
Abstract
Accident tolerant fuel (ATF) cladding is a new type of nuclear fuel cladding designed to improve the safety and performance of nuclear reactors. In this paper, the kinetics and degradation mechanisms during high-temperature oxidation in steam of the three most promising ATF cladding materials, i.e., chromium-coated zirconium alloys, FeCrAl alloys, and silicon carbide-based composites, are described. Each system has its own degradation mechanisms leading to different maximum survival temperatures. After providing general information and data to understand the oxidation and degradation processes, illustrative examples obtained at the Karlsruhe Institute of Technology are given for each type of cladding. The maximum temperatures at which the barrier effect of the cladding can be maintained for a reasonable period of time during nuclear accident scenarios are 1200–1300 °C for Cr-coated Zr alloys, 1400 °C for FeCrAl alloys, and 1700 °C for SiC-based composite claddings.
Similar content being viewed by others


Introduction
At least since the accidents at Fukushima Daiichi, the need to develop new accident-tolerant materials for nuclear reactors has become obvious. One focus of international research is on accident-tolerant fuel (ATF) cladding materials, since the fuel cladding is an important barrier against the release of fission products and is in direct contact with the cooling water during operation and high-temperature steam during accident scenarios.
The classical zirconium alloy (Zry) cladding materials, which have been continuously developed over the last decades, perform excellently under operating conditions. The fuel failure rate in the water-cooled power reactors currently in operation is very low and tends to decrease [1]. However, in loss of coolant accident scenarios with temperatures above 1000 °C, the strong reaction of Zr alloys with water vapor leads to the loss of the barrier effect of the Zr-based cladding tubes as well as to a significant release of hydrogen and heat, which in turn influences accident progression [2,3,4]. The chemical heat released by the oxidation of zirconium can exceed the residual nuclear decay heat after reactor shutdown, and the hydrogen released has the potential to cause oxyhydrogen explosions as was dramatically seen in Fukushima Daiichi. In this context, the development of ATF cladding materials has focused on significantly improved high-temperature oxidation resistance, which reduces the effects described above and extends the coping time for accident response [5,6,7].
In general, the high-temperature oxidation resistance of metals, alloys, and non-oxide ceramics is based on the formation of a protective oxide layer, in particular of one of the three oxides alumina (Al2O3), chromia (Cr2O3), or silica (SiO2) [8, 9]. Consequently, after a decade of research, the three most promising candidates for ATF coatings are Cr-coated zirconium alloys, FeCrAl alloys, and silicon carbide (SiC)-based ceramic composites that form chromium oxide, aluminum oxide, and silicon oxide, respectively, at high temperatures in oxidizing atmospheres.
In recent years, an enormous amount of work has been published on the development and characterization of ATF cladding materials. The authors of this paper have mainly focused on the investigation of the oxidation and degradation behavior of these promising ATF cladding materials at very high temperatures [10,11,12,13,14,15,16,17,18,19,20,21]. Based on this experience, the purpose of this paper is to summarize the high-temperature oxidation and degradation mechanisms of the three main ATF cladding concepts and to derive the maximum “survival temperatures” under accident conditions. The information provided here are complementary to another, recently published overview paper by Kane et al. [22], which focuses on LOCA burst behavior of the three ATF cladding materials. Information on ATF cladding behavior at normal operation conditions are provided by other reviews, e.g., [23]. For each material, the main oxidation and degradation mechanisms will first be briefly discussed, and then illustrative examples of the results obtained at KIT will be given. This overview does not contain new experimental results. For further details, the cited papers are recommended.
General Considerations on High-Temperature Oxidation-Resistant Materials
Materials that are designed for application at high temperatures in oxidizing atmosphere are usually protected by the formation of a solid oxide, which separates the material from direct contact with the atmosphere. This oxide should be thermodynamically stable as well as provide low kinetics for the transport of reactants through the solid layer. In fact, only three oxides fulfill these conditions in steam atmosphere, namely chromia, alumina, and silica [8, 9]. Hence, ATF cladding should consist of materials that form one of these three oxides during high-temperature oxidation in steam atmosphere.
High-temperature oxidation of materials is often described by parabolic correlations, i.e., the exponent n in Eq. 1 equals 0.5. X can be the thicknesses of oxide scale, the mass gain or produced hydrogen, k is the rate coefficient, and t the time of isothermal oxidation. The exponent n is around one in the case of the formation of non-protective oxides and may take other values depending on the predominant mechanism of the oxidation reaction. Over a temperature regime with a consistent rate limiting mechanism, the temperature dependence of k can follow an Arrhenius equation with the pre-exponential factor k0 and activation energy EA (Eq. 2).
The diagram in Fig. 1 compares the parabolic oxidation rate constants of selected materials and shows 2–4 orders of magnitude lower oxidation rates of potential ATF cladding materials compared to zirconium alloys and classical reactor steel. The high-temperature oxidation resistance of the formed oxides increases in the order Cr2O3 < Al2O3 < SiO2 by approximately one order of magnitude each.

Oxidation kinetics of potential ATF cladding materials forming Cr2O3, Al2O3, and SiO2, respectively, in comparison with Zr alloy and stainless steel [6]
From a thermochemical point of view, all oxides discussed here are stable at high temperatures, as shown by the Gibbs free energy curves in Fig. 2. Zirconia and alumina are the most stable, followed by silica and chromia, and iron oxide is by far the least stable oxide. A similar order applies to the reaction enthalpy values for the oxidation of metals and SiC. The oxidation of zirconium alloys is accompanied by a considerable release of chemical energy equivalent to half the calorific value of mineral oil (Zr-steam: 6 MJ/kgZr; mineral oil: 12 MJ/kgoil). The ΔHR values for the oxidation of aluminum are quite similar to those for the oxidation of zirconium. The oxidation of chromium, iron, and silicon carbide is significantly less exothermic, with almost no heat being released during the oxidation of iron by steam.

