
Abstract
Number densities of oxygen atoms, nO, in Ar-O2 mixtures with small initial O2 fractions, \({x}_{{O}_{2}}\) < 1%, flowing through a dielectric-barrier discharge (DBD), are calculated using a plug-flow reactor model, presuming that dissociation and excitation of oxygen species are solely driven by energy-transfer from long-lived excited Ar species, collectively denoted as Ar*. The rate by which Ar* species are generated is calculated from the volume density of power dissipated in the DBD. To obtain extended post-discharge (PD) regions with large nO, experiments were performed with \({x}_{{O}_{2}}\) = 100 ppm. For such low O2 fractions, the time-dependence of nO in the DBD and the early PD can be calculated by a closed equation. Calculations are compared with optical emission spectroscopic (OES) results, utilizing the proportionality of O-atom emission intensity at 777.4 nm to nO. O-atom densities in the PD are made accessible to OES using a tandem setup with a second DBD as sensing discharge. Model testing by experiment is based on the functional dependence of nO on DBD-residence time and PD-delay time, respectively. Wall losses of O atoms in asymmetrical DBD reactors are calculated by an alternative to Chantry’s equation. The agreement between O-atom densities attained at the DBD exit and experimental results is generally good while the speed of rise of nO in the discharge is overestimated, due to the assumption of a constant wall-loss frequency, kW. Compared with literature data, kW is orders of magnitude higher in the DBD and at least one order of magnitude lower in the PD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The conspicuous bright afterglows of electrical discharges in molecular nitrogen (“active nitrogen” [1]) have been studied, since the first scientific paper appeared in 1865 [2], in thousands of experimental and theoretical investigations until today. In contrast, “active oxygen” has found much less interest until recently, in parts due to the lack of strong optical emission. The large recombination energy of N atoms (9.75 eV) results in a variety of excited states, some of which are strongly emissive, while there are no allowed transitions from excited O2 molecules resulting from recombination of O atoms (recombination energy 5.1 eV) [3]. For this reason, the application of optical emission spectroscopy (OES) for the determination of atom concentrations in post-discharge regions of O-atoms-producing discharges from the emission of excited oxygen species has not become a standard method. An alternative method to measure number densities of oxygen atoms, titration with nitrogen dioxide, NO2, which has frequently been applied at low pressures (typically below 1 mbar) is no longer applicable beyond about 10 mbar, due to the difficulty of rapid mixing of the gases [4].
In recent years several optical methods have been developed to measure densities of ground-state oxygen atoms. Aside from absorption measurements in the vacuum-ultraviolet (VUV) wavelength region, using the transition O(2p4 3P → 2p33s 3S) at 130 nm (see references in Ref. [5]), fluorescence from the O(3s 3S) state in the VUV [5] or more often from the O(3p 3P) state in the near infrared region [6,7,8,9,10], excited by absorption of two UV photons (two-photon absorption laser-induced fluorescence, TALIF), has frequently been utilized for measurements on various kinds of discharges and flames [11]. However, the equipment required for such studies is quite costly and the calibration, generally by adding known quantities of suitable noble gases, is not a trivial task.
The experiments reported here emerged within the context of studies of metal oxidation by O2-containing gas mixtures in dielectric-barrier discharges (DBDs) and post-discharges thereof,Footnote 1 on which we will report separately. In these studies, we generally use O2 fractions well below 1%, in order to achieve a large O-atom number density and a low recombination rate in the post-discharge, i.e., a large usable length downstream from a discharge were substrates can be placed. The ability to provide a continuous variation of O-atom number density along a substrate region exposed to the discharge effluent is of high interest for the preparation of “continuous libraries” of oxidized surfaces for high-throughput combinatorial investigations [12].
For such investigations the knowledge of absolute O-atom number densities in the DBDs and the DB-PDs are highly desirable. In the present contribution we report on a simplified chemical-kinetic model in which the discharge volume is regarded as a uniform source of long-lived excited argon species, Ar*, generated at a rate gAr* (number per volume and time).
We also present a method to measure atom number densities in DB-PDs, utilizing a tandem-DBD configuration with a second discharge to sample the optical emission from excited atoms. In case of atmospheric-pressure discharges producing oxygen atoms in a large excess of argon, the sensing discharge serves to excite ground-state O atoms by energy transfer from Ar* species, namely Ar atoms in metastable (1s5 and 1s3)Footnote 2 and resonant (1s4 and 1s2) states with excitation energies between 11.55 and 11.83 eV [13], as well as excited argon dimers (excimers), Ar2(3Σu+) (9.8 eV [14]), formed from Ar(1s5) atoms in three-body reactions with two ground-state Ar atoms. Singlet Ar excimers, Ar2(1Σu+), formed from Ar(1s4) atoms, play virtually no role in this respect, due to their small radiative lifetime. In contrast to the situation at low pressures, the fraction of energy transfer reactions with Ar(1s5) atoms is relatively unimportant, due to the rapid reaction of the latter to Ar2(3Σu+) [15]. Energy transfer from the listed species to molecular oxygen and ozone (Ediss = 1.13 eV [16]), respectively, results in dissociation. By energy transfer from metastable Ar(1s5) and Ar(1s3), probably also from Ar(1s4) and possibly from Ar(1s2), oxygen atoms are excited to the state O(3p 3P) [17], responsible for the emission at 844.6 nm (3 s 3S0 ← 3p 3P) and, via collisional deactivation to O(3p 5P), for the emission at 777.4 nm (3s 5S0 ← 3p 5P) [18]. With an energy of 10.99 eV [13], O(3p 3P) cannot be produced by dissociative excitation from O2, O2(a), or O3 upon energy transfer from Ar(1s2…5) states. Therefore, the emissions at 777.4 and 844.6 nm do not depend on the number densities of these molecular oxygen species. Energy transfer from argon excimers (9.8 eV) to oxygen atoms directly populates O(3s 3S0) (9.52 eV) but it does not contribute to emission at 777.4 or 844.6 nm [19].
In the following section we first report experimental details. In Section "Calculation of oxygen-atom number density profiles along the gas-flow direction", a plug-flow model of a DBD in Ar-O2 with a small O2 content, \({x}_{{O}_{2}}\) ≤ 1%, is presented. For the very small O2 fractions used in this study, \({x}_{{O}_{2}}\) = 0.01%, an analytical equation for the spatial number density profile of oxygen atoms is derived, covering the discharge and the early post-discharge region. This equation enables an automatic non-linear curve fitting of experimental results. To calculate the wall-loss frequency for a parallel-plate DBD and afterglow reactor bounded by two different materials with significantly different loss coefficients we introduce an alternative to Chantry’s formulation [20] (3.2). Numerical and analytical solutions are compared in Section "Comparison of numerical and analytical calculations". In Section "Optical emission from excited O atoms as a measure of ground-state O number density", the use of the 777 nm OES line for the determination of nO is justified. Then results of experiments are reported and discussed. The final section summarizes conclusions and gives an outlook.
Experimental Section
Materials
Process gases were mixed from argon and oxygen with 6.0 and 5.0 purity, respectively, obtained from Linde AG, Germany. Borosilicate glass (Borofloat®) was from Schott AG, Germany.
Instrumentation
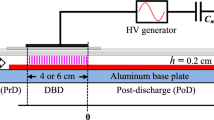
Experiments reported in this paper were performed at ambient pressure using two types of DBD configurations: Parallel-plate setups with an asymmetric electrode arrangement from a grounded aluminum base plate and a 0.2-cm thick borosilicate glass dielectric (configuration “AG”), as shown schematically in Fig. 1, as well as reactors with two dielectrics from borosilicate glass (configuration “GG”), obtained by placing glass strips on the Al base plate. The basic construction is a flat flow channel with inner dimensions of 18 cm length and 2.05 cm width (to accommodate 2.0-cm-wide samples in oxidation experiments to be reported separately). In the AG setups, gas-gap heights, h, of 0.11 or 0.25 cm are defined by side walls from borosilicate glass strips with the corresponding thickness placed between the Al base plate and the glass top plate. With a 0.25-cm high side wall and 0.175- or 0.11-cm thick and 2-cm wide glass strips placed on the metal base plates, GG configurations with h = 0.075 or 0.14 cm were obtained. Symbols like “AG-0.25” to denote a configuration, e.g. in the first column of Table 1, are also used to refer to series of measurements run with this setup. It is important to note that an aluminum oxide film is present on the aluminum base plate. This oxide film has a thickness of at least 3 nm and reaches 5–10 nm thickness after longer use of the reactor in Ar-O2 mixtures, as seen in oxidation experiments to be reported separately.

Scheme of the DBD-reactor with an aluminum base plate as ground electrode and borosilicate dielectric as top boundary of the flow channel. The sensing DBD may be run simultaneously with the main discharge to quantify the oxygen-atom number density at different positions in the post-discharge of the main DBD. By inserting 0.11-cm- or 0.175-cm-thick glass strips on the bottom of the reactor channel with a gap of h = 0.25 cm, the configuration was changed from aluminum-glass (“AG”) to glass-glass (“GG”) with 0.14 and 0.075 cm gap widths, respectively
To power the main DBD shown in Fig. 1, a high-voltage generator is connected to a rectangular metal electrode with a length LM (in gas-flow direction) of 1 or 4 cm, and 2.05 cm width, glued onto a rectangular 0.07-cm-thick piece of borosilicate glass as a carrier that can be slid on the borosilicate top plate of the flow reactor in order to vary the distance Δ between the discharges, see below.
