
Abstract
A comparative study of two plasma sources (floating-electrode dielectric barrier discharge, DBD, Drexel University; atmospheric pressure argon plasma jet, kINPen, INP Greifswald) on cancer cell toxicity was performed. Cell culture protocols, cytotoxicity assays, and procedures for assessment of hydrogen peroxide (H2O2) were standardized between both labs. The inhibitory concentration 50 (IC50) and its corresponding H2O2 deposition was determined for both devices. For the DBD, IC50 and H2O2 generation were largely dependent on the total energy input but not pulsing frequency, treatment time, or total number of cells. DBD cytotoxicity could not be replicated by addition of H2O2 alone and was inhibited by larger amounts of liquid present during the treatment. Jet plasma toxicity depended on peroxide generation as well as total cell number and amount of liquid. Thus, the amount of liquid present during plasma treatment in vitro is key in attenuating short-lived species or other physical effects from plasmas. These in vitro results suggest a role of liquids in or on tissues during plasma treatment in a clinical setting. Additionally, we provide a platform for correlation between different plasma sources for a predefined cellular response.
Similar content being viewed by others

Introduction
In the study of plasma medicine, partially ionized gases and their physico-chemical effectors are investigated for beneficial biological responses [1–3]. The observed beneficial effects in wound healing [4–6] and cancer [7–9] have significantly spurred research efforts and novel findings in recent years. Promising in vitro research investigations are an ethical and scientific necessity for translation of plasma applications to in vivo models and eventually to humans. However, there are technical and methodological challenges for direct plasma applications, especially with regard to different types of plasma sources and comparison of results. Major among them is the extent to which in vitro plasma effects depend on long-lived species or other effectors of the multicomponent system plasma, such as UV-radiation or electrical fields. Therefore, two main types of plasma sources, a floating-electrode dielectric barriers discharge [10] and an atmospheric pressure argon plasma jet [11] were compared with regard to cell growth reduction and its dependence on main plasma active components. The sources were chosen because they have been of relevance in plasma medical research for more than a decade and thus were subject to extensive physical characterization [12]. To compare plasma effects across labs which is easy to perform, cheap and could be applicable for clinical device calibration, a simple biological read-out was chosen.
Plasma medical research inevitably involves experiments on reactive species because these were found to be central effectors in a number of biological targets exposed to plasma, such as skin cells [13–15], immune cells [16–18], and cancer cells [19–52]. In many instances, hydrogen peroxide (H2O2) was an important mediator in these in vitro experiments [53–55]. H2O2 is not necessarily toxic by itself but rather exerts its biological effects through secondary processes, for example, Fenton reaction [56], by acting as substrate for oxygenases [57], and in redox signaling events enabling the translation of redox events into distinct biological responses [58]. We selected CT26 murine colon cancer cells for this work because H2O2 has been previously identified as inducing apoptotic but not the necroptotic cell death pathway in these cells [59, 60]. CT26 colon cancer monolayer cultures were exposed to either DBD or argon jet plasma. Plasma source dependent, our results demonstrate that H2O2 correlates with inhibition of CT26 metabolic activity. It plays a central but not exclusive role in plasma-induced cell toxicity.
Materials and Methods
Cell Culture
Murine CT26 colon cancer cells were maintained in cell culture flasks in Rosswell Park Memorial Institute 1640 (RPMI1640) cell culture medium supplemented with 10% fetal bovine serum (FCS), 2% glutamine, and 1% penicillin/streptomycin (all Sigma). For culturing and experiments, cells were maintained in standard incubation conditions at 37 °C with 95% humidified atmosphere and 5% carbon dioxide.
DBD Plasma Treatment
Treatment of cells with the DBD plasma system was performed in the absence of liquid unless otherwise specified. The DBD electrode used was 1.3 cm in diameter and fit into the wells of a 24-well plate. Cells were treated with plasma as previously described [61]. Briefly, CT26 cells were seeded at 1.5 × 105 cells per well in 0.5 ml of fully supplemented cell culture medium and incubated overnight at 37 °C with 5% CO2. Prior to plasma treatment, cells were washed twice with PBS, and PBS from the second wash was removed immediately before plasma exposure. Plasma was generated by applying a high voltage pulse to the DBD electrode 1 mm above the cells in the well. The pulse was generated with a nanosecond pulser (FPB-20-05NM, FID GmbH, Burbach, Germany) and the frequency of pulses was controlled with an external function generator (TTi, TG5011 LXT, Philadelphia, PA, USA). Treatment time was fixed at 10 s unless otherwise specified. In some experiments, either treatment time or pulse frequency was altered to deliver a fixed plasma treatment energy over different times. For the DBD comparative study, a microsecond pulse (0.07 mJ/pulse) was also used. The energy per pulse from both system was measured as previously described [62, 63] and total plasma energy delivered to the cells for both systems were calculated from treatment time, pulse frequency and energy per pulse. Complete media was added to each well after plasma treatment and cells were incubated overnight before viability was measured. Pulse parameters of both the nanosecond- and the microsecond-pulsed DBD system are summarized in Table 1.