Gibbs free energy of reaction and enthalpy of reaction for the oxidation of relevant oxides by water vapor. Calculated with HSC Chemistry [24]
A factor in the maximum operating and survival temperatures of cladding are the melting temperatures of the cladding and corresponding oxides, Table 1. Most materials have higher melting temperatures than Zr alloys, with the exception of FeCrAl alloys and the potentially formed iron oxide FeO. The latter can react eutectically with the UO2 and cause premature liquefaction of the fuel (see Sect. Main results of the bundle experiment QUENCH-19).
Although not directly related to oxidation, the volatility of protective oxides at high temperatures in steam can affect the oxidation resistance of materials. The formation of volatile oxides, hydroxides, and oxyhydroxides can lead to a regression of the oxide scales and thus to a reduction in the protective effect and to higher oxidation rates than would be expected based on parabolic oxidation kinetics. For zirconia, this effect is negligible up to very high temperatures, but the volatility in a water vapor-containing atmosphere is orders of magnitude higher for the other protective oxides, in the order Al2O3 < SiO2 < Cr2O3 [25,26,27]. Generally, oxide volatility at high temperatures is more relevant for long exposure times, e.g., in turbines, but for short accident scenarios, volatility becomes inconsequential.
Mechanical effects, i.e., the formation of tensile and compressive stresses during the growth of oxide scales may also influence their protectiveness. In most cases, the specific volume of the formed oxide is not the same as that of the consumed metal. The sign of the stress in the oxide is related to the Pilling–Bedworth ratio (PBR), which is defined by Eq. 3.
with Vox being the molar volume of the formed oxide and Vm the molar volume of the consumed metal. As is shown by the values in Table 2, all oxides relevant here take up a larger volume than the consumed metals and should build up compressive stresses during their growth. Details are discussed in the sections below.
Other causes of growth stresses discussed by Birks [9] include epitaxial stresses, compositional changes in the alloy or scale, recrystallization stresses, stresses due to oxide formation within the scale, and stresses caused by specimen geometry deviating from planar geometry. The latter should apply to cladding tubes, where the compressive stresses caused by the PBR are partially compensated by the convex shape of the rod-shaped geometry.
In summary, the protective effect of the oxides formed on the materials depends on their thermodynamic stability, on their ability to act as diffusion barriers for oxygen (or other oxidizing species) and metal ions, and thus on the kinetics of the oxidation reaction as well as on other factors like adhesion and mechanical behavior. In the following sections, the oxidation behavior of the three most promising ATF cladding types, i.e., Cr-coated Zr alloys, FeCrAl alloys, and SiC-based composite materials will be discussed in more detail.
Chromium-Coated Zirconium Alloys
Cr-coated zirconium alloys are the near-term solution for ATF cladding materials, which should be associated with only moderate changes in technology and licensing procedures. They are developed worldwide according to different coating methods with typical Cr coating thicknesses of 5–30 µm. Thicker layers would have a negative impact on the neutron economy and increase the risk of thermomechanical stresses between the Cr coating and the Zry bulk. In general, well-produced Cr coatings provide very good performance under harsh operating conditions and result in a 1–2 orders of magnitude reduction in high-temperature oxidation kinetics compared to zirconium alloys. The first lead test assemblies (LTAs) are already being used in commercial reactors in the US and Europe. Recent reviews on Cr-coated cladding have been published, for example, by Bischoff [28], Brachet [29, 30], Maier [31], Yeom [32], and Yang [33].
High-Temperature Oxidation and Degradation of Cr Coatings on Zr Alloys
The oxidation by water vapor of the chromium results in the formation of a well-adherent and protective Cr2O3 scale according to Eq. 4. This is the intended effect; however, it also causes consumption of the thin coating layer.
Further degradation mechanisms of the protective effect of Cr coatings can be easily derived from the binary Zr–Cr phase diagram [34], Fig. 3. According to this, the interaction between chromium coating and zirconium bulk results in the formation of the intermetallic compound ZrCr2 and the diffusion of Cr into the Zr bulk. Especially, the solubility of Cr in the high-temperature β-Zr phase is considerable. Both effects lead to the consumption of the Cr coating. Furthermore, also according to the Zr–Cr phase diagram, the eutectic interaction between Zr and Cr leads to the formation of melt at 1332 °C and should characterize the ultimate upper limit for the stability of Cr coatings.

Phase diagram of Zr–Cr (adapted from [34]) and its implications on Cr coating degradation
The volatilization of the protective oxide scale may have to be taken into account at very high temperatures and for longer durations, especially for chromium oxide with the highest volatilization rate among the high-temperature oxidation-resistant oxides [27]. Volatilization of chromium trioxide, CrO3 (Eq. 5), under dry oxidizing conditions and of chromium oxyhydroxides, e.g., CrO2(OH)2 (Eq. 6), in steam-rich atmospheres may cause formation of thinner oxide scales and higher metal recession than expected from the parabolic growth kinetics [35]. However, Brachet [30] investigated and discussed the volatilization of Cr2O3 and concluded that it can be neglected at least up to 1300 °C in steam atmosphere.
One reason for this favorable behavior is that the partial pressure of the Cr-containing gas species in pure water vapor is 2–3 orders of magnitude lower than in oxygen-containing atmospheres, since both chemical reactions require oxygen as a starting product (Eqs. 5 and 6) [36]. Consequently, significant volatilization of chromium oxide in water vapor (relevant for the short durations of nuclear accident scenarios) starts only at temperatures beyond 1400°C (Fig. 4) and should play a role only at temperatures where other degradation processes cause the protective effect to fail. However, chromia volatilization should be considered in air ingress scenarios and in analyzes of severe accidents of fuel assemblies with ATF cladding tubes, since volatile Cr-containing species can affect fission product chemistry in the circuit and in the containment.

Total gas partial pressure of chromium-containing species over chromia in water vapor, oxygen, and mixed atmosphere. Calculated with HSC Chemistry [24]
The oxidation kinetics of (bulk) chromium is mainly determined by the diffusion of metal cations through the growing oxide scale to the surface. Inward transport of oxygen ions along grain boundaries is also observed, resulting in compressive stresses by oxide formation at the metal-oxide interface or within the scale [9, 37, 38]. The parabolic rate constants for the oxidation of chromium in oxygen, air, and steam scatter widely. This could be caused by the different contributions of inward oxygen diffusion through oxide layers with different grain sizes and textures of the investigated materials. Brachet [30] summarized literature data on the parabolic rate coefficients for the oxidation of chromium in different atmospheres and illustrated the scatter over several orders of magnitude, but also recommended a correlation for the parabolic oxidation kinetics in steam based on recent data from CEA and Framatome, see Table 3 in the discussion section.
As long as the Cr coating is protective, a four-layer structure is found in agreement by all research groups, consisting of bulk-Zry, ZrCr2 interlayer, remaining Cr coating, and formed Cr2O3 layer. With increasing oxidation time and temperature, chromium diffuses into the Zry bulk forming ZrCr2 precipitates during cooling. These precipitates are preferentially found in the former β-Zr phase. The protective Cr coating effectively hinders oxygen (and hydrogen) diffusion into the bulk material [30, 39].
Detailed investigations of the Cr coating degradation mechanisms resulting in transition to non-protective coating were made by various research groups [11, 12, 30, 40]. Depending on temperature and Cr coating thickness and morphology, they show the transition from protective to non-protective behavior at 1200 °C after 30 min to 3 h and at 1300 °C after a few tenths of minutes. In addition to the above-discussed degradation effects, the main mechanism for this transition is the diffusion of zirconium along chromium grain boundaries toward the Cr/Cr2O3 interface and its oxidation by redox reaction with the Cr oxide, Eq. 7. Finally, a through-going network of zirconia is formed, which allows fast oxygen transport to the bulk Zry leading to the formation of oxygen-stabilized α-Zr(O) and, after its saturation, of ZrO2 as illustrated in Fig. 5. At this state, also the intermetallic phase ZrCr2 is oxidized according to Eq. 8.