The first 5 cm of the flat channel have the purpose to provide a laminar gas flow; the afterglow or post-discharge region downstream of the DBD with length LM is used for studies of the interaction of oxygen atoms with surfaces, for example the oxidation of metal films. The average residence time tres of an Ar-O2 gas mixture in the main DBD is controlled by the average gas flow velocity, vav, and LM: tres = LM/vav. In order to study the number density of atomic oxygen in the post-discharge region a second DBD (“sensing DBD”) is generated at a distance Δ from the main DBD, using another movable metal electrode, with a length, LS, of 1 cm, glued onto a borosilicate carrier plate of 0.07 cm thickness, attached to the 0.2-cm-thick borosilicate wall. The afterglow delay time is controlled by vav and Δ: tdel = Δ /vav. For the present studies, we used the same high-voltage (HV) generator to power the main DBD and the sensing DBD, but, in order to decrease the power density in the sensing DBD relative to the main discharge, a 0.3 MΩ resistor was installed between the voltage source and the sensing-DBD electrode.
A model 7020 UZ generator from SOFTAL electronics GmbH (Hamburg, Germany) was used to power the DBDs. For electrical characterization of the discharge the sinusoidal voltage applied to the discharge arrangement, Ua(t), as well as the transferred charge q(t) were measured using a high-voltage probe (Tektronix P6015A) and a series capacitor CM (560 pF), respectively. An oscilloscope (LeCroy WR604Zi) was used to monitor Ua(t) and the voltage drop UC(t) at the capacitor to calculate the dissipated power P. The measured frequency was between 15.8 and 16.2 kHz. The power density, P/V, (plasma volume V = 2.05 cm × LM × h) was applied to calculate the generation rate of Ar* species, gAr*, using results of a calculation with Bolsig + [21]Footnote 3 for a reduced field typical for a filamentary discharge in argon [22], see Section "Calculation of oxygen-atom number density profiles along the gas-flow direction".
Optical emission spectra were recorded using a QEPro spectrometer from Ocean Insights (Duiven, The Netherlands), equipped with a 10 µm slit and a grating with 600 lines/mm, covering the spectral range from 530 to 900 nm with an optical resolution of 0.4 nm. As shown in Fig. 1, spectra were measured (i) at the trailing edge of the main-DBD electrode to study the dependence of the emission from excited O atoms on tres or (ii) at the leading edge of the sensing-DBD electrode to investigate the emission as a function of tdel. An optical fiber from Ocean Optics, type P400-2-SR, was used for the measurements. It was oriented at an angle of 30° relative to the normal with a distance of 5 mm from the discharge edge. The acquisition time for a single scan was between 0.3 and 1 s, 10 scans were averaged. According to general experience with OES on DBDs the resulting intensities (band areas) may vary, under nominally identical conditions, within about a range of ± 5%. Spectral line intensities reported here were obtained by importing the spectra into OriginPro (Additive, Friedrichsdorf/Ts., Germany) and numerical integration. The 1130-nm emission from atomic oxygen (see Section "Optical emission from excited O atoms as a measure of ground-state O number density") was recorded under typical conditions during a single test measurement using an OceanFX spectrometer (10-µm slit, grating with 600 lines/mm), kindly provided by Ocean Insights.
Experiments
Experimental parameters used for the series measurements reported in Section "Model testing using residence-time variations" (variation of residence time tres in the main DBD) and 5.3 (variation of the delay time tdel in the DB-PD) are summarized in Table 1:
Ua,0 is the amplitude of the applied voltage, (P/V)M the power density in the main discharge. For each configuration denoted in the first column, the same primary voltage and frequency settings at the HV generator were applied for series with varying tres and series with varying tdel, attempting to achieve similar power densities in the main discharge, regardless of its length, the gas velocity and the presence of the sensing discharge. The electrical parameters in Table 1 are averages obtained from data measured and stored during the OES measurements, but evaluated after the experiments. That is, why the numbers in the 2nd and the 4th columns as well as the 3rd and the 5th column are not the same, as they should be ideally. Power densities in the main DBD were measured with the sensing DBD off. Power densities in the sensing DBD, (P/V)S, were not measured separately.
Numerical and Analytical Results
Calculation of Oxygen-Atom Number Density Profiles Along the Gas-Flow Direction
Outline of the Applied Model
Oxygen-atom number density profiles nO(x) in the DBD and downstream of the discharge were calculated using a plug flow reactor (PFR) model, based on spatially zero-dimensional kinetics of the relevant chemical reactions. We assume that the DBD represents a uniform source of long-lived excited argon species Ar* which are “driving” the chemical reactions in the gas mixture, flowing through the DBD with a gas velocity independent of the height coordinate (z). Diffusion along the gas flow direction (x) is neglected. Species number densities n considered in this section are averages over z, while the z-dependence itself is considered separately in Section "O-atom number density profiles nO(z) across the gas-flow direction, wall losses" in which wall losses in asymmetric DBDs with AG configuration are considered.
In Ar-O2 mixtures with small fractions of molecular oxygen (\({x}_{{O}_{2}}\) ≤ 1%) the dissociation of O2 by electron impact via reactions RI and RII [23] is negligible compared with dissociation via energy transfer from Ar atoms in any of the long-lived Ar(1si) states (2 < i < 5) or from Ar2(3Σu+) excimers, i.e. (RIII) followed by (RIV):
“Ar*” in (RIII) and (RIV) represents any element of the set containing Ar atoms in one of the four 1si states as well as triplet Ar2 excimers (later abbreviated as “Ar2*”), indirectly formed mainly via three-body collisions of Ar(1s5) with two Ar atoms. With an effective reduced electric field E/n of 200 Td, taken as characteristic of a filamentary DBD in Ar [22], a calculation with Bolsig + [21] gives rate coefficients kRI, kRII, and kRIII of 0.14, 0.18, and 0.78 × 108 cm3 s−1, respectively. With an energy loss fraction of 58%, reaction RIII consumes a major part of the dissipated electrical energy; using a composite decay frequency for Ar* of 2.4 × 105 s−1 [15] and a rate coefficient of 2.1 × 10–10 cm3 s−1 for (RIV) [24], it becomes evident that O2 dissociation via generation of Ar* followed by reaction (RIV) is a much more efficient way to generate atomic oxygen, in Ar-O2 mixtures with small O2 fractions, than via reactions RI and RII. In 52% (2%) of reactions between O2 and a 1:9 mixture of metastable Ar(1s3) and Ar(1s5) one O atom is formed in the excited 1D (1S) state, along with a ground-state 3P atom, according to Balamuta and Golde [25]. In a more recent paper by Fiebrandt et al. [26] the same percentages were assumed to hold for Ar(1s2) and Ar(1s4), too. For low-pressure surface wave discharges in Ar-O2 mixtures, to take another example, dissociation by electron collision and dissociation by energy transfer from Ar* are about equally frequent at a molecular-oxygen fraction \({x}_{{O}_{2}}\) of 10% [27]. Therefore, and in view of the kinetic data specified above in this section, the direct electron-collision contributions to the dissociation of O2 can be neglected in Ar-O2 mixtures with less than about 1% O2 without introducing an error beyond a few percent.
Model calculations were performed with a set of reactions including, in addition to Ar and Ar*, three ground-state oxygen species: Oxygen atoms in the ground state, O(2p4 3P) (in the following usually abbreviated as “O”), molecular oxygen (dioxygen), O2, and ozone, O3. In addition, excited oxygen atoms in the O(2p4 1D) state, the set of atoms in O(3s 3S0), O(3p 3P) or O(4s 5S0) states (summarized as “O*”), and excited metastable O2 molecules, O2(a 1Δg) and O2(b 1Σg+), are included. (See the diagram with excited states of atomic oxygen in the range from 9 to 12.5 eV in Fig. 5, Section "Optical emission from excited O atoms as a measure of ground-state O number density"). In the following, abbreviated names O(1D), O2(a), and O2(b) are applied in this section while the names of individual atoms in the O* set are retained in Section "Optical emission from excited O atoms as a measure of ground-state O number density" in order to preclude confusion. The reactions considered in the present work and kinetic data are shown in Table 2.
The model was kept as simple as possible without losing predictive value. At low molecular-oxygen fractions (\({x}_{{O}_{2}}\) ≤ 1%), only Ar must be considered as a (third) partner in collisional deactivation and recombination reactions. As defined above, “Ar*” denotes any Ar species with enough lifetime to play a role in reactions R3…R10, including Ar2 triplet excimers. Regarding formation of the latter species, the equation for R1 is a short-hand notation. From the power density, P/V, the generation rate of Ar* species, gAr*, was calculated by multiplication with 3.15 × 1017 J−1 (= 1 per 19.8 eV). This figure was determined from the energy loss fraction of 0.58 as calculated using Bolsig + [21] for a reduced field of 200 Td [22].
In view of the substantial share of O atoms among oxygen species obtained by dissociation it is required to include quenching by O atoms as a sink for Ar* in the model. So far, only the reaction between O atoms and a mixture of metastable Ar(1s5) and Ar(1s3) has been studied in detail, to the author’s knowledge. This process results mainly in excitation of O to O(3p 3P) [17]. For R3 in Table 2 it is assumed that the reported rate coefficient can also be applied, without introducing a too large error, for reactions of O with Ar2* as well as with Ar(1s4) and Ar(1s2), resulting in formation of O(4s 5S0) and O(3s 3S0), respectively. The three excited states are summarized by the symbol “O*”. As the only sinks for O*, quenching by collisions with Ar atoms (R30) and radiative decay (R31) are assumed, with a rate coefficient and frequency, respectively, as reported for O(3p 3P) in argon [43]. For the degree of dissociation resulting from the present model, the correct choice of this figure is not important. More details about states of oxygen atoms excited by Ar* are given in Section "Optical emission from excited O atoms as a measure of ground-state O number density".