Jet Plasma Treatment
Treatment with the genotoxically-safe [64–66] atmospheric pressure argon plasma jet kINPen 11 (similar in construction to the kINPen MED that has received accreditation as medical device in Germany; neoplas control, Germany) operated at a feed gas flux of three standard liters per minute (SLPM) of Argon gas (Air Liquide, France) was performed as described previously [67]. It is a DBD-like jet, with a central pin electrode shielded against an outer electrode by a dielectric, powered by 2–6 kV at 1 MHz. Briefly, 1 × 105 CT26 cells in 1 ml of fully supplemented cell culture medium were added to each well of a 24-well plate, and incubated overnight. In some experiments, 5 µg/ml of the H2O2-scavenging enzyme catalase (Sigma) was added prior to plasma treatment. A layout was programmed for a computer-driven xyz-table (CNC, Germany) hovering the plasma jet over the center of each well at a predetermined distance for the indicated treatment time. To compensate for evaporation of liquid, a predetermined amount of double-distilled water was added after plasma treatment.
Cytotoxicity
Cytotoxicity was assessed as a measure of total metabolic activity. After treatment, cells were incubated for 21 h at 37 °C. The medium was replaced with 1 ml of fully supplemented cell culture medium without phenol red and containing 100 µM of resazurin (Alfa Aesar, USA). Metabolically active cells generate NADPH that is used by intracellular enzymes to create highly fluorescent resorufin from resazurin. Both compounds freely diffuse through cell membranes for a convenient readout in cell culture supernatants. After incubation for 3 h at 37 °C, 4 × 200 µl of each well were transferred to a flat-bottom 96-well plate. Fluorescence was read at λex 535 nm and λem 590 nm using a microplate reader. Background fluorescence of cell culture medium alone containing resazurin was subtracted from all sample and control readings. Relative fluorescence of samples was then normalized to that of untreated controls cells.
Hydrogen Peroxide
H2O2 was quantified in double-distilled water (Drexel) or PBS (INP) using the amplex red detection reagent (Thermo, USA). If plasma does not acidify the solution, double-distilled water and PBS are in principle interchangeable. The pH of distilled water did not change under any of the treatment conditions used in this study for DBD therefore peroxide measurements were made in water. Plasma treated water or PBS was diluted 1:20 in amplex red reagent (5 µM) in PBS supplemented with horseradish peroxide (10U/ml). After incubation for 15 min in dark, fluorescence was read at λex 535 nm and λem 590 nm using a microplate reader. Relative fluorescence units were quantified against a linear regression delineated from a H2O2 standard curve (5000–78 nM), and multiplied by dilution factors to retrieve actual concentrations.
Software
Graphing, calculation of mean and standard deviation (S.D.), and linear regressions were done using prism software (Graphpad, USA). One-way analysis of variances was performed using prism as well.
Results
DBD and Jet Growth Inhibition of CT26 Cells and Relationship to H2O2 Deposition in Liquids
CT26 cells were treated with DBD (Fig. 1a) or jet (Fig. 1b) plasma, and metabolic activity was assessed after 24 h. At constant treatment times of 10 s, DBD toxicity increased with higher energy doses, giving a 50% inhibitory concentration (IC50) at about 740 mJ (Fig. 2a). The kINPen plasma cannot be tuned electrically as settings are fixed. Hence, dosimetry and increase in ‘energy’ can only be achieved by extending the treatment time. As expected, the latter was proportional to cellular toxicity with an IC50 of 100 s (Fig. 2b). At IC50 treatment conditions, H2O2 deposition by DBD and jet plasma was 50 µM (Fig. 2c) and 115 µM (Fig. 2d), respectively. In both cases, H2O2 levels increased with energy or time of exposure. Therefore, toxicity increased proportionally with increased energy and/or plasma treatment time, and concentrations of plasma-generated H2O2 correlated with that.