The whole mechanism, especially the redox reaction, Eq. 7, causes the (at a first glance surprising) effect of growth and reduction with time of the Cr2O3 layer, on the one hand, and the consumption and subsequent growth of metallic Cr layer, on the other hand [12, 41].
Various authors discuss the formation of blisters/bubbles/voids during high-temperature oxidation of Cr coatings. This could be related to the compressive stresses in the oxide because of the high Pilling–Bedworth ratio for the chromium oxidation [29], Table 2. Other mechanisms for the formation of pores of various sizes discussed are vacancy condensation and the reaction between Zr and Cr2O3 [12], segregation of Sn and Cr in the surface region of the substrate close to the Cr coating [42], and the Kirkendall effect due to the different diffusion rates of the atoms involved [13, 30, 43].
Single-Rod Experiments with Cr-Coated Fuel Rod Simulators
Single-rod oxidation and quench experiments were conducted in cooperation between Westinghouse, EPRI (US) and KIT (Germany) [10]. 10-cm long Optimized ZIRLO™ samples with cold-sprayed (CS) and PVD coating of different thicknesses (25 µm and 10 µm, respectively) were inductively heated in steam atmosphere under transient and isothermal conditions and quenched by a rising cylinder filled with water. For illustration, Fig. 6 shows such samples as coated, during testing in the QUENCH-SR (single-rod) facility with inductive heating, and after a transient test.

Cr-coated samples before (left), during (mid), and after transient experiment at up to > 1500 °C
Generally, less oxidation, measured online by mass spectrometry of the released hydrogen, was observed for the coated samples compared to ZIRLO reference samples. At 1100 °C, all coating remained protective for at least 1 h. At 1200 °C, only the thicker CS coating remained intact for 1 h, whereas the thinner PVD coating started transition to non-protective behavior after approximately 20 min as can be seen in Fig. 7 (left). Toward the end of these tests, the oxidation rate of the PVD Cr-coated sample reached the same level as that of the non-coated reference sample, however, the integral hydrogen release remained much lower for both coated samples. The failure of the Cr coating according to the mechanisms described above resulted in the formation of ZrO2 underneath the remaining Cr/Cr2O3 layers, see Fig. 8.

Hydrogen release during isothermal tests at 1200 °C (left) and transient tests with 10 K/min heating rate at up to > 1500 °C (right) with Cr-coated fuel rod simulators

Micrographs of Cr-coated ZIRLO cladding after 1 h isothermal oxidation at 1200 °C and after transient tests (10 K/min) up to ca. 1550 °C in steam. Taken from [10]
The post-test ductility was also much better for the coated sample: The uncoated sample easily broke just during handling. The Cr coating layer revealed excellent thermal shock resistance and adherence, without cracking and spallation.
In transient tests from 800 to > 1500 °C, lower oxidation rates were observed, as expected, for the coated samples compared to the uncoated one up to approx. 1350 °C. At higher temperatures, the oxidation rate of the (formerly) coated samples became significantly higher than for the uncoated one, see Fig. 7 (right). This rapid increase in oxidation rate should be caused by the failure of the Cr coating after passing the eutectic temperature in the Cr–Zr system. The intermediate decrease in the oxidation kinetics is caused by the formation of zirconia. The second peak in oxidation kinetics at around 1500 °C is due to the phase transition from tetragonal to cubic zirconia [3, 44]. In a recent study, this behavior was confirmed, and a strong correlation was found between the acceleration of the reaction rate after coating failure and the heating rate [45]. The cladding tubes were severely oxidized after the transient oxidation tests as seen in Fig. 8, but remained in shape with the typical “crocodile skin” surface, Fig. 6.
Another result of this series of experiments was that pre-damage of the Cr coating by scratches caused locally increased oxidation only where the scratches passed through the Zr alloy. No delamination of the adjacent Cr scale layer was observed, and the scratches had little effect on the overall hydrogen release [10].
FeCrAl Alloys
FeCrAl alloys are ferritic body-centered cubic (bcc) iron-based alloys with 12–24 wt% Cr and 3–6 wt% Al contents. Beside their potential application to ATF cladding [46,47,48], they have long been used for for high-temperature applications such as for heating elements in industrial furnaces and for gas burners [49].
Nuclear-grade iron-based FeCrAl alloys with optimized composition to perform under both normal and off-normal conditions have been mainly developed in the US [46, 50, 51] and Japan [48, 52, 53]. They usually have lower Cr contents (12–13 wt%) compared to commercial Kanthal alloys (20–22 wt%) to reduce neutron irradiation-induced embrittlement during operation [54]. The Al content is 5–6 wt%. In addition, FeCrAl alloys may contain minor alloying elements to improve oxidation resistance (by e.g., reactive elements), microstructure, mechanical properties, and processability as well as ODS (oxide dispersed strengthened) particles [48, 55, 56].
Oxidation of FeCrAl Alloys at High Temperatures
The superior oxidation resistance of FeCrAl alloys at high temperatures relies on the formation of a slowly growing and highly protective α-Al2O3 scale. A common understanding is that α-Al2O3 forms on FeCrAl alloys at temperatures exceeding 900–1000 °C.
Generally, all three elements of FeCrAl alloys can be oxidized by water vapor resulting in the formation of the corresponding oxides; see Eq. 9.
Al2O3 and Cr2O3 are the only stable solid oxides in the corresponding binary metal–oxygen systems. In the Fe–O system, Fe3O4 (magnetite) and FeO (wustite) with lower oxygen contents are known beside the sesquioxide Fe2O3 (hematite). All sesquioxides form solid solutions with each other with corundum or spinel-type structure [57]. Furthermore, ternary compounds can be formed of which the spinel-type compounds AB2O4 are worth mentioning [58].
The thermodynamic stability of the oxides is in the order Al2O3 > Cr2O3 > Fe2O3, i.e., alumina is the most stable oxide forming at the lowest oxygen partial pressure, which promotes the selective oxidation of aluminum at high temperatures. Alumina formation is also supported by Cr, as can be seen in Fig. 9. This so-called third-element effect reduces the amount of aluminum necessary to form alumina from 9 wt% in the binary system to 4 wt% with 10–15 wt% chromium added [59, 60]. Several metastable aluminum oxides are known, which may also be formed during oxidation of FeCrAl alloys [61]. All forms of alumina, except for α-Al2O3, are termed transient aluminas, which are thermodynamically meta-stable and fast-growing and hence have fairly poor protective properties [50, 62, 63].