For the important dissociation reactions of O2, O2(a), O2(b), and O3, (R4 to R10 in Table 2), a rate coefficient of 2.1 × 10–10 cm3s−1 was adopted, in agreement with other publications [27, 29, 30]. Originally this figure was reported by Velazco et al. [24] for the quenching of Ar atoms in the metastable Ar(1s5) state (= Ar(3P2)) by O2 at 300 K. In low-pressure plasmas, Ar(1s5) is generally the predominant Ar* species while in Ar-rich plasmas at atmospheric pressure its share of Ar* is the smallest, due to the already mentioned three-body reactions with ground-state O atoms to the triplet excimer Ar2*. Rate coefficients for quenching of other Ar(1s) species and of Ar2* by O2 are not very different from 2.1 × 10–10 cm3s−1: In the cited paper one finds values of 2.5, 2.4, and 3.1 × 10–10 cm3s−1 for Ar(1s4), Ar(1s3), and Ar(1s2), respectively. For the reaction of Ar2* with O2, Oka et al. report a value of 2.6 × 10–10 cm3s−1 [31]. The formation of O(2p4 1S) in 2% of reactions between Ar* and O2 [25] was neglected. In the reactions of Ar* with O2(a) and O2(b), respectively, the formation of O(3P) and O(1D) atoms is allowed for in Table 2, R6 to R9. However, due to the lack of kinetic data, the same branching ratios were assumed as for O2.
As a result of energy transfer from Ar* to O3 (R10), dissociation into ground-state O2 and an O(1D) atom is assumed, following Gentile [32]. In this context it is interesting to consider product channels of O3 photolysis at 157.6 nm (7.8 eV) where the dissociation into three O atoms is about as frequent as the formation of (singlet) O2 and O(1D) [33]. In view of the about 4 eV higher energy of Ar* it would appear reasonable to assume that complete dissociation into atoms prevails under our experimental conditions. For the small O2 fraction used in the present experiments (100 ppm), however, the 3-atom channel gives just about 3% more O atoms in the discharge than the 1-atom alternative, if the steady state with a constant Ar* generation rate of 1018 cm−3 s−1 is considered, see below. With higher O2 fractions, however, this channel would result in increasingly larger overall O yields than the 1-atom channel.
Another still open question is the product branching following the recombination of two ground-state oxygen atoms. In mechanisms applied in published work the formation of O2, O2(a), and O2(b) from two O atoms with O2 as a third collision partner was in the ratios of 0.50:0.33:0.17 [27, 34], or 0.88:0.06:0.06 [30], or 0.88:0.10:0.02 [35]. In Ref. [36] a branching into the “Herzberg states” O2(c, A, A’) and into O2(a), respectively, in the ratio 0.25:0.75 was deduced from experiments. In the present work a factor f in R11 of 0.33 is used, a value in agreement with the first two references and between the latter two. The choice of f has a relatively small effect on steady-state O atom densities calculated with parameters typical in the present context: With f = 0 and 1, respectively, these densities are decreased by 1.3% and increased by 3.3%, respectively, relative to the result with f = 0.33. O2(a) densities themselves increase about linearly when f is increased from 0.1 to 1. Any formation of O2(b) in R11 is neglected because its share in the more recently published models was only 2 or even 0%.
For the calculation of k14, the wall-loss frequency of O atoms, an equation was used which enables taking into account different loss probabilities γ on the two major walls of the flat channel, see Section "O-atom number density profiles nO(z) across the gas-flow direction, wall losses". In addition to dissociation by Ar* in the discharge, reaction with O atoms (R13), thermal decomposition (R15), and reaction with O2(a) (R25) are major sinks of ozone. Note that at room temperature, however, the contributions by R15 are negligible compared with others. Several reactions of O(1D) are included in R16 to R21 because O(1D) is a precursor of metastable O2(a) and O2(b) (R17, R18, R21). Under the present conditions, however, O(1D) number densities are very small, typically 109 cm−3, owing to rapid quenching by Ar (R16), and O2(a) is more efficiently formed via R11, unless f = 0. For O2(b) only collisional deactivation by Ar (R26) and by O3 (R27) are included, because of the large number density of Ar and the large value of k27. Quenching by O2 and by O atoms is much slower, with rate coefficients of 4.1 × 10–17 cm3 s−1 and 8.0 × 10–14 cm3 s−1, respectively [30], and therefore not taken into account. In R30 and R31, O(3p 3P) is taken as representative of excited states of O atoms, O*, which are formed by energy transfer from Ar* in R3. The decay of O(3p 3P) is dominated by the quenching reaction with Ar (R30), being, under the present conditions, about an order of magnitude faster than the radiative deactivation (R31).
The system of coupled rate equations derived from the reactions R1 to R31 was solved using the CVODE option of the freely available XPP softwareFootnote 4 .
Analytical Equation for n O(t)
A consideration of the rate coefficients for reactions involving Ar* and oxygen species, respectively, shows that it should be possible, as a good approximation, to separate generation and reactions of Ar* (R1…R10) from the major reactions of O atoms with O atoms (recombination), molecular oxygen, O2, and ozone, O3, (R11…R13) as well as the diffusion-controlled wall loss of O atoms, assumed here to be a first-order reaction (R14). Therefore, an approach using a quasi-steady-state approximation (QSSA) is obvious to calculate nAr* as a “fast variable”, rapidly adapting to the number densities of major oxygen species [44].
Analytical kinetic modelling of O2 dissociation and recombination is rendered more difficult, compared with other diatomic molecular gases, by the existence of a metastable excited state of substantial lifetime, singlet O2(a), as a major product of the recombination of O atoms, and ozone, O3, as product of the reaction of O atoms with O2 (with Ar as a third partner, R12). It can be shown, by considering the steady state for O3 in the discharge, that at sufficiently low input fractions of O2, \({x}_{{O}_{2}}\), the fraction of O3 formed in the discharge stays substantially below that of O atoms, due to its dissociation by Ar* (R10).
Therefore we restrict the present analytical treatment of the dissociation process to O2 fractions in the order of \({x}_{{O}_{2}}\) = 0.01%, whereas the key assumption of the model outlined above, the negligibility of processes induced by direct collision of electrons with oxygen species, holds up to about \({x}_{{O}_{2}}\) = 1%. However, in the present studies the fraction of molecular oxygen was always 0.01% because this choice provides substantial number densities of O atoms over several centimeters of the post-discharge region at moderate volume flows of the gas mixture.
Using the symbol ki,j for the sum ki + kj and making use of the presumed equalities of rate coefficients k4 = k6 = k8, k5 = k7 = k9, and k10 = k4,5, respectively, the number density of Ar* in the quasi-steady state, resulting from rapid formation with the rate gAr* and rapid quenching by O, O2, O2(a), O2(b), and O3 can be written:
S2 is the starting number density of O2 at t = 0. It is calculated from the input molecular-oxygen fraction \({x}_{{O}_{2}}\) using the ideal gas law, S2 = \({x}_{{O}_{2}}\) × NA × p/(RT). The simple final expression in Eq. (1) is obtained by taking into account the conservation of the total number density of oxygen atoms contained in different oxygen species, 2 × S2, and replacing, in the initial expression, \({n}_{{O}_{3}}\) with \({n}_{{O}_{3}}\) × 3/2. The error resulting by this approximation, a lowering of nAr*(t), is generally below 3%, due to the smallness of \({n}_{{O}_{3}}\) in the discharge, unless (P/V)M < 0.8 Wcm−3, the smallest power density used in this study. The resulting error in steady-state oxygen-atom densities is even smaller, typically below 2%.
The steady-state expression for nAr*(t) is inserted into the rate equation for nO(t):
The intermediate formation of O(1D) by R5 is neglected in the present analysis, i.e., R5, R7, R9, and R10 are assumed to result directly in formation of ground-state O atoms only. This assumption is reasonable because, in atmospheric-pressure argon, O(1D) is rapidly quenched to the ground state (R16), resulting in average number densities typically below 1010 cm−3. Therefore, O(1D) plays virtually no role in formation of O2(a), being much more efficient by R11 than by R17 and R21.
The non-linear dependence of nAr* on nO, see Eq. (1), generally precludes a straightforward closed solution of Eq. (2). This dependence can, however, be neglected in a first approximation, if the degree of dissociation keeps small or, as it is the case here, if the rate coefficient for quenching of Ar* by the atoms is about half as large as that for the molecules: k3 ≈ k4,5/2: Therefore, and due to the presence of the term k2, accounting for losses by radiation, the denominator in Eq. (1) decreases, upon full dissociation of O2, only from 7.7 × 105 s−1 to 6.2 × 105 s−1, i.e., by 20%, if S2 = 2.5 × 1015 cm−3 (\({x}_{{O}_{2}}\) = 100 ppm). This systematic error can be reduced further to about one third by appropriately transforming the denominator and using the well-known Taylor expansion of the function f(x) = (1 + x)−1 into a binomial series, f(x) = 1-x + x2 + … Here, x ≡ (k3-k4,5/2) × n0(t)/(k2 + k4,5 × S2). The result was used up to the quadratic term for the first term in Eq. (3) while only the linear term can be used in the product k4,5 × nAr*, in order to avoid cubic terms.
The resulting differential equation for the time-dependence of the average number density of O atoms in a differential control volume travelling through the reactor channel is
Its solution is [45]:
At steady state, the oxygen-atom number density becomes \(n_{O} (\infty ) = \left( {w - b} \right)/\left( {2c} \right)\).