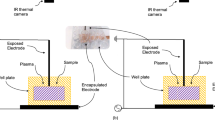
DBD and jet plasma. a DBD treatment of cells in a 24-well plate. b Atmospheric pressure argon plasma jet treatment of cells in a 24-well plate

IC50 and generation of H2O2. a DBD treatment of CT26 cells at different energy dosages. b Atmospheric pressure argon plasma jet treatment of CT26 cells at different treatment times. c H2O2 deposition of DBD plasma at different energy dosages. d H2O2 deposition of jet plasma at different treatment times. In (a) and (b), metabolic activity was assessed after 24 h and normalized to that of untreated control cells. Data are presented as mean with SD
Energy Input and Amount of Liquid were Imperative for DBD Plasma Toxicity
CT26 cells were treated with varying nanosecond-pulsed DBD plasma treatment times having an overall constant energy input of IC50-related 700 mJ. Overall toxicity was very similar under all treatment conditions (Fig. 3a). Corroborating these results, similar H2O2 concentrations were measured for all treatment times having the same total energy input (Fig. 3b). To understand the impact of plasma pulse duration, the DBD plasma was operated at microsecond instead of nanosecond pulsing. Toxicity correlated with increasing energy dosages yielding an IC50 of 768 mJ (Fig. 3b). This energy generated about 47 µM of H2O2 (Fig. 3d). Both the IC50 (768 mJ) and H2O2 generation (47 µM) in microsecond plasma mode were very similar to that of the nanosecond plasma (742 mJ and 50 µM, respectively). These observations suggest that the impact of the pulsing frequency and treatment time on toxicity were negligible as long as the total plasma energy was maintained. In addition, H2O2 seemed to correlate well with metabolic attenuation of cells. This was, at least in part, supported by the finding that with increasing cell densities, IC50 values began to increase for cell densities exceeding 1.25 × 105/well (Fig. 4a). Specifically, metabolic inhibition of cell densities below 2 × 105 differed significantly although statistical comparison between all other groups was non-significant. Nonetheless, this result indicates the involvement of additional contributors to plasma toxicity other than H2O2 in the DBD treatment to satisfy stoichiometry. Next, the impact on liquid presence on top of the cells during DBD plasma treatment was evaluated. In all DBD plasma treatments above, cell culture medium was removed prior to exposure. Here, varying amounts of PBS were added prior to treatment. Only 50 µl of liquid halved overall toxicity from 50 to 25% (Fig. 4b). Presence of 200 µl fully abrogated it. Therefore, we conclude that total energy input and amount of liquid are key parameters in determining overall DBD plasma toxicity in 2D cell cultures in vitro.

Electrical parameters of DBD plasma on cell viability and H2O2 production. a Different treatment times with varying energy were applied onto CT26 cells so that total energy deposition was kept constant at 700 mJ. b H2O2 concentrations for treatment regimens applied in (a). µs instead of ns pulsing of DBD plasma at different energies applied to CT26 cells. d H2O2 concentrations for treatment regimens applied in (c). In a and c, metabolic activity was assessed after 24 h and normalized to that of untreated control cells. Data are presented as mean with SD

Toxicity in dependence on cell density and amount of liquid present. a Varying concentrations of CT26 cells in 24-well plates were exposed to the IC50 dose of DBD plasma. b 1 × 105 cells per well in 24-well plates with varying amounts of PBS were exposed to the IC50 dose of DBD plasma. In all experiments, metabolic activity was assessed after 24 h and normalized to that of untreated control cells. Data are presented as mean with SD; statistical analysis was performed using one-ways analysis of variances comparing all group means with Tukey post-testing
Importance of H2O2 in CT26 Toxicity
The role of H2O2 in DBD plasma toxicity was further investigated by comparing plasma-IC50 and its respective H2O2 concentration to that of exogenously added H2O2. With DBD plasma, about 50 µM were found to correlate to the IC50 (Fig. 2c) whereas a dilution series of H2O2 alone in 500 µl final volume identified an IC50 of 2062 µM (Fig. 5a). In contrast, for the jet plasma IC50, corresponding peroxide concentrations were 115 µM whereas a dilution series of H2O2 alone in 1000 µl final volume revealed an IC50 of 125 µM (Fig. 5b). These data suggested that H2O2 was central in cellular effects from jet plasma and only partially in DBD plasma toxicity, with a contribution of additional effectors in the latter system. This was expected because direct DBD operation is associated with a significant amount of charged and short-lived chemically active species. To underline this finding for the jet plasma, CT26 cells were treated for 150 s with argon gas alone, plasma, or plasma in presence of the H2O2-scavenging enzyme catalase. Plasma treatment alone markedly reduced cell viability compared to argon gas control, whereas presence of catalase abolished plasma toxicity (Fig. 5c).