Oxide map of the Al-Cr-Fe system at 1000 °C, after Scheil [64]
Even if alumina formation is thermodynamically preferred, other oxides may form due to kinetic limitations during the initial transient phase and in the case of too low aluminum concentration in the alloy or due to Al depletion in the bulk alloy during oxidation [65]. After the initial phase of oxidation, the formation of a protective α-alumina scale is determined by the competition between the oxidation rate governed by diffusion of Al and O through the oxide scale and the diffusion of aluminum in the substrate to the interface [66]. Al is much more mobile than Fe and Cr in FeCrAl alloys at high temperatures [67], which supports the formation of protective Al2O3 also on alloys with relatively low Al content.
Recent results on oxidation mechanisms and kinetics of FeCrAl alloys show only a moderate dependence of the parabolic rate constants on the alloy composition as long as a protective alumina scale is formed in the temperature range of approximately 1000–1400 °C [16, 17, 47]. A recommended correlation based on data by Pint [5] is given in Table 3. At lower temperatures, the oxidation kinetics is determined by the formation of Al-rich transient oxides, and it is therefore 2–3 orders of magnitude higher than the extrapolated correlation for α-alumina, see Fig. 10. However, this has only a limited effect on accident scenarios because of the generally low level of oxidation rates between 600 and 900 °C. Of greater relevance in this context is the tendency of nuclear-grade FeCrAl alloys with 10–13 wt% Cr to enter catastrophic oxidation at temperatures as low as 1300–1400 °C, i.e., at lower temperatures than the commercial APM and APMT Kanthal alloys. As shown in Fig. 10, the by five orders of magnitude higher oxidation rates under these conditions correlate with the kinetics of iron oxide formation [17], and this usually results in fast and complete oxidation of FeCrAl samples. The onset of catastrophic oxidation is dependent on alloy composition and heating rate with earlier transition for high heating rates [16].

Parabolic rate constants of nuclear-grade alloys B136Y3 (13Cr-6Al) and C26M2 (12Cr–6Al–Mo–Si–Y) as a function of temperature with the parabolic correlations of Fe-oxide, α-alumina, and θ-alumina. Taken from [17]
The volatilization of aluminum oxide in steam is very low and should not play a significant role as long as the oxidation is determined by a protective α-alumina scale [27].
Main Results of the Bundle Experiment QUENCH-19
The first large-scale bundle test with ATF cladding material, namely with the FeCrAl alloy B136Y3 (Century Tubes Inc. /ORNL), was conducted in the QUENCH facility at KIT in cooperation with ORNL, USA, in 2018 [68, 69]. This facility is a large-scale bundle facility with 24 electrically heated, > 2 m long fuel rod simulators using zirconia annular pellets as fuel simulator [2], see Fig. 11 (left). It is extensively instrumented with high-temperature thermocouples, pressure gauges, level meters, etc., and coupled with a mass spectrometer for analysis of hydrogen and other gases. Usually, experiments are completed by reflooding (which gave the facility the name). The QUENCH-19 experiment with FeCrAl cladding was run with a very similar scenario as test QUENCH-15 with uncoated ZIRLO cladding with respect to bundle geometry and power input [70]. The FeCrAl experiment was run 2000s longer at the highest power input, until first local melting of the FeCrAl cladding was expected. Hence, the FeCrAl bundle was quenched 2000s later than the ZIRLO bundle.

Schematic of the QUENCH facility during the reflood phase (left), and post-test view of the FeCrAl bundle after experiment QUENCH-19
As expected, the hydrogen release was much less in the FeCrAl experiment: Until the time when the QUENCH-15 test was terminated by reflood, 100 × less hydrogen was produced in the QUENCH-19 experiments (0.4 g and 40 g, respectively), see Fig. 12. Even at the end of the QUENCH-19 test after a 2000s longer heating time at the highest power level, only 9 g hydrogen were measured compared to 47 g in the shorter test with ZIRLO.

Hydrogen release during experiment QUENCH-19 (FeCrAl), and reference test QUENCH-15 (ZIRLO)
Some melt formation was seen in the bundle after the test, which also came from stainless steel (SS 304) thermocouple sheaths. Rupture of some cladding tubes was observed to most probably have been caused by strong shrinking during the quench phase due to the 2 × higher thermal expansion coefficient of FeCrAl compared to Zr alloys, Fig. 11. Only low oxidation of the FeCrAl surfaces unaffected by melt attack was seen along the whole bundle. The post-test examinations of the QUENCH-19 bundle revealed another interesting phenomenon. At positions where oxidized FeCrAl melt was formed, a strong interaction between FeO and the ZrO2 pellets occurred due to the low eutectic temperature of 1332°C in the FeO–ZrO2 system [71]. This is of interest because the phase diagram of the FeO–UO2 system is very similar with a eutectic temperature of 1335 °C [72], which poses the potential risk of fuel relocation at very low temperatures compared to the melting temperature of uranium oxide (2865 °C).
Post-test calculations of the QUENCH-19 experiment in the framework of the IAEA project ACTOF resulted in much too low values of the hydrogen release during the test [73]. One important reason for this is that the codes used only one correlation for the oxidation of FeCrAl alloy in the solid state resulting in an underestimation of the hydrogen release below and above the validity temperature region for α-alumina formation as shown in Fig. 10.
Silicon Carbide Composites
Silicon carbide ceramic composites are intensively studied for application as heating elements and high-temperature structural materials e.g., for gas turbines, space travel, and in the nuclear industry. They offer excellent high-temperature mechanical properties, low density, high oxidation and irradiation resistance as well as very low interaction with thermal neutrons. Silicon carbide composites are considered the most promising long-term candidates for ATF cladding tubes in LWRs with regard to oxidation resistance at very high temperatures [19, 74, 75].
High-Temperature Oxidation and Degradation of Silicon Carbide
The oxidation of SiC is more complex than that of zirconium alloys due to the formation of volatile reaction products and its stronger dependence on boundary conditions. In general, its high-temperature oxidation resistance is based on the formation of a protective SiO2 layer [19]. Many different phases of SiC as well as of SiO2 exist, which is not discussed further here.
In dry atmospheres, like oxygen or air, passive oxidation with the formation of a protective silica scale (Eq. 10) or active oxidation with the formation of gaseous silicon monoxide (Eq. 11) may take place, depending on temperature and oxygen partial pressure. The rate-controlling process for passive oxidation is the diffusion of oxygen through the growing oxide scale [76]. It is observed at high oxygen partial pressure and lower temperature [77, 78]. Active oxidation may be an issue for instance for high-temperature application of SiC components in technical helium cooling gas with oxidizing impurities like in high-temperature gas-cooled reactors [20].
A protective silica scale is also produced during oxidation of silicon carbide in steam-containing atmosphere, see Eq. 12 and Table 3. Hydrogen is released in addition to carbon-containing gases. At very high temperatures, the formation of volatile hydroxides or oxyhydroxides may become significant, e.g., according to Eq. 13 and Eq. 14. Formation and volatilization of a silica scale at the same time finally lead to paralinear oxidation kinetics as illustrated in Fig. 13.