O-Atom Number Density Profiles n O(z) Across the Gas-Flow Direction, Wall Losses
The wall-loss frequency kW = k14 (see Table 2) is calculated from the first non-zero positive root, α1, of the equation
γj (j = 1 or 2) are the loss coefficients for the major walls in a flat reactor with rectangular cross section (height h < < length l and width w), R and M are the universal gas constant and the atomic mass of the considered species, respectively. vth is the average thermal velocity, vth = [8RT/(πM)]1/2. In the present case, for T = 293 K, we have vth = 6.23 × 104 cm/s, D = DO,Ar = 0.38 cm2/s [46] and M = MO = 16 g/mol. With γ1 = γ2 = 2 × 10–4 for a symmetric reactor with two dielectrics from a borosilicate glass [47], g1 = g2 = 8.20 cm−1 while γ1 = 0.05 (g1 = 2100 cm−1) is applied for the Al2O3 surface in a DBD reactor with one borosilicate dielectric and a metallic electrode, here being the aluminum base plate, carrying an ultrathin passivating oxide film [48].
Equation (5) follows from the solution of the diffusion equation for nO(z,t) in a flat channel with the height variable z, in analogy to heat conduction in solids bounded by two parallel planes [49]. Including a first-order decay reaction with the rate coefficient kr, the general solution for the concentration profile is
where the αi are calculated from Eq. (5). The coefficients Ai can be determined from the number density distribution at t = 0 by series expansion or by a non-linear fit procedure.
Equation (5) is an alternative to the frequently used approximation by Chantry for the wall-loss time, τW = 1/kW, calculated as the sum of surface-loss and diffusion-loss times, τS and τD [20] (see also [50]). For a flat channel with distance h between the two main walls, Chantry’s equation reads:
Note that the correction factor (1 − γ/2) introduced by Motz and Wise [51] is an approximation; an improved correction function, depending on the wall-loss probability or sticking coefficient γ and the reactant mass fraction near the surface, can be found in Ref. [52]. It should be emphasized that, strictly speaking, Eq. (7) is not valid for the calculation of wall losses in situations where the volume reaction is not purely first order, such as the recombination reactions in post-discharges.
If γ1 = γ2, one can compare results of Eqs. (5) and (7) with each other, see Fig. 2.

In the “knee” of the blue curve at γ1 = γ2 = 7.6 × 10–4, where the deviation between results from the two equations is the largest, Chantry’s approximate equation results in a wall loss time which is about 5% larger than the exact value. However, the main advantage of the transcendental Eq. (5) over its alternative, Eq. (7), is that it covers cases with unequal loss coefficients on the two main walls. Today it can be solved easily and with arbitrary accuracy with the aid of a suitable graphing software, by looking up the intersections of the graphs of its l.h.s. and its r.h.s. Alternatively a mathematics software or a web sourceFootnote 5 may be used. In the present case the first three roots for h = 0.11 cm, γ1 = 0.05, and γ2 = 2 × 10–4, as an example, are α1 = 18.1 cm−1, α2 = 44.3 cm−1, and α3 = 72.1 cm−1. The corresponding loss frequencies are calculated from the equation \(k_{W,i} = D\;\alpha_{i}^{2}\) (Eq. 6). For the fundamental diffusion mode with i = 1 the result is kW = kW,1 = 124 s−1; i. e., the wall loss time is 8.1 ms. The diffusion mode with i = 2 decays within 1.3 ms. For h = 0.25 cm, α1 = 9.2 cm−1 and kW = 32.2 s−1.
In cases where either (i) g1g2 > > α12 or (ii) g1g2 < < α12, the r.h.s. of Eq. (5) can be approximated and two different averages γav can be calculated. As (i) α12/(g1g2) or (ii) g1g2/α12 goes to zero, these averages approach an “effective” αeff, i. e., the loss coefficient which would be required on both walls of a symmetrical reactor in order to give the same wall loss time as in the asymmetric case:
γavh and γava are the harmonic and arithmetic averages of γ1 and γ2, respectively. The approximation used in Eq. (8) can be applied for relatively large products γ1γ2: With h = 0.11 cm, γ1 = γ2 = 0.05 for example (two alumina dielectrics), the approximation results in an error of only 0.02%, if the harmonic mean is used. In Table 3, values of kW are compiled for a number of configurations with practical relevance at atmospheric pressure, with small h (0.075 up to 0.25 cm). Entries #4, 7, 10, 15, signified by italic, refer to the given situation in the DBD in experiments with the four different configurations run in this publication.
For low-pressure plasmas, on the other hand, for example in a situation at 1 mbar with h = 5 cm, DO,Ar = 380 cm2/s, γ1 = 5 × 10–4 and γ2 = 1 × 10–4 (g1 = 0.0205, g2 = 0.0041) the first positive root of Eq. (5) is α1 = 0.069 while α1 = 0.070 is calculated with the arithmetic mean value γava = 3 × 10–4 (Eq. 9).
Comparison of Numerical and Analytical Calculations
As shown below (Fig. 3), the solution of Eq. (4) for a set of parameters, typical of the experiments reported here, agrees within a few percent with a numerical calculation, including the full set of reactions in Table 2.

Comparison of Ar* and O number densities as calculated from Eqs. (1) and (4) (blue lines), and number densities of O*, O3, and O2(a) (red lines) obtained by assuming validity of QSSA, Eqs. (10), (11), and (12), respectively, with results of numerical calculations (dot-dashed black lines). The parameter set is typical for the experimental conditions
Equation (4) is used below for nonlinear curve-fitting of the OES measurements. Once the temporal dependence of nAr*(t) and nO(t) is known, one can attempt to calculate number densities of excited O*, O2(a) and O3, using steady-state assumption for these species: The red curve for the excited O atoms, O*, shown in Fig. 3 agrees very well with the numerical result, as expected in view of short time scales involved, cp. Equation (10).
The two interdependent Eqs. (11) and (12) for O2(a) and O3 are calculated using only leading reactions of these species, i.e., neglecting R22, R23, and R24 in Eq. (11) and R15, R19, R20, R21, and R27 in Eq. (12). They can be used to calculate \({n}_{{O}_{2}{(a)}}\) and \({n}_{{O}_{3}}\) separately as functions of nAr* and nO. The comparison with the numerical calculation reveals that for these two species the steady state number densities are lower than the numerical results by factors approaching 1.1 and 1.3, respectively, for long residence times. At least a rough estimate of these number densities seems possible but this point has not been investigated in greater depth, due to the availability of a numerical alternative. Note that the number density of Ar* is virtually independent of the degree of dissociation, an important prerequisite in the argument outlined in Section "Optical emission from excited O atoms as a measure of ground-state O number density".
n O(t) in the Post-discharge, Approximation for Small Delay Times
For the special case, that gAr* = 0 (w = b), Eq. (4) represents the decrease of nO(t) in the post-discharge region, due to first- and second-order volume and first-order wall losses. It can be rewritten as
S1 is the number density at the downstream end of the discharge, where the afterglow delay time is zero, S1 ≡ nO(tdel = 0). For small delay times tdel, approximate expressions may be used which are helpful for the evaluation of experimental data. Figure 4 shows graphs of nO(tdel)/S1 as well as of the inverse, S1/nO(tdel), together with linear approximations, which can be used during the first few milliseconds, nO(tdel)/S1 = 1–(β + κS1) × tdel, and S1/nO(tdel) = 1 + (β + κS1) × tdel. The latter approximation works slightly better, up to about 3–4 ms under typical conditions. Please note, however, that Eq. (4) was based on the neglect of reactions with O3. In the DBD region this approximation can be justified by its relatively small number densities, due to dissociation by Ar* (R10). In the DB-PD, the validity of Eq. (13) is generally limited to the early post-discharge region, under the conditions used in the present paper typically up to tdel = 5 or 10 ms.

Graphs of nO(tdel)/S1 and of S1/nO(tdel), calculated from Eq. (13) with typical values of β, κ and S1: 80 cm−1, 5 × 10–14 cm3 s−1, and 1 × 1015 cm−3, respectively (black and red bold curve, respectively). The thin straight lines are linear approximations, valid for small tdel, see text (Color figure online)
Optical Emission from Excited O Atoms as a Measure of Ground-State O Number Density
In this section we present an argument why the emission from excited O atoms, O(3p 5P), at 777.4 nm can be used to derive a relative measure of the number density, nO, of ground-state oxygen atoms, O(2p4 3P). Figure 5 shows a diagram with energies of long-lived states of argon and of oxygen, respectively, in the energy range from 9.0 to 12.5 eV [13, 18]. Optical emissions at 777.4 and at 844.6 nm from discharges in oxygen-containing gases have their origins in the O(3p 5P) and O(3p 3P) state, respectively. In the frequently applied actinometric determination of O-atom densities in plasmas, where trace concentrations of Ar or other noble gases are added to an oxygen-containing gas under study (see, e.g., Ref. [53] and citations therein), excitation processes are, in general, predominantly due to direct electron impact. For Ar-O2 mixtures with very small fractions of O2 at atmospheric pressure, like in the present study, excited states of oxygen species are virtually exclusively populated by energy transfer from long-lived Ar(1si) states and Ar2 excimers, Ar2*, for similar reasons as outlined above for the dissociation of O2. Possible energy pathways from Ar* to O are indicated by the blue arrows in Fig. 5. Among the states of O shown, Ar2* populates O(3s 3S0), resulting in emission of three VUV lines between 130 and 131 nm [19]. As noted already in Section "Introduction", Ar* species cannot generate O(3p 3P) by dissociative excitation of O2, O2(a), or O3.

Under the conditions used here, for an undissociated mixture of Ar with 100 ppm O2, Ar2* is the species with the highest time- and space-averaged fraction within the Ar* set, with a share of about 0.35, while the fraction of Ar(1s2) is 0.25. These figures are results of calculations, using a simple analytical model which was developed for the plasmachemical kinetics of Ar(1si) states and Ar2 excimers in mixtures of argon with hexamethyldisiloxane, HMDSO [15]. The approach gives also reasonable results, e.g., for the fractions of the five individual Ar* species in Ar-TMS (tetramethylsilane), as seen in a comparison with published model results [54].