Experimentally added H2O2 on CT26 viability. a Varying concentrations of H2O2 were prepared in 50 µl PBS and added to CT26 cells at Drexel lab. Immediately after that, solutions were diluted with 450 µl of cell culture medium, which was a similar procedure to DBD plasma treatment. b Varying concentrations of H2O2 were prepared in 1000 µl PBS and added to CT26 cells at INP Greifswald. c CT26 cells were treated with 150 s of argon gas alone, 150 s of plasma, or 150 s of plasma in presence of catalase. In all experiments, metabolic activity was assessed after 24 h and normalized to that of untreated control cells. Data are presented as mean with SD
Discussion
With regard to in vitro cell culture experiments, jet plasmas and most DBDs differ in one important parameter: the use of external feed gas. Once in direct contact with cells, feed gas—even in the plasma-off mode—immediately necrotizes cells due to drying effects (unpublished observation). Therefore, plasma jet experiments with large gas fluxes require sufficient amounts of liquid that—even if parts of liquid are blown aside—shields cells from dehydration. Excess presence of liquids, however, offers competing non-cellular targets to react with short-lived species, leading to dominant effects of long-lived oxidants on cells. In contrast, DBD plasmas streamers come in direct contact with cells. Compared to jet plasmas, this increases the contribution of (low range) UV-radiation, electrical fields, and short-lived species to any biological effect observed. The importance of excess liquid as “scavenger” was exemplified by our results showing full abrogation of DBD plasma effects with addition of 200 µl or more.
Reactive molecules are known to be crucial regulators of cellular activity in health and disease [68–70]. Undoubtedly, reactive species are central mediators of biological plasma effects [71]. Many types have been identified in plasma-treated liquids in the past years [72]. For the kINPen plasma jet, this includes for example nitrite, nitrate, hydrogen peroxide, superoxide, hydroxyl radical, peroxynitrite, and singlet oxygen [72–74]. Plasma gas phase analysis suggests the presence of many others [75–77]. Yet, if organic target molecules and/or cells are not present at the site of species creation or deposition, short-lived species yield more stable products such as hydrogen peroxide or hypochlorous acid [73].
Our results indicated a dominant and a partial role of peroxide for the jet and DBD toxicity, respectively. The latter was especially illustrated by a lower IC50 H2O2 deposition by the DBD compared to the jet, and the non-linear correlation between cell number and DBD cytotoxicity. For the DBD, this suggested additional cytotoxic effectors at work. As 200 µl of liquid on top of cells fully abrogated DBD plasma toxicity, these effectors may be for instance poration, UV-radiation, charged particles or short-lived species being decomposed in absence of target cells. For example, the DBD plasma may create nanopores [78] allowing the entry of species into cells by a process similar to aquaporins [79], a route potentially counteracted by excess liquid. Also, short wavelength UV radiation is efficiently scavenged by a few hundred nanometers of liquid layer [80]. A combination of different plasma properties is also possible as seen with bacteria [81–83]. Corroborating results of the jet plasma in the present work, presence and concentration of H2O2 strongly correlated with cytotoxic effects of the kINPen [84–86] and other jet and DBD plasma sources [87–90], especially plasma-treated liquids [91–93]. Yet, in a helium/oxygen plasma jet, we previously demonstrated cytotoxicity correlating with short-lived species supporting the generation of hypochlorous acid in aqueous media [94].
H2O2 itself is a relatively non-reactive molecule with low reaction-constants with biomolecules [95]. Its effects mainly depend on two factors: the presence of catalysts and/or other oxidants, and the enzymatic profile of the target cells handling oxidants. It is well known the reaction of H2O2 and iron generates highly toxic hydroxyl radicals, by a process known as the Fenton reaction [96]. Hence, iron close to cell membranes, intracellular iron, and/or shuttling of H2O2 through membranes would be important denominators for plasma effects. Moreover, synergistic effects of plasma-generated H2O2 with nitrite have been proposed [97] that may act in concert with membrane-based oxidases to form peroxynitrite and other toxic species [98]. Extracellular trap formation following kINPen plasma treatment was not replicated by addition of hydrogen peroxide alone either [99]. In addition, redox enzymes and antioxidant defenses guide diverse cell fates when two different cell types are subjected to the same plasma treatment [100].