Paralinear mass change during HT oxidation of SiC in steam due to formation and volatilization of a superficial SiO2 scale
Many authors observed formation of bubbles in the silica scale especially during high-temperature oxidation in steam-containing atmospheres [18, 79,80,81,82]. Pressure can build up by the formation of CO and H2 at the SiC/SiO2 interface if the permeability of inward-diffusing oxidizing species is higher than that of the outward-moving gaseous reaction products. The tendency to bubble formation increases with temperature and increasing water vapor partial pressure. However, the effect of bubble formation on the overall oxidation kinetics does not seem to be too important according to the experience of the authors, e.g., bubble formation during isothermal tests with SiCf/SiC cladding segments in steam at 1700 °C did not affect the oxidation kinetics appreciably [21]. Another mechanism of degradation and bubble formation at the SiC/SiO2 interface is the reaction of both compounds resulting in the formation of gaseous monoxides SiO and CO, Eq. 15. However, this reaction takes place only at temperatures above approximately 1800 °C according to the thermodynamic calculations [24].
Impurities in the furnace, the sample, or the oxidizing atmosphere might strongly affect oxidation kinetics [26]. This is one of the reasons why published data on high-temperature oxidation of silicon carbide are so scattered. The oxidation process is accelerated by the presence of impurities and residual sintering additives, which affect the microstructure of the protective SiO2 scale.
SiC ceramic matrix composites (CMC) themselves, consisting of SiC fibers typically coated with pyrolytic carbon (PyC) in a SiC matrix have only a limited high-temperature oxidation resistance [81, 83, 84]. The carbon layer is easily oxidized and the CMC provides a large surface for oxidation. Therefore, all SiC CMC-based cladding tube designs contain a superficial monolithic SiC environmental barrier coating (EBC) or seal coat [75, 81, 85].
An upper temperature limit for a reasonable oxidation resistance should be given by the melting temperature of the formed silica scale at around 1725 °C, but for a short period, also higher temperatures have been reported without severe degradation of SiC-based cladding tube segments [18].
Single-Rod Experiments with SiC CMC Cladding Segments
Isothermal oxidation tests and a transient experiment in steam until failure were conducted with SiC CMC cladding tube segments produced by CEA (France) in the QUENCH-SR facility at KIT [21, 86]. The samples were filled with graphite rods acting as susceptor for inductive heating, see Fig. 14.

SiC CMC samples as received (left), X-ray tomography showing end plugs and graphite core (mid), and after 1 h steam oxidation at 1700 °C (right)
The isothermal tests ran at 1700 °C for one and three hours, respectively, and were terminated by quenching with water. Even though formation of bubbles was observed during the experiments, all samples survived the harsh test conditions with only slight modification of the surface, Fig. 14. The surface was covered by 1–3 µm thick silica scales. Mechanical properties were only slightly degraded [21].
The release of gaseous reaction products, mainly H2, CO2, and CO, was measured in-situ by mass spectrometry. Rather constant low release of all these gases established soon (1–3 min) after initiating the steam injection. This indicated a quick establishment of an equilibrium of silica formation (parabolic kinetics) and volatilization (linear kinetics) under these conditions. Based on the hydrogen release data, the SiC recession rate was estimated to be 15–25 µm/h under the conditions of these experiments (T = 1700 °C, P = 1 bar, PH2O = 0.65 bar, and Fgas = 5 cm/s).
A transient test with 10 K/min heating rate in steam was conducted from 1400 °C until failure of the sample, defined by strongly increasing gas release rates and formation of white smoke. As shown in Fig. 15, the gas release rates (i.e., oxidation rate) increased up to approximately 1500 °C and then remained nearly constant up to 1700 °C, before increasing dramatically by a factor of 20 at about 1820 °C. This transition was most probably caused by the (local) failure of the protective SiC seal coat and the steam attack of the fiber-matrix composite, as can be seen in Fig. 16.

Temperature and gas release during the transient test

Post-test appearance of the SiC-CMC sample after the transient test up to > 1800 °C
Discussion
The high-temperature oxidation resistance of the three most promising ATF cladding concepts, i.e., Cr-coated Zr alloys, FeCrAl alloys, and SiC composites, relies on the formation of one of the protective oxides chromia, alumina, and silica. This agrees with the generally accepted knowledge of the high-temperature oxidation behavior of materials. The growth kinetics of these oxides can be described by parabolic correlations according to Equs.1 and 2.
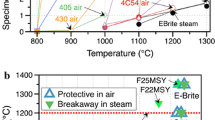
Table 3 and Fig. 17 provide data for the parabolic rate constants, here especially for oxide scale growth, according to Eqs. 16 and 17 (with dox oxide thickness, t time, K0 pre-exponential constant, EA activation energy, gas constant R = 8.314 J/Kmol, and T temperature) selected from the literature for comparison. These correlations are not universally valid, because they cover only a certain temperature range or a special material, but they illustrate the improved oxidation kinetics of the ATF cladding materials compared to Zry and can be used for a first estimation of the expected oxide thickness at a given temperature and time. Furthermore, these correlations do not take into account the volatilization of oxides, especially of silica [87] and possibly of chromia for temperatures higher than the protective behavior of Cr coatings [88]. The volatilization kinetics of oxides is strongly dependent on the thermal–hydraulic boundary conditions, and its detailed discussion is beyond the scope of this paper. Typical oxide thicknesses on Zr alloys under severe accident conditions are > 100 µm, about 10 µm chromia, and around 1 µm alumina or silica, see also Fig. 18.

Arrhenius plot of the parabolic rate constants for the oxidation of ATF cladding materials. Correlations taken from Table 3

