.org/10.1093/jxb/erx438
Jamil M, Kountche BA, Haider I, Wang JY, Aldossary F, Zarban RA, Jia KP, Yonli D, ShahulHameed UF, Takahashi I, Ota T, Arold ST, Asami T, Al-Babili S (2019) Methylation at the C-3′ in D-ring of strigolactone analogs reduces biological activity in root parasitic plants and rice. Front Plant Sci. https://doi.org/10.3389/fpls.2019.00353
Johnson AW, Gowda G, Hassanali A, Knox J, Monaco S, Razavi Z, Rosebery G (1981) The preparation of synthetic analogues of strigol. J Chem Soc Perkin Trans 1:1734–1743. https://doi.org/10.1039/P19810001734
Kitahara S, Tashiro T, Sugimoto Y, Sasaki M, Takikawa H (2011) First synthesis of (±)-sorgomol, the germination stimulant for root parasitic weeds isolated from Sorghum bicolor. Tetrahedron Lett 52:724–726. https://doi.org/10.1016/j.tetlet.2010.12.010
Kondo Y, Tadokoro E, Matsuura M, Iwasaki K, Sugimoto Y, Miyake H, Takikawa H, Sasaki M (2007) Synthesis and seed germination stimulating activity of some imino analogs of strigolactones. Biosci Biotechnol Biochem 71:2781–2786. https://doi.org/10.1271/bbb.70398
Kountche BA, Jamil M, Yonli D, Nikiema MP, Blanco-Ania D, Asami T, Zwanenburg B, Al-Babili S (2019) Suicidal germination as a control strategy for Striga hermonthica (Benth.) in smallholder farms of sub-Saharan Africa. Plants People Planet 1:107–118. https://doi.org/10.1002/ppp3.32
Kurt B, Ozleyen A, Antika G, Yilmaz YB, Tumer TB (2020) Multitarget profiling of a strigolactone analogue for early events of alzheimer’s disease. In vitro therapeutic activities against neuroinflammation. ACS Chem Neurosci 11:501–507. https://doi.org/10.1021/acschemneuro.9b00694
Lachia M, Dakas PY, De Mesmaeker A (2014a) Asymmetric synthesis of the four stereoisomers of 5-deoxystrigol. Tetrahedron Lett 55:6577–6581. https://doi.org/10.1016/j.tetlet.2014.10.040
Lachia M, Wolf HC, De Mesmaeker A (2014b) Synthesis of strigolactones analogues by intramolecular [2+2] cycloaddition of ketene-iminium salts to olefins and their activity on Orobanche cumana seeds. Bioorganic Med Chem Lett 24:2123–2128. https://doi.org/10.1016/j.bmcl.2014.03.044
Lachia M, Wolf HC, Jung PJM, Screpanti C, De Mesmaeker A (2015) Strigolactam: new potent strigolactone analogues for the germination of Orobanche cumana. Bioorganic Med Chem Lett 25:2184–2188. https://doi.org/10.1016/j.bmcl.2015.03.056
Lombardi C, Artuso E, Grandi E, Lolli M, Spirakys F, Priola E, Prandi C (2017) Recent advances in the synthesis of analogues of phytohormones strigolactones with ring-closing metathesis as a key step. Org Biomol Chem 17:8218–8231. https://doi.org/10.1039/C7OB01917C
López-Ráez JA, Charnikhova T, Gómez-Roldán V, Matusova R, Kohlen W, De Vos R, Verstappen F, Puech-Pages V, Bécard G, Mulder P, Bouwmeester H (2008) Tomato strigolactones are derived from carotenoids and their biosynthesis is promoted by phosphate starvation. New Phytol 178:863–874. https://doi.org/10.1111/j.1469-8137.2008.02406.x
López-Ráez JA, Shirasu K, Foo E (2017) Strigolactones in plant interactions with beneficial and detrimental organisms: the Yin and Yang. Trends Plant Sci 22:527–537. https://doi.org/10.1016/j.tplants.2017.03.011
Lumbroso A, Villedieu-Percheron E, Zurwerra D, Screpanti C, Lachia M, Dakas PY, Castelli L, Paul V, Wolf HC, Sayer D, Beck A, Rendine S, Fonné-Pfister R, De Mesmaeker A (2016) Simplified strigolactams as potent analogues of strigolactones for the seed germination induction of Orobanche cumana Wallr. Pest Manag Sci 72:2054–2068. https://doi.org/10.1002/ps.4268
Macías FA, García-Díaz MD, Pérez-De-Luque A, Rubiales D, Galindo JCG (2009) New chemical clues for broomrape-sunflower host—parasite interactions: synthesis of guaianestrigolactones. J Agric Food Chem 57:5853–5864. https://doi.org/10.1021/jf900870j