Using the 777.4-nm line for a relative measure of the ground-state O-atom number density was motivated by its larger intensity and better separation from neighboring Ar lines than the 844.6-nm alternative. While Ar2* is able to dissociate O2, it does not contribute to emission in the visible or near-infrared region: The emission at 844.6 nm originates from O(3p 3P) which is generated by energy transfer from metastable Ar atoms in Ar(1s5) and Ar(1s3) states [18] and presumably also from resonant Ar(1s4). The O(3p 5P) state responsible for the emission line at 777.4 nm is populated from O(3p 3P) in 28% of collisional quenching processes with Ar [55], (green arrow in Fig. 5). With this number, a radiative lifetime of 35 ns, and a quenching rate coefficient by Ar of 1.4 × 10–11 cm3 s−1 for O(3p 3P) [43], it can be seen that quenching from O(3p 3P) to O(3p 5P) is more frequent than radiative transition to O(3s 3S0). In addition to processes driven by species Ar(1s3,4,5), near-resonant energy transfer from Ar(1s2) to O(4s 5S0) and radiative cascade to O(3p 5P) appears as a plausible, additional pathway. In fact, the emission at 1130 nm from O(4s 5S0) has been observed in one preliminary experiment, with a different spectrometer as normally used, under typical conditions of this study. Attempts to relate the intensity of this emission to the 777-nm line were not made. Unfortunately there is a lack of data concerning energy transfer rate coefficients from the resonant Ar states to O atoms as well as of quenching and branching data for O(4s 5S0). That is, why a quantitative kinetic model of the involved processes is not possible as yet.
For the present study it is sufficient that a strictly linear relation between the 777-nm emission from O(3p 5P) and nO is maintained while the Ar-O2 mixture passes the DBD. Therefore, it is required that the fractions of Ar2* and Ar(1s2) within the set of Ar* species remain constant while the gas composition changes due to dissociation. In general, changing the number density of a quenching species changes the fractions of individual Ar* species [15]. In the present case, however, this effect is relatively small: Reducing \({x}_{{O}_{2}}\) to 50 ppm, for example, increases the excimer fraction by about 2% and decreases the fraction of Ar(1s2) by 5%. Additionally it must be noted that the dissociation replaces one O2 molecule by two O atoms with a lower quenching rate coefficient: k3 = 6.7 × 10–11 cm3s−1 for quenching by O as compared to k4 + k5 = 2.1 × 10–10 cm3s−1 for O2. Therefore, the change in nAr* upon dissociation of O2 is comparably small (see Fig. 3) and the assumption of constant fractions of the individual Ar* species is reasonable: The three pathways initiated by energy-transfer reactions of O with (i) Ar2*, not contributing to emission at 777 nm, (ii) Ar(1s2), contributing via O(4s 5S0) and (iii) Ar(1s3,4,5), contributing via O(3p 3P), stay in a fixed ratio.
To account for the linear relation between power densities and Ar* number densities it is required to divide the emission line intensity through a quantity proportional to P/V in order to arrive at a relative measure of ground-state O-atom number densities. That is why we have chosen to divide the whole spectra through the peak intensity of the neighboring Ar transition Ar(1s5 ← 2p7) at 772.4 nm before determination of the line area in order to arrive at what is named, in this paper, I777. This should eliminate effects due to variations in the geometry of the experimental arrangement and in the dissipated electrical power.
Experimental Results
Introductory Note: Absolute O-Atom Number Densities from Relative Measurements of Growth or Decay Curves
A consideration of equations for species generation and decay by reactions of first and second order, respectively, shows that in the first case the number density n(t) can be separated into a factor containing the generation rate g and/or the initial number density n0, and another, time-dependent factor, which is independent of g and n0. For a second-order reaction (or a combination of first- and second-order reactions) this separation is no longer possible: The time-dependence of n(t) now contains information about g and/or n0, see Eqs. (14) to (16) for the pure first-order case and Eqs. (17) to (18) for the pure second-order case with rate coefficients k1 and k2, respectively:
Reactions of second order frequently dominate in post-discharge regions, unless first-order wall losses are much faster. That is why, for example, an absolute quantification of atom number densities in flowing nitrogen post-discharges is possible with measurements of the delay-time dependence of lines in the nitrogen 1st Positive System, the relative spectral intensities of which are proportional to the squared atom number density, nN2 [56,57,58].
Model Testing Using Residence-Time Variations
The model outlined in Section "Calculation of oxygen-atom number density profiles along the gas-flow direction" results in the time-dependent O-atom number density, nO(t), in an Ar-O2 mixture with \({x}_{{O}_{2}}\) ≤ 0.01 while it passes through a DBD and the early DB-PD, respectively. In the following, these results are compared with experimental data obtained with varied residence times in the DBD, tres, or (see Section "Oxygen-atom number densities in the post-discharge from delay-time variations") delay times in the DB-PD, tdel. The comparison is not based on absolute measurements of nO by any of the established methods mentioned in the introduction but on OES measurements of the 777-nm line whose suitably normalized intensity, I777, is proportional to nO, as argued in Section "Optical emission from excited O atoms as a measure of ground-state O number density". Thus both, nO and I777, have the same dependence on time, which depends on the absolute value of nO, following the information given in Section "Introductory note: Absolute O-atom number densities from relative measurements of growth or decay curves". In the following, a scaling factor, fscal, defined as fscal ≡ nO/I777, is determined from experiments with varied residence time, using non-linear curve fitting to the analytical expression Eq. (4) which can be applied to the present experiments with \({x}_{{O}_{2}}\) = 100 ppm.
Figure 6 shows values of I777 from optical emission spectra taken from the discharge at the trailing edge of a 1-cm-long electrode, under an angle of 30° relative to the normal. The experiments were run with four different reactor geometries and power densities as specified in the figure. While the intensities for the two GG series are not too different, in spite of the substantial difference in power densities, I777 approaches a steady-state value more than twice as large in the wider AG channel, despite a somewhat lower power density. Qualitatively one can conclude from these observations that there is substantial wall recombination of the oxygen atoms in experiments, beyond what is expected from literature data for glass and Al2O3 used above.

Experimental results of OES measurements at the trailing edge of a 1-cm-long electrode (data points) and results of calculations, using Eq. (4) fitted to experimental data under conditions specified in the figure. For AG-0.25, two measurements were made at tres = 40 and 64 ms, respectively. Figures at the curves are steady-state values of nO, nO(∞), from Eq. (4)
Using non-linear curve fitting, the experimental data for each configuration were fitted to analytical functions nO(tres). In principle it should be possible to determine, in this way, two fit parameters: the wall-loss frequency kW (= k14 in the model), entering into Eq. (4) via the variable b, as well as the scaling factor fscal, relating the intensities I777 with the oxygen-atom number densities, fscal = nO/I777. According to what was described in Section "Optical emission from excited O atoms as a measure of ground-state O number density", one should expect a single value of fscal to result for all fits.
With the present experimental data, however, this is virtually impossible due to (i) the small number of data points per configuration combined with the statistical error of a single intensity measurement and (ii) the fact that different pairs of suitably chosen kW and fscal have similar results so that the effects of these quantities cannot be separated as it was observed in model calculations. Therefore, kW had to be entered as a constant by presuming an effective wall-loss probability, γeff, i.e., the value to be inserted into Chantry’s Eq. (7) in order to obtain the same wall-recombination frequency as with the transcendental Eq. (5).
It turned out that diffusion-controlled wall loss (τS < < τD in Eq. (7), see also Section "Discussion") had to be assumed in the discharge in order to account for the large difference in I777(tres) between the two AG series. Simultaneously this choice gives good agreement with experimental results for the O-atom density at the DBD exit presented in the following section. Therefore, γeff = 0.05 was chosen, resulting in the wall-loss frequencies given in rows #4, 7, 10, and 15 of Table 3. (Note that beyond about γeff = 0.05, kW becomes virtually independent of γeff). Then fscal was calculated by curve fitting, resulting in figures shown in Fig. 6. Within the GG and AG configurations, fscal is virtually the same, within experimental errors, as it should be. However, the average fscal for AG setups is by a factor of 2 smaller than fscal for GG setups; a possible reason is discussed later.
A comparison of calculated curves with experimental data points reveals that for all configuration the measured intensities I777 increase slower with tres than calculated within the present model. The reason for this discrepancy is most probably the way how wall losses are accounted for: the assumption of a constant wall-loss frequency kW in the rate equation for oxygen atoms is not applicable to the situation with a rapid change of nO within milliseconds, as shown in Section "Discussion".
Figures at the solid lines in Fig. 6 are steady-state oxygen-atom number densities, nO(∞), calculated from Eq. (4) under the specified conditions. (In the following section, Table 4, these numbers are compared with results from experiments with delay-time variation.) These densities are in the range from 1.1 to 2.1 × 1015 cm−3, which means that between about 20% and 40% of the O2 molecules fed into the DBD (100 ppm corresponding to 2.5 × 1015 cm−3) are dissociated at steady state.
Oxygen-Atom Number Densities in the Post-discharge from Delay-Time Variations
There are, in principle, two ways to study the delay-time dependence of O-atom number densities in a DBD post-discharge, using OES from a sensing DBD: (1) At constant distance Δ between main DBD and sensing DBD (see Fig. 1), by varying the average gas flow velocity, vav, or (2) by a variation of Δ at constant vav. While the first approach is experimentally easier, it has the drawback that the residence time in the sensing discharge varies with the gas flow velocity, resulting in different extents of O2 dissociation. Therefore, the alternative method was applied, with a constant average gas velocity of 400 cm/s, varying Δ between 1.0 (in one case 0.5) and 8.0 cm.