Examples of oxide scale formation during high-temperature oxidation in steam for ATF cladding materials and Zircaloy-4 for comparison. The corresponding thicknesses of ZrO2 under the same conditions as for the ATF materials would be 200 µm, 350 µm, and 2400 µm, respectively
The degradation mechanisms, and with it the maximum application/survival temperatures, significantly differ between the three ATF cladding concepts. The protective effect of the Cr-coated zirconium alloys is not only affected by the oxidation of the chromium but also by interactions between Cr coating and Zry bulk as discussed above. The ultimately limiting mechanism is the eutectic melt formation in the system Cr–Zr at 1332 °C. At lower temperatures, the thickness of the Cr coating plays an important role; thicker Cr layers provide a higher chromium reservoir and usually withstand high-temperature conditions for a longer time without losing their protective effect [90]. A recent study [91] showed the preservation of the protective effect of a 90 µm thick Cr coating at 1300 °C for 1 h, while thinner and for ATF cladding application more prototypic coatings showed a transition to non-protective behavior at 1200 °C already after about 30 min [10, 12]. Beside the thickness of the Cr coating, its quality and morphology do also affect the protectiveness of the coating [29, 90,91,92,93,94]. Chromia volatility seems not to play a role for the degradation of the coating for the temperature range discussed here and the relatively short durations of hours considered for nuclear accident scenarios. A serious problem for any type of coating on zirconium alloys can be a possible temperature runaway after loss of the protective effect of the coating and the consequent intense Zr-steam reaction. The investigation of this effect will be one of the objectives of the OECD-NEA Joint Undertaking QUENCH-ATF [95].
The high-temperature oxidation resistance of FeCrAl alloys is based on the formation of a protective α-alumina scale. This forms after initial formation of transient oxides of all three metals in the alloy in the temperature range between 1000 and 1400 °C. Nuclear-grade FeCrAl alloys should have low Cr and Al contents for optimized radiation resistance. Low Cr and Al contents can impair the HT oxidation resistance; therefore, a compromise must eventually be found. Nuclear-grade FeCrAl alloys with 10–13 wt% Cr generally show a better oxidation resistance in steam than commercial Kanthal alloys at intermediate temperatures of 1000–1300 °C, but tend to suffer catastrophic oxidation at higher temperatures of around 1400 °C [17, 51]. The transition temperature to catastrophic oxidation is quite stochastic even under very similar conditions and needs further investigation. It is dependent on the composition, morphology, and surface finish of the cladding as well as on the heating rate and other boundary conditions.
The high-temperature oxidation resistance of SiC-based composites for nuclear cladding tubes is based on the formation of a protective silica layer on the monolithic outer SiC layer. The fiber matrix composite itself has a low resistance to oxidation mainly due to the interphase between fiber and matrix that is usually made of carbon. The limited number of publications on the oxidation behavior of SiC-based cladding at very high temperatures seems to confirm high oxidation resistance up to at least 1700 °C lasting for several hours. At that high temperature, volatilization of the formed silica scale as well as bubble formation must be taken into account as degradation mechanisms. Generally, the oxidation of SiC materials is strongly dependent on impurities (in material and atmosphere) as well as on experimental conditions (pressure, gas flow rates, gas composition). This complicates the comparison of data obtained in different labs and explains the wide scatter of published data.
Conclusions
This paper summarized oxidation and degradation of the three most promising ATF cladding concepts, discussed the limiting mechanisms for protection at very high temperatures, and provided illustrative examples of experimental work at KIT.
All ATF claddings discussed in this paper provide strongly improved high-temperature oxidation resistance compared to classical zirconium alloys. The examples provided in the paper and from the literature (Fig. 18) give impressive proof of the significantly improved high-temperature oxidation resistance of the discussed ATF cladding materials compared to zirconium alloys. They could thus help to reduce the risk of temperature escalation and hydrogen detonation in severe accidents as well as increase the coping time for accident management measures.
The maximum survival temperatures for the ATF cladding materials derived in this paper are:
-
1200–1300 °C for Cr-coated Zry
-
around 1400 °C for nuclear-grade FeCrAl alloys with 10–13 wt% Cr
-
1700 °C for SiCf-SiC composites with monolithic external SiC layer.
However, various issues with all these materials must be solved for successful introduction in NPP fuel elements. Among these, potential temperature runaway after loss of the Cr coating protective effect, interaction of oxidized FeCrAl with the fuel at low temperatures, and hydrothermal corrosion and leak tightness of silicon carbide should be mentioned. Current national research activities especially in France, Japan, and the USA, as well as international research collaborations in the framework of the OECD-NEA (QUENCH-ATF) [95], IAEA (ATF-TS) [97], and the European Union (IL TROVATORE [98], SCORPION) will help to resolve the open questions toward the application of ATF cladding in nuclear reactors.
Data Availability
Experimental data produced at KIT will be made available on request.
References
V. Inozemtsev, and V. Onufriev, in LWR Fuel Performance Meeting, Top Fuel 2013 (2013).
M. Steinbrück, M. Große, L. Sepold, and J. Stuckert, Nuclear Engineering and Design 240, 1714 (2010).
M. Steinbrück, in Encyclopedia of Materials: Metals and Alloys, vol. 1, ed. F. G. Caballero (Elsevier, Oxford, 2022), p. 454.