Nonetheless, in vitro cell cultures are only model systems that may guide in vivo studies. It is important to considered that the biomolecule to liquid ratio in tissues is much lower than in vitro systems. There are several challenges before the translation of in vitro plasma effects to in vivo effects becomes intuitive. The species variety is unique to cold plasmas with their concentration depending on the plasma source. Methods to directly detect short-lived species in tissues are currently unavailable. Modeling studies suggest that plasma in direct contact with a target alters species deposition and cell membrane oxidation [101–103]. Experimentally, plasma-derived oxidants have been shown to be deposited on cells and liquids through plasma-treated agarose membranes [104] and micropores [105], which may facilitate the deciphering effects of jet plasma short-lived species in cells.
While this study was not comprehensive and has limitations, it is the first attempt to find common and divergent themes between different plasma sources intended for future medical applications. Only one type of DBD and jet plasma source was investigated. A straightforward biological read-out of metabolic/growth inhibition was chosen for comparison because its no-wash-one-step approach minimizes lab-to-lab variation. We offer protocols for testing other plasma sources to “standardize” their biological effects by relatively easy means.
In conclusion, we provide a biological response model (IC50 of CT26 colorectal cancer cells) that allows for easy comparison between very different plasma discharges.
References
Fridman G, Friedman G, Gutsol A, Shekhter AB, Vasilets VN, Fridman A (2008) Plasma Process Polym 5:503
Weltmann KD, von Woedtke T (2017) Plasma Phys Control Fusion 59:014031
Laroussi M (2009) IEEE Trans Plasma Sci 37:714
Heinlin J, Isbary G, Stolz W, Morfill G, Landthaler M, Shimizu T, Steffes B, Nosenko T, Zimmermann J, Karrer S (2011) J Eur Acad Dermatol Venereol 25:1
Bekeschus S, Schmidt A, Weltmann K-D, von Woedtke T (2016) Clin Plasma Med 4:19
Emmert S, Brehmer F, Hänßle H, Helmke A, Mertens N, Ahmed R, Simon D, Wandke D, Maus-Friedrichs W, Däschlein G (2013) Clin Plasma Med 1:24
Hirst AM, Frame FM, Arya M, Maitland NJ, O’Connell D (2016) Tumour Biol 37:7021
Yan D, Sherman JH, Keidar M (2016) Oncotarget 2016, 5
Miller V, Lin A, Fridman A (2016) Plasma Chem Plasma Process 36:259
Dobrynin D, Fridman G, Friedman G, Fridman A (2009) New J Phys 11:115020
Weltmann KD, Kindel E, Brandenburg R, Meyer C, Bussiahn R, Wilke C, von Woedtke T (2009) Contrib Plasma Phys 49:631
Fridman G, Shereshevsky A, Jost MM, Brooks AD, Fridman A, Gutsol A, Vasilets V, Friedman G (2007) Plasma Chem Plasma Process 27:163
Arndt S, Landthaler M, Zimmermann JL, Unger P, Wacker E, Shimizu T, Li YF, Morfill GE, Bosserhoff AK, Karrer S (2015) PLoS ONE 10:e0120041
Kim KC, Piao MJ, Madduma Hewage SR, Han X, Kang KA, Jo JO, Mok YS, Shin JH, Park Y, Yoo SJ, Hyun JW (2016) Int J Mol Med 37:29
Schmidt A, von Woedtke T, Bekeschus S (2016) Oxid. Med. Cell. Longev. 2016:9816072
Bekeschus S, Rödder K, Schmidt A, Stope MB, von Woedtke T, Miller V, Fridman A, Weltmann K-D, Masur K, Metelmann H-R, Wende K, Hasse S (2016) Plasma Process. Polym 13:1144