The first experiments of this kind were made with the same high-voltage generator feeding both DBDs with the same voltage, resulting in virtually the same power densities. It turned out that the OES signal, the normalized line area I777, after a first rapid decline with increasing Δ, due to O-atom recombination, soon turned off into a region with a much smaller slope. We interpreted this observation as the beginning dominance of emission from excited O atoms, resulting from O2 dissociation within the sensing DBD and, in a second step, excitation of the fragments. Therefore, in order to decrease the power density P/V in the sensing DBD relative to that in the main DBD, a 0.3-MΩ resistor was installed between the sensing DBD and the high-voltage generator. With increasing applied voltage, the main discharge ignited first. Then the voltage was increased further until the sensing discharge ignited, too, and could be run stably.
Data used for Figs. 7 and 8 were obtained by OES measurements on DBDs in the same channel reactors as used for the experiments with variation of tres reported in the previous section. However, in order to guarantee that steady-state atom densities were attained at the chosen gas velocity (400 cm/s), the length of the main electrode, LM, was increased to 4 cm. Normalized band intensities were calculated from emission spectra measured at the leading edge of the top electrode, feeding the sensing DBD (Fig. 1), under an angle of 30° from the normal, as above. For each distance Δ, two spectra were taken: One with only the main DBD running and a second with both DBDs on. Data used to calculate the I777 values in Figs. 7 and 8 were obtained after subtracting the first spectrum from the second, in order to correct for any backscattering of radiation from the main DBD.

Reciprocal of the normalized intensities of the 777-nm line, 1/I777, as a function of the delay time measured for four different reactor channels; see text for the nomenclature used. Data points for 2.5 ≤ tdel/ms ≤ 5 (7.5 in case of the 0.11-cm channel) were used for a linear fit, resulting in the intercepts and the ratios of slopes and intercepts given in the figure. Data points marked by the arrow were obtained with the main DBD off

Data from Fig. 7, normalized by dividing through I777(0), the reciprocals of the intercepts shown in Fig. 7. Black curves are values of nO(tdel)/nO(0), calculated numerically with the model described above; the corresponding values of nO(0) are shown in the upper left. Power densities of the main DBDs are given together with the legends
In Fig. 7, reciprocals, 1/I777, are plotted against the delay time, calculated from the average gas velocity and the distance Δ, tdel = Δ/vav. According to the considerations in Section "Comparison of numerical and analytical calculations" (cp. Figure 4), linear fits should be possible to the initial data points, up to a few milliseconds. In fact, this linearity is evident up to 5 ms, except for the series AG-0.11 were only one point falls into this interval. Linear curve-fitting to the first two data points of this series and to the data points measured at Δ = 1, 1.5, and 2 cm (tdel = 2.5, 3.75, and 5 ms) for the other series resulted in linear equations, whose right-hand sides are shown in the figure. Note that the second figures in the brackets are the ratios of slope and intercept with the ordinate which, following Section "Comparison of numerical and analytical calculations", should equal.
Interestingly, the two GG measurement series result virtually in the same intercept with the ordinate and similar slopes, irrespective of the difference in channel height h which should influence the slope/intercept ratio via the wall loss frequency, kW, and result in a larger slope/intercept ratio for h = 0.075 cm, in contrast to what is observed. Therefore one can conclude that the wall-recombination frequency kW does not significantly contribute to β; the post-discharge wall-loss probability γPD for the GG channels should be well below 1 × 10–4, cp. Table 3. For the AG configuration the narrower channel with h = 0.11 cm results in an intercept of 1/I777 about twice as large as the wider channel with h = 0.25 cm, indicating an O-atom number density roughly half as large, as already found in Section "Model testing using residence-time variations". Here, the slope/intercept ratio is also much smaller for h = 0.11 (44 s−1) than for h = 0.25 cm (107 s−1). The wall-loss frequency, as calculated from literature data for SiO2 and Al2O3 and a channel width of 0.11 cm, should already be larger than 100 cm−1, see entry #6 in Table 3. Again, the wall losses appear to be negligible, compared with volume losses by recombination and by reaction with O2.
Therefore, neglecting wall losses, one can calculate nO(0) for the four reactor configurations by subtracting \(\tilde{k}_{12} \times S_{2}\) = 30 s−1 from the slope/intercept ratios and dividing by κ = 5 × 10–14 cm3 s−1. Results are shown in Table 4, column A. The results in the B column of Table 4 were obtained by comparison of the experimental data with numerically calculated decay curves: A set of such curves was generated, assuming a vanishingly small wall-loss probability γPD (1 × 10–8), by varying the rate of Ar* generation, gAr*, normalizing the results by dividing through nO(tdel = 0), and comparing the resulting curves with correspondingly normalized experimental results. Figure 8 shows that good agreement is obtained for post-discharges with O-atom number densities at the discharge exit between 0.52 and 1.8 × 1015 cm−3.
Comparison of columns A and B in Table 4 shows generally satisfying agreement of the two experimental methods, except for AG-0.11—probably due to the lack of data at small delay times. Data in columns C and D are steady-state values, nO(tres = ∞), obtained by numerical calculation of O-atom production in the 4-cm- and 1-cm-long main DBD, respectively, with somewhat different power densities for each configuration. One should expect that nO(tres = ∞) = nO(tdel = 0) for given parameters. Very good agreement between experimental and calculated values is obtained for the wider channels AG-0.25 and GG-0.14. Stronger deviations for AG-0.11 are probably due to lack of experimental data, like the deviations between A and B. The discrepancy for GG-0.075 is probably due to the strong effect of wall losses in the narrow channel, which are no longer accounted for by the assumptions on which Eq. (4) is based, see the discussion section.
Beyond delay times of about 10 ms (Δ > 4 cm) experimental data begin to deviate from the calculated curves towards larger values and seem to become virtually constant at 20 ms. We attribute this observation to the generation of oxygen atoms in the sensing discharge. Interestingly, in all series, except in AG-0.25 (probably due to incidental deviation), the normalized intensities at 20 ms (Δ = 8 cm), I777(20 ms), are significantly larger than the intensities measured in the “fresh” Ar-O2 mixture, with the main DBD off. Corresponding results are shown in Figs. 7 and 8 behind the break in the time axis and marked by a black arrow. Evidently the same residence time in the sensing DBD results in a larger amount of O atoms in a gas which passed the main DBD. Tentatively we attribute this observation to the formation of O3 in the post-discharge: Fig. 9 shows calculated time evolutions of the gas composition after passage through the main DBD.

Number densities of reactive species O, O3, and O2(a) during and after passage through DBDs, calculated numerically for 6 different parameter sets. Power densities are the same as in Fig. 8. The magenta and light brown curves illustrate the effect of changing γPD from 1 × 10–5 to 1 × 10–4 and to 1 × 10–8, respectively, for a 0.11-cm channel
In the post-discharge it takes about 10 to 20 ms for the ozone number density to reach a constant value around 4 to 5 × 1014 cm−3 while the number density of O2(a) begins to decline eventually. Note that during the passage through the detection volume of the sensing DBD a certain number of Ar atoms in the Ar-O2 mixture is converted to Ar*, and a given number of Ar* species is able to produce different numbers of O or O(1D) atoms, depending on the reaction partner and the dissociation mechanism:
Therefore, the formation of O3 from O2 should result in an increase of the number of O atoms formed by repeated dissociation of the “plasma-treated” gas mixture if the reaction between Ar* and O3 actually forms three atomic species instead of one, as assumed in R10 so far. This consideration is supported by numerical calculation using start number densities of species as characteristic for the post-discharge after 20–30 ms. Increasing the rate coefficient k10, on the other hand, does not result in increased production of O atoms.
Figure 9 also shows the effects of wall-loss probabilities in the discharge and in the post-discharge, γDBD and γPD, respectively. Experiments show that γPD must be significantly smaller than 1 × 10–4 but it cannot be decided experimentally if γPD is smaller than 1 × 10–5. In the discharge, γDBD must have a value much larger than calculated based on literature data, in order to account for the observation that for both, AG and GG series, narrowing the channel can nearly compensate (GG-0.14 and GG-0.075 in Fig. 9) or even override (AG-0.25 and AG-0.11 in Fig. 9, experiments in Fig. 6) the effect of an increased power density on the degree of dissociation.
Discussion
The experimental data support the expectation that it should be possible to quantify number densities of atomic oxygen in Ar-O2 post-discharges by optical-emission measurements of the spatial decay of nO. In the wider channels of type AG or GG, discrepancies less than 15% are obtained between measurements and numerical calculations, applying a simplified model in which the discharge volume is considered as a uniform source of Ar* atoms. In view of the simplifications made in the kinetic model such as the use of a composite species Ar*, the agreement of model calculations with experiments is fully satisfactory.
It turned out, however, that the frequencies of heterogeneous recombination in the DBD and in the PD differ greatly: Effective recombination probabilities of O atoms on the reactor walls, calculated with literature data for SiO2 and Al2O3 were too small by two orders of magnitude to provide a reasonable fit of experimental data from the discharge. On the other hand, smaller values than expected had to be postulated for the walls of the post-discharge region.