S. Zinkle, K. Terrani, J. Gehin, L. Ott, and L. Snead, Journal of Nuclear Materials 448, 374 (2014).
B. Pint, K. Terrani, Y. Yamamoto, and L. Snead, Metallurgical and Materials Transactions E 2, 190 (2015).
K. Terrani, Journal of Nuclear Materials 501, 13 (2018).
C. Tang, M. Stueber, H. Seifert, and M. Steinbrueck, Corrosion Reviews 35, 141 (2017).
D. J. Young, High Temperature Oxidation and Corrosion of Metals, (Elsevier Ltd., Amsterdam, 2016).
N. Birks, G. Meier, and F. Pettit, Introduction to the High Temperature Oxidation of Metals, 2nd ed (Cambridge University Press, Cambridge, 2006).
M. Steinbrück, U. Stegmaier, M. Große, L. Czerniak, E. Lahoda, R. Daum, and K. Yueh, Journal of Nuclear Materials 559, 153470 (2022).
J. Liu, C. Tang, M. Steinbrück, J. Yang, U. Stegmaier, M. Große, D. Yun, and H. Seifert, Corrosion Science 192, 109805 (2021).
J. Liu, M. Steinbrück, M. Große, U. Stegmaier, C. Tang, D. Yun, J. Yang, Y. Cui, and H. Seifert, Corrosion Science 202, 110310 (2022).
J. Yang, U. Stegmaier, C. Tang, M. Steinbrück, M. Große, S. Wang, and H. Seifert, Journal of Nuclear Materials 547, 152806 (2021).
J. Liu, R. Meng, M. Steinbrück, M. Große, U. Stegmaier, C. Tang, J. Yang, and D. Yun, Journal of Nuclear Materials 573, 154144 (2023).
E. Kashkarov, D. Sidelev, N. Pushilina, J. Yang, C. Tang, and M. Steinbrueck, Corrosion Science 203, 110359 (2022).
C. Tang, A. Jianu, M. Steinbrueck, M. Grosse, A. Weisenburger, and H. Seifert, Journal of Nuclear Materials 511, 496 (2018).
C. Kim, C. Tang, M. Grosse, Y. Maeng, C. Jang, and M. Steinbrueck, Journal of Nuclear Materials 546, 153696 (2022).
V. A. Avincola, M. Grosse, U. Stegmaier, M. Steinbrueck, and H. Seifert, Nuclear Engineering and Design 295, 468 (2015).
H. Pham, M. Kurata, and M. Steinbrueck, Thermo 1, 151 (2021).
M. Steinbrueck, V. A. Avincola, I. J. Markel, U. Stegmaier, U. Gerhards, and H. J. Seifert, Journal of Nuclear Materials 517, 337 (2019).
M. Steinbrück, M. Große, U. Stegmaier, J. Braun, and C. Lorrette, Coatings 12, 875 (2022).
K. Kane, S. Bell, N. Capps, B. Garrison, K. Shapovalov, G. Jacobsen, C. Deck, T. Graening, T. Koyanagi, and C. Massey, Journal of Nuclear Materials 574, 154152 (2023).
NEA, inState-of-the-Art Report on Light Water Reactor Accident-Tolerant Fuels, Nuclear Science (OECD Publishing, Paris, 2018).
Outotec, HSC Chemistry 10, (PPORI, 1974–2023).
E. J. Opila, Materials Science Forum 461–464, 765 (2004).
E. Opila and N. Jacobson, in Ceramics Science and Technology. eds. R. Riedel and I. W. Chen (Wiley, Hoboken, 2013).
P. Meschter, E. Opila, and N. Jacobson, Annual Review of Materials Research 43, 559 (2013).
J. Bischoff, C. Delafoy, C. Vauglin, P. Barberis, C. Roubeyrie, D. Perche, D. Duthoo, F. Schuster, J.-C. Brachet, E. Schweitzer, and K. Nimishakavi, Nuclear Engineering and Technology 50, 223 (2018).
J.-C. Brachet, I. Idarraga-Trujillo, M. Flem, M. Saux, V. Vandenberghe, S. Urvoy, E. Rouesne, T. Guilbert, C. Toffolon-Masclet, M. Tupin, C. Phalippou, F. Lomello, F. Schuster, A. Billard, G. Velisa, C. Ducros, and F. Sanchette, Journal of Nuclear Materials 517, 268 (2019).
J.-C. Brachet, E. Rouesne, J. Ribis, T. Guilbert, S. Urvoy, G. Nony, C. Toffolon-Masclet, M. Le Saux, N. Chaabane, H. Palancher, A. David, J. Bischoff, J. Augereau, and E. Pouillier, Corrosion Science 167, 108537 (2020).
B. Maier, H. Yeom, G. Johnson, T. Dabney, J. Walters, J. Romero, H. Shah, P. Xu, and K. Sridharan, JOM 70, 198 (2018).
H. Yeom, B. Maier, G. Johnson, T. Dabney, M. Lenling, and K. Sridharan, Journal of Nuclear Materials 526, 151737 (2019).
J. Yang, M. Steinbrück, C. Tang, M. Große, J. Liu, J. Zhang, D. Yun, and S. Wang, Journal of Alloys and Compounds 895, 162450 (2022).
D. Arias and J. Abriata, Bulletin of Alloy Phase Diagrams 7, 237 (1986).
M. Stanislowski, E. Wessel, K. Hilpert, T. Markus, and L. Singheiser, Journal of the Electrochemical Society 154, A295 (2007).
B. Pint, Oxidation of Metals 95, 335 (2021).
C. Cotell, G. Yurek, R. Hussey, D. Mitchell, and M. Graham, Oxidation of Metals 34, 173 (1990).
C. Cotell, G. Yurek, R. Hussey, D. Mitchell, and M. Graham, Oxidation of Metals 34, 201 (1990).
J. Klaisnerová, L. Křivský, P. Gávelová, and J. Krejčí, Manufacturing Technology 22, 703 (2022).
X. Han, C. Chen, Y. Tan, W. Feng, S. Peng, and H. Zhang, Corrosion Science 174, 108826 (2020).
X. Han, J. Xue, S. Peng, and H. Zhang, Corrosion Science 156, 117 (2019).
X. Hu, C. Dong, Q. Wang, B. Chen, H. Yang, T. Wei, R. Zhang, W. Gu, and D. Chen, Journal of Nuclear Materials 519, 145 (2019).
A. Fazi, M. Sattari, K. Stiller, H.-O. Andrén, and M. Thuvander, Journal of Nuclear Materials 576, 154268 (2023).
M. Steinbrück, Oxidation of Metals 70, 317 (2008).
D. Kim, M. Steinbrück, M. Grosse, C. Tang, and Y. Lee, Journal of Nuclear Materials 583, 154538 (2023).
S. Dryepondt, K. Unocic, D. Hoelzer, C. Massey, and B. Pint, Journal of Nuclear Materials 501, 59 (2018).
K. Field, M. Snead, Y. Yamamoto, and K. Terrani, in Handbook on the Materials Properties of FeCrAl Alloys for Nuclear Power Production Applications (ORNL/SPR-2018/905, 2018).
K. Sakamoto, and Y. Miura, in Proceedings of the TOPFUEL 2018 Conference (Prague, Czech Republic, 2018).
E. Gulbransen and K. Andrew, Journal of The Electrochemical Society 106, 294 (1959).
Y. Yamamoto, B. Pint, K. Terrani, K. Field, Y. Yang, and L. Snead, Journal of Nuclear Materials 467, 703 (2015).
B. Pint, Corrosion Reviews 35, 167 (2017).
F. Nagase, K. Sakamoto, and S. Yamashita, Corrosion Reviews 35, 129 (2017).
K. Sakamoto, Y. Miura, S. Ukai, A. Kimura, A. Yamaji, K. Kusagaya, and S. Yamashita, in GLOBAL 2019—International Nuclear Fuel Cycle Conference and TOP FUEL 2019 - Light Water Reactor Fuel Performance Conference (2020).
K. Field, X. Hu, K. Littrell, Y. Yamamoto, and L. Snead, Journal of Nuclear Materials 465, 746 (2015).
B. A. Pint, A. J. Garratt-Reed, and L. W. Hobbs, Materials at High Temperatures 13, 3 (1995).
D. Naumenko, B. Pint, and W. Quadakkers, Oxidation of Metals 86, 1 (2016).