Kaushik NK, Kaushik N, Min B, Choi KH, Hong YJ, Miller V, Fridman A, Choi EH (2016) J Phys D Appl Phys 49:084001
Miller V, Lin A, Fridman G, Dobrynin D, Fridman A (2014) Plasma Process Polym 11:1193
Ishaq M, Kumar S, Varinli H, Han ZJ, Rider AE, Evans MD, Murphy AB, Ostrikov K (2014) Mol Biol Cell 25:1523
Brulle L, Vandamme M, Ries D, Martel E, Robert E, Lerondel S, Trichet V, Richard S, Pouvesle JM, Le Pape A (2012) PLoS ONE 7:e52653
Bekeschus S, Rödder K, Fregin B, Otto O, Lippert M, Weltmann KD, Wende K, Schmidt A, Gandhirajan RK (2017) Oxid Med Cell Longev 2017:4396467
Ahn HJ, Kim KI, Hoan NN, Kim CH, Moon E, Choi KS, Yang SS, Lee JS (2014) PLoS ONE 9:e86173
Attri P, Kumar N, Uhm HS, Choi EH, RSC Adv 2014
Binenbaum Y, Ben-David G, Gil Z, Slutsker YZ, Ryzhkov MA, Felsteiner J, Krasik YE, Cohen JT (2017) PLoS ONE 12:e0169457
Chang JW, Kang SU, Shin YS, Kim KI, Seo SJ, Yang SS, Lee J-S, Moon E, Baek SJ, Lee K (2014) Arch Biochem Biophys 545:133
Chang JW, Kang SU, Shin YS, Seo SJ, Kim YS, Yang SS, Lee JS, Moon E, Lee K, Kim CH (2015) Sci Rep 5:18208
Chernets N, Kurpad DS, Alexeev V, Rodrigues DB, Freeman TA (2015) Plasma Process Polym 12:1400
Conway GE, Casey A, Milosavljevic V, Liu Y, Howe O, Cullen PJ, Curtin JF (2016) Br J Cancer 114:435
Duan J, Lu X, He G (2017) J Appl Phys 121:013302
Han D, Cho JH, Lee RH, Bang W, Park K, Kim MS, Shim JH, Chae JI, Moon SY (2017) Sci Rep 7:43081
Hou J, Ma J, Yu KN, Li W, Cheng C, Bao L, Han W (2015) BMC Genom 16:435
Ikeda J, Tsuruta Y, Nojima S, Sakakita H, Hori M, Ikehara Y (2015) Plasma Process Polym 12:1370
Kang SU, Cho JH, Chang JW, Shin YS, Kim KI, Park JK, Yang SS, Lee JS, Moon E, Lee K, Kim CH (2014) Cell Death Dis 5:e1056
Kim SJ, Chung TH (2015) Appl Phys Lett 107:063702
Kumar N, Attri P, Yadav DK, Choi J, Choi EH, Uhm HS (2014) Sci Rep 4:7589
Lee HJ, Shon CH, Kim YS, Kim S, Kim GC, Kong MG (2009) New J Phys 11:115026
Lee S, Lee H, Bae H, Choi EH, Kim SJ (2016) Sci Rep 6:30005
Ma Y, Ha CS, Hwang SW, Lee HJ, Kim GC, Lee KW, Song K (2014) PLoS ONE 9:e91947
Mizuno K, Yonetamari K, Shirakawa Y, Akiyama T, Ono R (2017) J Phys D Appl Phys 50(12L):T01
Nguyen NH, Park HJ, Yang SS, Choi KS, Lee JS (2016) Sci Rep 6:29020
Panngom K, Baik KY, Nam MK, Han JH, Rhim H, Choi EH (2013) Cell Death Dis 4:e642
Park SB, Kim B, Bae H, Lee H, Lee S, Choi EH, Kim SJ (2015) PLoS ONE 10:e0129931
Schmidt A, Bekeschus S, von Woedtke T, Hasse S (2015) Clin Plasma Med 3:24
Siu A, Volotskova O, Cheng X, Khalsa SS, Bian K, Murad F, Keidar M, Sherman JH (2015) PLoS ONE 10:e0126313
Suzuki-Karasaki Y (2016) Int J Mol Med 38:S50
Tabuchi Y, Uchiyama H, Zhao QL, Yunoki T, Andocs G, Nojima N, Takeda K, Ishikawa K, Hori M, Kondo T (2016) Int J Mol Med 37:1706
Tanaka H, Mizuno M, Toyokuni S, Maruyama S, Kodera Y, Terasaki H, Adachi T, Kato M, Kikkawa F, Hori M (2015) Phys Plasmas 22:122004
Torii K, Yamada S, Nakamura K, Tanaka H, Kajiyama H, Tanahashi K, Iwata N, Kanda M, Kobayashi D, Tanaka C, Fujii T, Nakayama G, Koike M, Sugimoto H, Nomoto S, Natsume A, Fujiwara M, Mizuno M, Hori M, Saya H, Kodera Y (2015) Gastric Cancer 18:635