The wall-recombination probability γ of O atoms on a silica or borosilicate glass surface is not a material property but strongly dependent on the environment. It has frequently been reported that the contact with a gas discharge increases γ of O atoms, see, for example Refs. [59,60,61]. An extreme example was reported by Cartry et al. [62]: In contact with a microwave plasma at 133 Pa, γ for a quartz tube at room temperature was 0.03, two orders of magnitude higher than typical for silica surfaces not exposed to a plasma. Values beyond 0.01 are otherwise only achieved at elevated temperature; so far it is not known how the exposure to a plasma in general and to a DBD in the present case enhances O atom recombination—by ion-induced formation of new active sites for O-atom attachment, by plasma-enhanced desorption, or by the incidence of electrons on the surface in every period of the applied voltage. Similar to the processes in filamentary DBDs in air [63], one can expect that surges of argon cations with energies of several tens of eV, sufficient to break Si–O bonds at the surface, are hitting the surface during a few nanoseconds in every period of the applied AC voltage. How these ions effect the silica or borosilicate surface remains to be studied.
Residence-Time Dependence of n O in the DBD
With γ > 0.01, the wall recombination in the discharge is predominantly controlled by diffusion. Criteria for diffusion- or surface-control in a channel with, e.g., h = 0.1 cm as a typical height, can be derived from Eq. (21). Neglecting the Motz-Wise correction factor, which is 0.995 and 0.99995 for γ = 0.01 and 1 × 10–4, respectively, the ratio of characteristic times for surface process and diffusion becomes:
Therefore, the heterogeneous process in the discharge should mainly be controlled by the diffusive transport of O atoms to the walls. With number densities in the range from 1 to 2 × 1015 cm−3, the volume reactions of O atoms are largely second order (three-body recombination at constant nAr). Unfortunately, Eqs. (5) and (6) are, strictly speaking, not applicable for this situation because they were derived for first-order volume and surface reactions. In the chemical literature, wall effects on number densities of reactive species in situations with second-order volume reactions have already been dealt with since the 1950s [64, 65] but an easily applicable equation for the wall-recombination frequency is not available in the literature, to the knowledge of the authors.
Another systematic error in the model is related to the rapid rise of nO as the Ar-O2 mixture enters into the DBD: 50% of the saturation value nO(∞) is typically achieved within less than 2 ms. Therefore, in order to justify the assumption of a constant wall-loss frequency based on the fundamental diffusion mode, kW = a12 × D, the diffusion mode with i = 2 (Eq. (6)) should have died away at least an order of magnitude faster. It is interesting to note that, in situations with γ = 0.05 on both walls, α2 differs from α1 only by a factor of 2 and the decay times of the first and the second mode only by a factor of 4. With kW = 100 s−1, for example, the decay time for the mode with i = 2 is 2.5 ms. Figure 10, to take another example, shows the exponential decrease of number density in the fundamental diffusion mode for a flat channel with h = 0.2 cm and walls with γ = 0.05 and γ = 2 × 10–4 at z = 0 and z = h, respectively, (red straight line) and, for comparison, the evolution of the normalized z-averaged number density as it follows from integration of Eq. (6), including 13 terms, starting from a virtually uniform distribution of atoms, n(z,0) = 1, at t = 0 (black curve) and kr = 0. Here, α1 = 11.0 cm−1 and α2 = 25.1 cm−1, see entry #11 in Table 3. During the first milliseconds the wall loss is significantly faster than at later times, t > 5 ms. The insert shows the number density distribution n(z,t) as a function of z at different moments in time t. Note that the inclusion of even as many as 13 terms with αI < 200 results only in a modest approximation to n(z,0) = 1. However, the residual ripple dies out within 0.2 ms.

Time evolution of the average number density of a species in a flat channel, lost at the walls with probabilities 0.05 at z = 0 and 2 × 10–4 at z = 0.2 cm, resp. The initial distribution is n(z,0) ≈ 1; the black curve is calculated from Eq. (6), with terms up to i = 13. The red curve is obtained if the fundamental diffusion mode (i = 1) is already established at t = 0. (D = 0.38 cm2 s−1)
These latter considerations explain the discrepancies between the observed rates of rise of I777 and the calculated nO as a function of tres in Fig. 6. The discrepancy for the very narrow channel with h = 0.075 cm in in Table 4, on the other hand, could be due to the non-justified assumption of a dominant first-order volume reaction to derive Eqs. (5), (6), and(7). Obviously, however, this does not compromise the agreement for larger channel heights, h = 0.14 or 0.25 cm, due to substantially smaller wall-losses; note that kW ~ h−2 (Eq. 7) if wall losses are diffusion-controlled.
At present, the differences in fscal between measurements in configurations AG and GG in Fig. 6 cannot be explained. In the model calculations constant temperature was assumed to be maintained in the gas phase during passage through the DBD. In the experiments, some temperature increase takes place which is higher for the GG configuration, under otherwise identical conditions, due to the lack of good thermal contact with the massive aluminum base plate. Possibly the relation between I777 and nO is temperature-dependent, beyond the mere gas-density effect. Further discussion of this topic is beyond the scope of this paper.
Measurements of n O(tdel) in the DB-PD
With a much smaller loss probability γ (< < 1 × 10–4) in the post-discharge than in the DBD, the heterogeneous reaction is largely controlled by the slow surface process. Errors introduced by non-steady diffusion or by the second-order nature of the volume reaction should not play a big role.
In order to evaluate separately the wall-loss frequency and nO(tdel), experiments should be performed with at least two different, suitably chosen gap widths, h, as it was done here to some extent. To achieve a situation where wall-recombination processes are slow, compared with volume reactions, channels should be used with widths of several mm, if feasible.
In order to suppress the formation of O atoms in the sensing DBD by dissociation of O2 as well as products O2(a) and O3 from the main DBD, the power density in the sensing DBD should be reduced as far as possibly. So far, a 0.3 MΩ resistor was used for this purpose, without optimization. Another possibility could be the use of an appropriately chosen inductivity or, most suitably, a separate voltage source.
Conclusions and Outlook
Number densities of oxygen atoms generated in highly diluted gas mixtures of O2 with argon during passage through a dielectric-barrier discharge can be calculated using a simplified kinetic model, assuming that the DBD volume is a uniform source of long-lived excited Ar species, Ar*, which transfer their energy to oxygen species O2, O2(a), O3, and O, resulting in dissociation and excitation, respectively. The corresponding rate equations can be solved numerically or, with a quasi-steady state approximation for Ar*, by an analytical equation for nO(t), with a deviation of less than 10% from the numerical approach for the experimentally studied low O2 fraction of 100 ppm. To cover reactor configurations with surfaces differing strongly with respect to the wall-recombination probability, an alternative to Chantry’s equation for the calculation of wall-loss frequencies is proposed. Model calculations were compared with experimental data from optical-emission spectroscopy, using the 777.4 nm-line of excited oxygen atoms to obtain a relative measure of its number density. Absolute O-atom number densities were derived by studies of the delay-time dependence of nO in the post-discharge, using a second DBD as a sensing discharge to excite O atoms produced in the main DBD. In view of the model simplifications, good agreement was obtained between model calculations and the latter experimental method for a double-dielectric channel with a 0.14 cm gap and for an asymmetrical channel with Al base plate and h = 0.25 cm, respectively. Wall recombination of O atoms on oxide surfaces was found to be substantially accelerated by exposure to the discharge.
At present the experimental method based on delay-time-dependent OES measurement using a sensing DBD at different distances from the O-producing DBD appears to be a trustworthy method relying on only a small number of manageable presuppositions: Proportionality between the suitably normalized emission intensity, I777, and the O-atom number density, nO, and validity of a small number of rate coefficients, see Eq. (13), and the wall loss frequency. The latter can be determined by experiments on DBD reactors with two or more different, suitably chosen channel heights, h.
To improve the method, a few issues deserve to be studied in greater detail: (i) It should be investigated, using numerical calculations, under what conditions Eq. (5), derived for the case of a first-order volume reaction, can be applied, without too large errors, to a situation dominated by the second-order recombination reaction of O atoms. (ii) The experiments require constant power density in the detection area of the sensing DBD while its distance from the main DBD is varied. So far, this has not yet been investigated; it was just assumed to be constant if the same applied voltage is used while varying Δ. (iii) The excitation kinetics of ground-state O atoms by individual Ar(1si) species should be studied. (iv) Last not least it is challenging to extend the analytical Eq. (4) to a model in which the composite species Ar* is resolved into its components: Individual Ar(1si) species as well as Ar2 excimers.
Notes
We use abbreviations “PD” and “DB-PD” for “post-discharge” and “dielectric-barrier post-discharge”, respectively.
Paschen notation.
Version from March 2016 with cross sections from LXcat, http://www.lxcat.laplace.univ-tlse.fr, downloaded on 4 Jun 2013.