Macías FA, Santana A, Durán AG, Cala A, Galindo JCG, Galindo JLG, Molinillo JMG (2013) Guaianolides for multipurpose molecular design. In: Beck JJ, Coats JR, Duke SO, Koivunen ME (eds) Pest management with natural products. American Chemical Society, Washington, pp. 167–188. https://doi.org/10.1021/bk-2013-1141.ch012
Malik H, Rutjes FPJT, Zwanenburg B (2010) A new efficient synthesis of GR24 and dimethyl A-ring analogues, germinating agents for seeds of the parasitic weeds Striga and Orobanche spp. Tetrahedron 66:7198–7203. https://doi.org/10.1016/j.tet.2010.06.072
Malik H, Kohlen W, Jamil M, Rutjes FPJT, Zwanenburg B (2011) Aromatic A-ring analogues of orobanchol, new germination stimulants for seed of parasitic weeds. Org Biomol Chem 9:2286–2293
Mangnus EM, Dommerholt FJ, de Jong RLP, Zwanenburg B (1992) Improved synthesis of strigol analogue GR24 and evaluation of the biological activity of its diastereomers. J Agric Food Chem 40:1230–1235. https://doi.org/10.1021/jf00019a031
Matsui J, Yokota T, Bando M, Takeuchi Y, Mori K (1999) Synthesis and structure of orobanchol, the germination stimulant for Orobanche minor. European J Org Chem 4:2201–2210. https://doi.org/10.1002/(sici)1099-0690(199909)1999:9%3c2201::aid-ejoc2201%3e3.0.co;2-q
Mori N, Nishiuma K, Sugiyama T, Hayashi H, Akiyama K (2016) Carlactone-type strigolactones and their synthetic analogues as inducers of hyphal branching in arbuscular mycorrhizal fungi. Phytochemistry 130:90–98. https://doi.org/10.1016/j.phytochem.2016.05.012
Morris JC, McErlean CSP (2016) Synthesis of highly enantio-enriched stereoisomers of hydroxy-GR24. Org Biomol Chem 14:1236–1238. https://doi.org/10.1039/c5ob02349a
Moujir L, Callies O, Sousa PMC, Sharopov F, Seca AML (2020) Applications of sesquiterpene lactones: A review of some potential success cases. Appl Sci 10:3001. https://doi.org/10.3390/app10093001
Mwakaboko AS, Zwanenburg B (2011a) Strigolactone analogs derived from ketones using a working model for germination stimulants as a blueprint. Plant Cell Physiol 52:699–715. https://doi.org/10.1093/pcp/pcr031
Mwakaboko AS, Zwanenburg B (2011b) Single step synthesis of strigolactone analogues from cyclic keto enols, germination stimulants for seeds of parasitic weeds. Bioorganic Med Chem 19:5006–5011. https://doi.org/10.1016/j.bmc.2011.06.057
Mwakaboko AS, Zwanenburg B (2016) Strigolactone analogues with a D-ring modified at C-2. Eur J Org Chem 2016:3495–3499. https://doi.org/10.1002/ejoc.201600576
Nejrotti S, Iannicelli M, Jamil SS, Arnodo D, Blangetti M, Prandi C (2020) Natural deep eutectic solvents as an efficient and reusable active system for the Nazarov cyclization. Green Chem 22:110–117. https://doi.org/10.1039/c9gc03465j
Nomura S, Nakashima H, Mizutani M, Takikawa H, Sugimoto Y (2013) Structural requirements of strigolactones for germination induction and inhibition of Striga gesnerioides seeds. Plant Cell Rep 32:829–838. https://doi.org/10.1007/s00299-013-1429-y
Pospíšil T (2021) Synthesis of simple strigolactone mimics. In: Cardinale F, Prandi C (eds) Strigolactones. Humana, New York, pp 31–36. https://doi.org/10.1007/978-1-0716-1429-7_4
Prandi C, Occhiato EG, Tabasso S, Bonfante P, Novero M, Scarpi D, Bova ME, Miletto I (2011) New potent fluorescent analogues of strigolactones: synthesis and biological activity in parasitic weed germination and fungal branching. Eur J Org Chem. https://doi.org/10.1002/ejoc.201100616
Reizelman A, Scheren M, Nefkens GHL, Zwanenburg B (2000) Synthesis of all eight stereoisomers of the germination stimulant strigol. Synth 13:1944–1951