Utsumi F, Kajiyama H, Nakamura K, Tanaka H, Hori M, Kikkawa F (2014) Springerplus 3:398
Yajima I, Iida M, Kumasaka MY, Omata Y, Ohgami N, Chang J, Ichihara S, Hori M, Kato M (2014) Exp Dermatol 23:424
Yan DY, Nourmohammadi N, Talbot A, Sherman JH, Keidar M (2016) J Phys D Appl Phys 49:274001
Yang H, Lu R, Xian Y, Gan L, Lu X, Yang X (2015) Phys Plasmas 22:122006
Golkowski M, Golkowski C, Leszczynski J, Plimpton SR, Maslowski P, Foltynowicz A, Ye J, McCollister B (1984) IEEE Trans Plasma Sci 2012:40
Kalghatgi S, Kelly CM, Cerchar E, Torabi B, Alekseev O, Fridman A, Friedman G, Azizkhan-Clifford J (2011) PLoS ONE 6:e16270
Girard PM, Arbabian A, Fleury M, Bauville G, Puech V, Dutreix M, Sousa JS (2016) Sci Rep 6:29098
Winterbourn CC (1995) Toxicol Lett 82–83:969
Winterbourn CC (1985) Biochim Biophys Acta 840:204
Hanschmann EM, Godoy JR, Berndt C, Hudemann C, Lillig CH (2013) Antioxid Redox Signal 19:1539
Peng X, Gandhi V (2012) Ther Deliv 3:823
Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, Delrue I, Taminau J, Wiernicki B, De Groote P, Garg AD, Leybaert L, Grooten J, Bertrand MJ, Agostinis P, Berx G, Declercq W, Vandenabeele P, Krysko DV (2016) Cell Rep 15:274
Lin A, Truong B, Pappas A, Kirifides L, Oubarri A, Chen S, Lin S, Dobrynin D, Fridman G, Fridman A, Sang N, Miller V (2015) Plasma Process Polym 12:1392
Ayan H, Fridman G, Gutsol AF, Vasilets VN, Fridman A, Friedman G (2008) IEEE Trans Plasma Sci 36:504
H. Ayan, D. Staack, G. Fridman, A. Gutsol, Y. Mukhin, A. Starikovskii, A. Fridman, G. Friedman (2009) J Phys D-Appl Phys 42:125202
Kluge S, Bekeschus S, Bender C, Benkhai H, Sckell A, Below H, Stope MB, Kramer A (2016) PLoS ONE 11:e0160667
Wende K, Bekeschus S, Schmidt A, Jatsch L, Hasse S, Weltmann KD, Masur K, von Woedtke T (2016) Mutat Res, Genet Toxicol Environ Mutagen 798–799:48
Schmidt A, Woedtke TV, Stenzel J, Lindner T, Polei S, Vollmar B, Bekeschus S (2017) Int J Mol Sci 18:868
Bekeschus S, Kolata J, Muller A, Kramer A, Weltmann K-D, Broker B, Masur K (2013) Plasma Med 3:1
Quijano C, Trujillo M, Castro L, Trostchansky A (2016) Redox Biol 8:28
Cortese-Krott MM, Koning A, Kuhnle GG, Nagy P, Bianco C, Pasch A, Wink DA, Fukuto J, Jackson AA, van Goor H, Olson KR, Feelisch M, Antioxid Redox Signal 2017
Prescott C, Bottle SE (2016) Cell Biochem Biophys 2016, 1
Graves DB (2012) J Phys D Appl Phys 45:263001
Jablonowski H, von Woedtke Th (2015) Clin Plasma Med 3:42
Wende K, Williams P, Dalluge J, Gaens WV, Aboubakr H, Bischof J, von Woedtke T, Goyal SM, Weltmann KD, Bogaerts A, Masur K, Bruggeman PJ (2015) Biointerphases 10:029518
Bekeschus S, Kolata J, Winterbourn C, Kramer A, Turner R, Weltmann KD, Broker B, Masur K (2014) Free Radic Res 48:542
Iseni S, Zhang S, van Gessel AFH, Hofmann S, van Ham BTJ, Reuter S, Weltmann KD, Bruggeman PJ (2014) New J Phys 16:123011
Schmidt-Bleker A, Winter J, Iseni S, Dunnbier M, Weltmann KD, Reuter S (2014) J Phys D Appl Phys 47:145201
Dunnbier M, Schmidt-Bleker A, Winter J, Wolfram M, Hippler R, Weltmann KD, Reuter S (2013) J Phys D Appl Phys 46:435203
Zerrouki A, Yousfi M, Rhallabi A, Motomura H, Jinno M (2016) Plasma Process Polym 13:633