See, for example, https://www.wolframalpha.com/
References
Wright AN, Winkler CA (1968) Active nitrogen. Academic Press, New York, London
Morren MA (1865) Ann Chem Phys 4:293
Slanger TG (1978) J Chem Phys 69(11):4779. https://doi.org/10.1063/1.436504
Mearns AM, Morris AJ (1970) J Phys Chem 74(22):3999. https://doi.org/10.1021/j100716a025
Tendo S, Kohguchi H, Yamasaki K (2018) Chem Phys Lett 710:96. https://doi.org/10.1016/j.cplett.2018.08.058
Niemi K, Schulz-von der Gathen V, Döbele HF (2001) J Phys D Appl Phys 34(15):2330. https://doi.org/10.1088/0022-3727/34/15/312
van Gessel AFH, van Grootel PH, Bruggemann PJ (2013) Plasma Sources Sci Technol 22(5):055010. https://doi.org/10.1088/0963-0252/22/5/055010
Klochko AV, Lemainque J, Booth JP, Starikovskaia SM (2015) Plasma Sources Sci Technol 24(2):025010. https://doi.org/10.1088/0963-0252/24/2/025010
Dvořák P, Mrkvičková M, Obrusník A, Kratzer J, Dědina J, Procházka V (2017) Plasma Sources Sci Technol 26(8):085002. https://doi.org/10.1088/1361-6595/aa76f7
Myers B, Barnat E, Stapelmann K (2021) J Phys D Appl Phys 54(45):455202. https://doi.org/10.1088/1361-6463/ac1cb5
Ding P, Ruchkina M, Del Cont-Bernard D, Ehn A, Lacoste DA, Bood J (2021) J Phys D Appl Phys 54(27):275201. https://doi.org/10.1088/1361-6463/abf61f
McGinn PJ (2019) ACS Comb Sci 21(7):501. https://doi.org/10.1021/acscombsci.9b00032
Kramida A, Ralchenko Y, Reader J, NIST ASD Team (2021) NIST atomic spectra database (ver 5.9), National Institute of Standards and Technology, Gaithersburg, MD. https://physics.nist.gov/asd, https://doi.org/10.18434/T4W30F
Kogelschatz U (2012) J Opt Technol 79(8):484. https://doi.org/10.1364/JOT.79.000484
Klages CP, Czerny AK, Philipp J, Becker MM, Loffhagen D (2017) Plasma Process Polym 14(12):1700081. https://doi.org/10.1002/ppap.201700081
Xie D, Guo H, Peterson KA (2000) J Chem Phys 112(19):8378. https://doi.org/10.1063/1.481442
Piper LG, Clyne MAA, Monkhouse PB (1982) J Chem Soc Faraday Trans 2(78):1373. https://doi.org/10.1039/F29827801373
King DL, Piper LG, Setser DW (1977) J Chem Soc Faraday Trans 2(73):177. https://doi.org/10.1039/F29777300177
Moselhy M, Stark KH, Schoenbach KH, Kogelschatz U (2001) Appl Phys Lett 78(7):880. https://doi.org/10.1063/1.1336547
Chantry PJ (1987) J Appl Phys 62(4):1141. https://doi.org/10.1063/1.339662
Hagelaar GJM, Pitchford LC (2005) Plasma Sources Sci Technol 14(4):722. https://doi.org/10.1088/0963-0252/14/4/011
Keller S, Rajasekaran P, Bibinov N, Awakowicz P (2012) J Phys D Appl Phys 45(12):125202. https://doi.org/10.1088/0022-3727/45/12/125202
Eliasson B, Kogelschatz U (1986) J Phys B Atom Mol Phys 19(8):1241. https://doi.org/10.1088/0022-3700/19/8/018
Velazco JE, Kolts JH, Setser DW (1978) J Chem Phys 69(10):4357. https://doi.org/10.1063/1.436447
Balamuta J, Golde MF (1982) J Phys Chem 86(14):2765. https://doi.org/10.1021/j100211a041
Fiebrandt M, Bibinov N, Awakowicz P (2020) Plasma Sources Sci Technol 29(4):045018. https://doi.org/10.1088/1361-6595/ab7cbe
Kutasi K, Guerra V, Sá P (2010) J Phys D Appl Phys 43(17):175201. https://doi.org/10.1088/0022-3727/43/17/175201
Klages CP (2020) Plasma Process Polym 17(8):2000028. https://doi.org/10.1002/ppap.202000028
Bogaerts A (2009) Spectrochim Acta Part B 64(11–12):1266. https://doi.org/10.1016/j.sab.2009.10.003
Van Gaens W, Bogaerts A (2013) J Phys D Appl Phys 46(27):275201. https://doi.org/10.1088/0022-3727/46/27/275201
Oka T, Kogoma M, Imamura M, Arai S, Watanabe T (1979) J Chem Phys 70(7):3384. https://doi.org/10.1063/1.437923
Gentile AC (1995) Kinetic processes and plasma remediation of toxic gases, dissertation, University of Illinois at Urbana-Champaign, Urbana, Illinois, USA
Taherian MR, Slanger TG (1985) J Chem Phys 83(12):6246. https://doi.org/10.1063/1.449573
Feoktistov VA, Mukhovatova AV, Popov AM, Rakhimova TV (1995) J Phys D Appl Phys 28(7):1346. https://doi.org/10.1088/0022-3727/28/7/011
Booth J-P, Chatterjee A, Guaitella O, Santos Sousa J, Lopaev D, Zyryanov S, Rakhimova T, Voloshin D, Mankelevich Y, de Oliveira N (2020) Plasma Sources Sci Technol 29(11):115009. https://doi.org/10.1088/1361-6595/abb5e7
Pershin AA, Torbin AP, Mikheyev PA, Kaiser RI, Mebel AM, Azyazov VN (2021) J Chem Phys 155(16):165307. https://doi.org/10.1063/5.0064361
Tsang W, Hampson RF (1986) J Phys Chem Ref Data 15(3):1087. https://doi.org/10.1063/1.555759
Hippler H, Rahn R, Troe J (1990) J Chem Phys 93(9):6560. https://doi.org/10.1063/1.458972
Atkinson R, Baulch DL, Cox RA, Crowley JN, Hampson RF, Hynes RG, Jenkin ME, Rossi MJ, Troe J (2004) Atmos Chem Phys 4(6):1461. https://doi.org/10.5194/acp-4-1461-2004
Dougherty EP, Rabitz H (1980) J Chem Phys 72(12):6571. https://doi.org/10.1063/1.439114
Collins RJ, Husani D, Donovan RJ (1973) J Chem Soc Faraday Trans 2(69):145. https://doi.org/10.1039/F29736900145
Azyazov VN, Torbin AP, Pershin AA, Mikheyev PA, Heaven MC (2015) Chem Phys 463:65. https://doi.org/10.1016/j.chemphys.2015.09.007
Niemi K, Schulz-von der Gathen V, Döbele HF (2005) Plasma Sources Sci Technol 14(2):375. https://doi.org/10.1088/0963-0252/14/2/021
Turányi T, Tomlin AS, Pilling MJ (1993) J Phys Chem 97(1):163. https://doi.org/10.1021/j100103a028
Ince EL (1956) Ordinary differential equations. Dover Publications, New York, pp 23–25
Yolles RS, Wise H (1968) J Chem Phys 48(11):5109. https://doi.org/10.1063/1.1668181
Marinov D, Teixeira C, Guerra V (2017) Plasma Process Polym 14(1–2):1600175. https://doi.org/10.1002/ppap.201600175
Guha J, Kurunczi P, Stafford L, Donnelly VM, Pu Y-K (2008) J Phys Chem C 112(24):8963. https://doi.org/10.1021/jp800788a
Carslaw HS, Jaeger JC (1959) Conduction of heat in solids. Oxford University Press, Oxford, p 144
Lieberman MA, Lichtenberg AJ (2005) Principles of plasma discharges and materials processing. Wiley, New Jersey, USA, p 312
Motz H, Wise H (1960) J Chem Phys 32(6):1893. https://doi.org/10.1063/1.1731060
Dorsman R, Kleijn CR (2007) Chem Vap Depos 13(2–3):91. https://doi.org/10.1002/cvde.200606515
Tsutsumi T, Greb A, Gibson AR, Hori M, O’Connell D, Gans T (2017) J Appl Phys 121:143301. https://doi.org/10.1063/1.4979855
Loffhagen D, Becker MM, Czerny AK, Klages CP (2021) Plasma Chem Plasma Process 41:289. https://doi.org/10.1007/s11090-020-10121-y
Caplinger JE, Perram GP (2020) Plasma Sources Sci Technol 29(1):015011. https://doi.org/10.1088/1361-6595/ab5e5f
Pointu AM, Ricard A, Odic E, Ganciu M (2008) Plasma Process Polym 5(6):559. https://doi.org/10.1002/ppap.200800016
Mazánkova V, Trunec D, Krčma F (2015) Plasma Phys Technol 2(1):50
Khosravi Z (2017) FTIR-spectroscopic studies of plasma nitrogenation of polyethylene surfaces, Dissertation, Technische Universität Braunschweig
Diamy AM, Legrand J-C, Andari JA (1997) New J Chem 21:977
Krištof J, Macko P, Veis P (2012) Vacuum 86(6):614. https://doi.org/10.1016/j.vacuum.2011.07.041
Marinov D, Guerra V, Guaitella O, Booth J-P, Rousseau A (2013) Plasma Sources Sci Technol 22(5):055018. https://doi.org/10.1088/0963-0252/22/5/055018
Cartry G, Duten X, Rousseau A (2006) Plasma Sources Sci Technol 15(3):479. https://doi.org/10.1088/0963-0252/15/3/025
Babaeva NY, Ning N, Graves DB, Kushner MJ (2012) J Phys D Appl Phys 45:115203. https://doi.org/10.1088/0022-3727/45/11/115203
Gomer R (1951) J Chem Phys 19(3):284. https://doi.org/10.1063/1.1748195
Jackson DP, Armstrong DA (1971) J Chem Phys 75(19):2883. https://doi.org/10.1021/j100688a001
Acknowledgements
The authors acknowledge funding of their work by the Deutsche Forschungsgemeinschaft (DFG, GZ: KL 1096/33-1). They thank Antje Jung and Axel Pregizer-Winter for technical assistance. For helpful discussions the authors are grateful to Detlef Loffhagen, Leibniz Institute for Plasma Science and Technology (INP), Greifswald. They also thank Dirk Langemann, Technische Universität Braunschweig, for a valuable clue to improving the solution of Eq. (2).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Klages, CP., Bröcker, L., Betz, M.L. et al. Atomic-Oxygen Number Densities in Ar-O2 DBDs and Post-discharges with Small Initial O2 Fractions: Plug-Flow Model and Experiments. Plasma Chem Plasma Process 43, 285–314 (2023). https://doi.org/10.1007/s11090-022-10293-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11090-022-10293-9