Samson E, Frischmuth K, Berlage U, Heinz U, Hobert K, Welzel P (1991) Synthesis of (+)-strigol, (+)-4′-epi-strigol, and their enanthiomers. Tetrahedron 47:1411–1416. https://doi.org/10.1016/S0040-4020(01)86417-6
Sanchez E, Artuso E, Lombardi C, Visentin I, Lace B, Saeed W, Lolli ML, Kobauri P, Ali Z, Spyrakis F, Cubas P, Cardinale F, Prandi C (2018) Structure–activity relationships of strigolactones via a novel, quantitative in planta bioassay. J Exp Bot 69:2333–2343. https://doi.org/10.1093/jxb/ery092
Seto Y, Sado A, Asami K, Hanada A, Umehara M, Akiyama K, Yamaguchi S (2014) Carlactone is an endogenous biosynthetic precursor for strigolactones. Proc Natl Acad Sci U S A 111:1640–1645. https://doi.org/10.1073/pnas.1314805111
Shiotani N, Wakabayashi T, Ogura Y, Sugimoto Y, Takikawa H (2021) Studies on strigolactone BC-ring formation: chemical conversion of an 18-hydroxycarlactonoate derivative into racemic 4-deoxyorobanchol/5-deoxystrigol via the acid-mediated cascade cyclization. Tetrahedron Lett 68:152922. https://doi.org/10.1016/j.tetlet.2021.152922
Shoji M, Suzuki E, Ueda M (2009) Total synthesis of (±)-5-deoxystrigol via reductive carbon-carbon bond formation. J Org Chem 74:3966–3969. https://doi.org/10.1021/jo9002085
Soto-Cruz FJ, Zorrilla JG, Rial C, Varela RM, Molinillo JMG, Igartuburu JM, Macías FA (2021) Allelopathic activity of strigolactones on the germination of parasitic plants and arbuscular mycorrhizal fungi growth. Agronomy 11:2174. https://doi.org/10.3390/agronomy11112174
Steiner U, Willhalm B (1952) Darstellung von l, l-Dirnethyl-cyclohexanon-(3) -carbonsaureester-(2). Helv Chim Acta 220:1752–1756
Sugimoto Y, Wigchert SCM, Thuring JWJF, Zwanenburg B (1998) Synthesis of all eight stereoisomers of the germination stimulant sorgolactone. J Org Chem 63:1259–1267. https://doi.org/10.1021/jo9718408
Takahashi A, Ogura Y, Enomoto M, Kuwahara S (2016) Enantioselective synthesis of the tricyclic core of (+)-strigol. Tetrahedron 72:6634–6639. https://doi.org/10.1016/j.tet.2016.08.078
Tanaka M, Sugimoto Y, Kuse M, Takikawa H (2013) Synthesis of 7-Oxo-5-deoxystrigol, a 7-oxygenated strigolactone analog. Biosci Biotechnol Biochem 77:832–835. https://doi.org/10.1271/bbb.130020
Thuring JWJF, Nefkens GHL, Zwanenburg B (1997a) Asymmetric synthesis of all stereoisomers of the strigol analogue GR24. Dependence of absolute configuration on stimulatory activity of Striga hermonthica and Orobanche crenata seed germination. J Agric Food Chem 45:2278–2283. https://doi.org/10.1021/jf960466u
Thuring JWJF, Nefkens GHL, Zwanenburg B (1997b) Synthesis and biological evaluation of the strigol analogue carba-GR24. J Agric Food Chem 45:1409–1414. https://doi.org/10.1021/jf960443f
Tokunaga T, Hayashi H, Akiyama K (2015) Medicaol, a strigolactone identified as a putative didehydro-orobanchol isomer, from Medicago truncatula. Phytochemistry 111:91–97. https://doi.org/10.1016/j.phytochem.2014.12.024
Ueno K, Ishiwa S, Nakashima H, Mizutani M, Takikawa H, Sugimoto Y (2015) Regioselective and stereospecific hydroxylation of GR24 by Sorghum bicolor and evaluation of germination inducing activities of hydroxylated GR24 stereoisomers toward seeds of Striga species. Bioorg Med Chem 23:6100–6110. https://doi.org/10.1016/j.bmc.2015.08.003
Ueno K, Nakashima H, Mizutani M, Takikawa H, Sugimoto Y (2018) Bioconversion of 5-deoxystrigol stereoisomers to monohydroxylated strigolactones by plants. J Pestic Sci 43:198–206. https://doi.org/10.1584/jpestics.D18-021