J. M. Amores, V. S. Escribano, and G. Busca, Materials Chemistry and Physics 60, 168 (1999).
K. Lipkina, D. Hallatt, E. Geiger, B. Fitzpatrick, K. Sakamoto, H. Shibata, and M. Piro, Journal of Nuclear Materials 541, 152305 (2020).
F. Stott, G. Wood, and J. Stringer, Oxidation of Metals 44, 113 (1995).
E. N. E. Airiskallio, M. Heinonen, I. Väyrynen, K. Kokko, M. Ropo, M. Punkkinen, H. Pitkänen, M. Alatalo, J. Kollár, B. Johansson, and L. Vitos, Physical Review B - Condensed Matter and Materials Physics 81, 033105 (2010).
K. Prasanna, A. Khanna, R. Chandra, and W. Quaakkers, Oxidation of Metals 46, 465 (1996).
G. Rybicki and J. Smialek, Oxidation of Metals 31, 275 (1989).
H. El Kadiri, R. Molins, Y. Bienvenu, and M. Horstemeyer, Oxidation of Metals 64, 63 (2005).
E. Scheil and E. Schulz, Archiv für das Eisenhüttenwesen 6, 155 (1932).
N. Israelsson, High Temperature Oxidation and Chlorination of FeCrAl Alloys, (Chalmers university of Technology, Göteborg, 2014).
B. Lesage, L. Maréchal, A. M. Huntz, and R. Molins, Defect and Diffusion Forum 194–199, 1707 (2001).
D. Rohrberg, K.-H. Spitzer, L. Dörrer, P. Tankeu, M. Podsiadlo, G. Borchardt, T. Markus, and R. Schmid-Fetzer, Materials at High Temperatures 25, 247 (2008).
J. Stuckert, M. Grosse, M. Steinbrueck, and K. Terrani, in GLOBAL 2019 - International Nuclear Fuel Cycle Conference and TOP FUEL 2019—Light Water Reactor Fuel Performance Conference (2020).
P. Doyle, J. Stuckert, M. Grosse, M. Steinbrück, A. Nelson, J. Harp, and K. Terrani, Journal of Nuclear Materials 580, 154433 (2023).
J. Stuckert, J. Birchley, M. Große, B. Jaeckel, and M. Steinbrück, Nuclear Engineering and Design 241, 3224 (2011).
S. Bechta, E. Krushinov, V. Almjashev, S. Vitol, L. Mezentseva, Y. Petrov, D. Lopukh, V. Khabensky, M. Barrachin, S. Hellmann, K. Froment, M. Fischer, W. Tromm, D. Bottomley, F. Defoort, and V. Gusarov, Journal of Nuclear Materials 348, 114 (2006).
S. Bechta, E. Krushinov, V. Almjashev, S. Vitol, L. Mezentseva, Y. Petrov, D. Lopukh, V. Khabensky, M. Barrachin, S. Hellmann, K. Froment, M. Fischer, W. Tromm, D. Bottomley, F. Defoort, and V. Gusarov, Journal of Nuclear Materials 362, 46 (2007).
IAEA, in Final Report: Analysis of Options and Experimental Examination of Fuels for Water Cooled Reactors with Increased Accident Tolerance (ACTOF) (IAEA-TECDOC-1921, 2020).
L. Snead, T. Nozawa, Y. Katoh, T.-S. Byun, S. Kondo, and D. Petti, Journal of Nuclear Materials 371, 329 (2007).
J. Braun, C. Sauder, J. Lamon, and F. Balbaud-Célérier, Composites Part A: Applied Science and Manufacturing 123, 170 (2019).
B. Deal and A. Grove, Journal of Applied Physics 36, 3770 (1965).
N. Jacobson, B. Harder, and D. Myers, Journal of the American Ceramic Society 96, 838 (2013).
B. Harder, N. Jacobson, and D. Myers, Journal of the American Ceramic Society 96, 606 (2013).
B. Schneider, A. Guette, R. Naslain, M. Cataldi, and A. Costecalde, Journal of Materials Science 33, 535 (1998).
E. Opila, Journal of the American Ceramic Society 82, 625 (1999).
K. Terrani, B. Pint, C. Parish, C. Silva, L. Snead, and Y. Katoh, Journal of the American Ceramic Society 97, 2331 (2014).
H. Pham, Y. Nagae, M. Kurata, D. Bottomley, and K. Furumoto, Journal of Nuclear Materials 529, 151939 (2020).
H. Kleykamp, V. Schauer, and A. Skokan, Journal of Nuclear Materials 227, 130 (1995).
C. Cabet, NATO Science for Peace and Security Series B: Physics and Biophysics, (Springer, Dordrecht, 2008), p. 351.
L. Chaffron, C. Sauder, C. Lorrette, L. Briottet, A. Michaux, L. Gélébart, A. Coupé, M. Zabiego, M. Le Flem, and J.-L. Séran, in EPJ Web of Conferences, Vol. 51 (2013), p. 01003.
M. Steinbrück, M. Große, U. Stegmaier, J. Braun, and C. Lorrette, in Proceedings of the TOPFUEL 2021 Conference (Santander, Spain, 2021).
V. A. Avincola, D. Cupid, and H. Seifert, Journal of the European Ceramic Society 35, 3809 (2015).
D. Young and B. Pint, Oxidation of Metals 66, 137 (2006).
J. Cathcart, R. Pawel, R. McKee, R. Druschel, G. Yurek, J. Campbell, and S. Jury, Zirconium metal-water oxidation kinetics IV. Reaction rate studies (ORNL/NUREG-17, ORNL USA, 1977).
E. Kashkarov, D. Sidelev, M. Syrtanov, C. Tang, and M. Steinbrück, Corrosion Science 175, 108883 (2020).
Q. Li, P. Song, R. Zhang, Z. Li, Y. Wang, P. Du, and J. Lu, Corrosion Science 203, 110378 (2022).
W. Wang, G. Zhang, C. Wang, T. Wang, and T. Li, Journal of Nuclear Materials 563, 153660 (2022).
Y. Wang, L. Wang, L. Shang, G. Bai, J. Li, F. Xue, and W. Gong, Corrosion Science 205, 110449 (2022).
W. Wang, G. Zhang, C. Wang, T. Wang, Y. Zhang, and T. Xin, Journal of Alloys and Compounds 946, 169385 (2023).
OECD-NEA, QUENCH-ATF project (2020). [Online]. Available: https://www.oecd-nea.org/jcms/pl_36597/quench-atf-project.
B. Pint, K. Unocic, and K. Terrani, Materials at High Temperatures 32, 28 (2015).
IAEA, Testing and Simulation for Advanced Technology and Accident Tolerant Fuels (ATF-TS), [Online]. Available: https://nucleus.iaea.org/sites/connect/NFEpublic/Pages/ATF-TS.aspx.
M. Grosse, K. van Loo, E. Frankberg, C. Tang, K. Lambrinou, F. Di Fonzo, and M. Steinbrück, in GLOBAL 2019—International Nuclear Fuel Cycle Conference and TOP FUEL 2019 (2020).
Funding
Open Access funding enabled and organized by Projekt DEAL. No special funding was obtained for this work. The experimental work was conducted in the framework Helmholtz (HGF) program NUSAFE at KIT.
Author information
Authors and Affiliations
Contributions
MS had the idea for the article, performed the literature search and data analysis, and wrote first draft of the manuscript. MS, MG, CT, and JS contributed to the experimental work, material preparation, data collection and analysis. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Steinbrueck, M., Grosse, M., Tang, C. et al. An Overview of Mechanisms of the Degradation of Promising ATF Cladding Materials During Oxidation at High Temperatures. High Temperature Corrosion of mater. (2024). https://doi.org/10.1007/s11085-024-10229-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11085-024-10229-y