Yan D, Talbot A, Nourmohammadi N, Sherman JH, Cheng X, Keidar M (2015) Biointerphases 10:040801
Jablonowski H, Bussiahn R, Hammer MU, Weltmann KD, von Woedtke T, Reuter S (2015) Phys Plasmas 22:122008
Lackmann JW, Schneider S, Edengeiser E, Jarzina F, Brinckmann S, Steinborn E, Havenith M, Benedikt J, Bandow JE (2013) J R Soc Interface 10:20130591
Pavlovich MJ, Sakiyama Y, Clark DS, Graves DB (1051) Plasma Process Polym 2013:10
Shimizu T, Nosenko T, Morfill GE, Sato T, Schmidt HU, Urayama T (2010) Plasma Process Polym 7:288
Winter J, Tresp H, Hammer MU, Iseni S, Kupsch S, Schmidt-Bleker A, Wende K, Dunnbier M, Masur K, Weltmannan KD, Reuter S (2014) J Phys D Appl Phys 47:285401
Bekeschus S, Iseni S, Reuter S, Masur K, Weltmann K-D (2015) IEEE Trans Plasma Sci 43:776
Winter J, Wende K, Masur K, Iseni S, Dunnbier M, Hammer MU, Tresp H, Weltmann KD, Reuter S (2013) J Phys D Appl Phys 46:295401
Brun P, Pathak S, Castagliuolo I, Palu G, Brun P, Zuin M, Cavazzana R, Martines E (2014) PLoS ONE 9:e104397
Priya Arjunan K, Morss Clyne A, Plasma Process Polym 2011, 8, 1154
Saito K, Asai T, Fujiwara K, Sahara J, Koguchi H, Fukuda N, Suzuki-Karasaki M, Soma M, Suzuki-Karasaki Y (2016) Oncotarget 7:19910
Balzer J, Heuer K, Demir E, Hoffmanns MA, Baldus S, Fuchs PC, Awakowicz P, Suschek CV, Oplander C (2015) PLoS ONE 10:e0144968
Adachi T, Kano A, Nonomura S, Kamiya T, Hara H (2016) Arch Biochem Biophys 606:120
Boehm D, Heslin C, Cullen PJ, Bourke P (2016) Sci Rep 6:21464
Adachi T, Tanaka H, Nonomura S, Hara H, Kondo S, Hori M (2015) Free Radic Biol Med 79:28
Bekeschus S, Wende K, Hefny MM, Rodder K, Jablonowski H, Schmidt A, Woedtke TV, Weltmann KD, Benedikt J (2017) Sci Rep 7:2791
Winterbourn CC (2008) Nat Chem Biol 4:278
Neyens E, Baeyens J (2003) J Hazard Mater 98:33
Girard F, Badets V, Blanc S, Gazeli K, Marlin L, Authier L, Svarnas P, Sojic N, Clement F, Arbault S (2016) Rsc Adv 6:78457
Bauer G, Graves DB (2016) Plasma Process Polym 2016
Bekeschus S, Winterbourn CC, Kolata J, Masur K, Hasse S, Broker BM, Parker HA (2016) J Leukoc Biol 100:791
Schmidt A, Rodder K, Hasse S, Masur K, Toups L, Lillig CH, von Woedtke T, Wende K, Bekeschus S (2016) Plasma Process Polym 13:1179
Norberg SA, Tian W, Johnsen E, Kushner MJ (2014) J Phys D Appl Phys 47:475203
Van der Paal J, Verlackt C, Yusupov M, Neyts E, Bogaerts A (2015) J Phys D Appl Phys 48:155202
Stoffels E, Sakiyama Y, Graves DB (2008) IEEE Trans Plasma Sci 36:1441
Oh JS, Szili EJ, Gaur N, Hong SH, Furuta H, Kurita H, Mizuno A, Hatta A, Short RD (2016) J Phys D Appl Phys 49:304005
Oh JS, Kojima S, Sasaki M, Hatta A, Kumagai S (2017) Sci Rep 7:41953
Acknowledgements
Funding at INP Greifswald was received from the German Federal Ministry of Education and Research (BMBF, Grant Numbers 03Z22DN11 and 03Z22DN12).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bekeschus, S., Lin, A., Fridman, A. et al. A Comparison of Floating-Electrode DBD and kINPen Jet: Plasma Parameters to Achieve Similar Growth Reduction in Colon Cancer Cells Under Standardized Conditions. Plasma Chem Plasma Process 38, 1–12 (2018). https://doi.org/10.1007/s11090-017-9845-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11090-017-9845-3