Wigchert SCM, Kuiper E, Boelhouwer GJ, Nefkens GHL, Verkleij JAC, Zwanenburg B (1999) Dose-response of seeds of the parasitic weeds Striga and Orobanche toward the synthetic germination stimulants GR 24 and Nijmegen 1. J Agric Food Chem 47:1705–1710. https://doi.org/10.1021/jf981006z
Woo S, McErlean CSP (2019) Total synthesis and stereochemical confirmation of heliolactone. Org Lett 21:4215–4218. https://doi.org/10.1021/acs.orglett.9b01402
Xiao J, Wang Y-W, Peng Y (2017) Nickel-promoted reductive cyclization cascade: a short synthesis of a new aromatic strigolactone analogue. Synth 49:3576–3581. https://doi.org/10.1055/s-0036-1588804
Xie X, Kisugi T, Yoneyama K, Nomura T, Akiyama K, Uchida K, Yokota T, McErlean CSP, Yoneyama K (2017) Methyl zealactonoate, a novel germination stimulant for root parasitic weeds produced by maize. J Pestic Sci 42:58–61. https://doi.org/10.1584/jpestics.D16-103
Xiong Z, Tian J, Xue P, Zhang X, Lv H (2019) Enantioselective synthesis of chiral multicyclic γ-lactones: via dynamic kinetic resolution of racemic γ-keto carboxylic acids. Org Chem Front 7:104–108. https://doi.org/10.1039/c9qo01047e
Yamamoto S, Atarashi T, Kuse M, Sugimoto Y, Takikawa H (2020) Concise synthesis of heliolactone, a non-canonical strigolactone isolated from sunflower. Biosci Biotechnol Biochem. https://doi.org/10.1080/09168451.2020.1734444
Yasui M, Ota R, Tsukano C, Takemoto Y (2017) Total synthesis of avenaol. Nat Commun 8:1–9. https://doi.org/10.1038/s41467-017-00792-1
Yoneyama K, Xie X, Yoneyama K, Kisugi T, Nomura T, Nakatani Y, Akiyama K, McErlean CSP (2018) Which are the major players, canonical or non-canonical strigolactones? J Exp Bot 69:2231–2239. https://doi.org/10.1093/jxb/ery090
Yoshimura M, Fonné-Pfister R, Screpanti C, Hermann K, Rendine S, Dieckmann M, Quinodoz P, De Mesmaeker A (2019) Total synthesis and biological evaluation of heliolactone. Helv Chim Acta. https://doi.org/10.1002/hlca.201900211
Yoshimura M, Dieckmann M, Dakas PY, Fonné-Pfister R, Screpanti C, Hermann K, Rendine S, Quinodoz P, Horoz B, Catak S, De Mesmaeker A (2020) Total synthesis and biological evaluation of zealactone 1a/b. Helv Chim Acta. https://doi.org/10.1002/hlca.202000017
Yoshino T, Ng F, Danishefsky SJ (2006) A total synthesis of xestodecalactone A and proof of its absolute stereochemistry: interesting observations on dienophilic control with 1,3-disubstituted nonequivalent allenes. J Am Chem Soc 128:14185–14191. https://doi.org/10.1021/ja064270e
Zorrilla JG, Cala A, Rial C, Mejías FJ, Molinillo JMG, Varela RM, Macías FA (2020) Synthesis of active strigolactone analogs based on eudesmane- and guaiane-type sesquiterpene lactones. J Agric Food Chem 68:9636–9645. https://doi.org/10.1021/acs.jafc.0c02361
Zwanenburg B, Mwakaboko AS, Reizelman A, Anilkumar G, Sethumadhavan D (2009) Structure and function of natural and synthetic signalling molecules in parasitic weed germination. Pest Manag Sci 65:478–491. https://doi.org/10.1002/ps.1706
Zwanenburg B, Ćavar Zeljković S, Pospíšil T (2016) Synthesis of strigolactones, a strategic account. Pest Manag Sci 72:15–29. https://doi.org/10.1002/ps.4105
Funding
This work was supported by the “Ministerio de Economía, Industria y Competitividad” (MINEICO), Spain, Project AGL2017-88–083-R.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflict of interest or competing financial interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zorrilla, J.G., Rial, C., Varela, R.M. et al. Strategies for the synthesis of canonical, non-canonical and analogues of strigolactones, and evaluation of their parasitic weed germination activity. Phytochem Rev 21, 1627–1659 (2022). https://doi.org/10.1007/s11101-022-09801-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11101-022-09801-8