
Abstract
Since the oxidation reactions in the process of steel production occur in harsh conditions (i.e., high temperatures and gas atmospheres), it is practically impossible to observe in situ the compositional changes in the steel and the formed oxide scale. Hence, a coupled thermodynamic-kinetic numerical model is developed that predicts the formation of oxide phases and the composition profile of the steel alloy’s constituents in a short time due to external oxidation. The model is applied to high-temperature oxidation of Fe–Mn alloys under different conditions. Oxidizing experiments executed with a thermogravimetric analyzer (TGA) on Fe–Mn alloys with different Mn contents (below 10 wt %) are used to determine kinetic parameters that serve as an input for the model. The mass gain data as a function of time show both linear and parabolic regimes. The results of the numerical simulations are presented. The effect of different parameters, such as temperature, Mn content of the alloy, oxygen partial pressure, and oxidizing gas flow rate on the alloy composition and oxide phases formed, is determined. It is shown that increasing the temperature and decreasing the oxygen partial pressure both lead to a thicker depleted area.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
High-temperature oxidation is a prominent phenomenon in several steps of steelmaking, such as hot rolling. Oxidation at high temperatures and high oxygen partial pressure can change the composition of the steel near the surface due to the depletion of the alloying elements. Furthermore, the oxide scale’s characteristics such as thickness and phase composition vary depending on the oxidation condition. Since it is challenging to study such rapid processes under such extreme conditions experimentally, models are needed to predict the composition at the substrate’s surface as well as that of the oxide scale.
To model the oxidation behavior, fundamental and comprehensive knowledge of thermodynamics and kinetics of the occurring process is required [1, 2]. Although numerical and analytical models have been developed to predict the oxidation behavior and the compositional change of alloys during thermal oxidation, their general application is limited. A number of these studies focused on finding a criterion to predict whether internal or external oxidation occurs [3,4,5,6,7]. Furthermore, mathematical models developed for external oxidation of binary and ternary alloys mostly consider only parabolic growth kinetics for the oxide layer [8,9,10,11,12], while the growth rate in the initial stages of oxidation is usually linear [8, 13, 14]. Moreover, in most cases, only selective oxidation of the alloying element forming the most stable oxide phase at low oxygen partial pressures is considered [4, 5, 15,16,17,18,19,20].
As an input for the oxidation simulations, the linear and parabolic kinetic constants of the oxidation reaction under different conditions of steel composition, temperature, and oxygen partial pressure are needed [8, 21,22,23,24]. For the hot-rolling process, high oxygen partial pressures and temperatures need to be considered. However, experimental research on the oxidation of iron alloys with manganese as the main alloying element is either focused on the annealing step, i.e., at low oxygen partial pressures [25,26,27,28,29] or are done for long exposure times [30,31,32].
In the present work, a coupled thermodynamic-kinetic numerical model is developed that also considers hot-rolling conditions, based on earlier works by Nesbitt [11], and Nijdam [8, 33]. The model is applicable to high-temperature external oxidation of different alloys in dry oxygen with different partial pressures and flows. Here the model is applied to short-time, high-temperature oxidation of iron-manganese binary alloys to predict the amount of formed oxides, the concentration profiles of iron and manganese within the substrate alloy, and their oxide/metal (O/M) interface concentration. Measuring such parameters is not simply possible experimentally, because during cooling diffusion and homogenization in both the oxide scale and the substrate alloy can occur. Furthermore, fast cooling of steels could evoke phase transformations (such as martensite formation). Therefore, the developed model could provide information on oxidation that is practically impossible to be measured.
The simulation consists of two main steps. In the first step, thermodynamic calculations are implemented in order to predict the stable phases. In the second step, the alloy’s composition change beneath the O/M interface is calculated using a recently developed modified flux equation for a lattice fixed frame of reference [34, 35].
There are several advantages of the current model over the previous analytical and numerical models for oxidation. First, the computations include the initial stages of oxidation, whereas previous models only considered long-time exposures. Second, there is no limit to the number of elements that can oxidize simultaneously, unlike previous models that considered the most stable oxide phase only. Finally, the computation time is shorter due to the implementation of the modified flux equation for diffusion.
As an input for the calculations, the kinetic constants of both linear and parabolic growth modes are needed. Therefore, a series of oxidation experiments using a thermogravimetric analyzer (TGA) were conducted. The iron-manganese alloys were exposed to a gas mixture with different oxygen partial pressures and different velocities. Then the kinetic constants were obtained from the mass gain data.
Coupled Thermodynamic-Kinetic Model
We have considered a single-phase Fe-Mn binary alloy reacting with oxygen at temperature T and pressure p.
where Me represents Fe or Mn.
The most stable oxide phases can be found by calculating and comparing the dissociation oxygen partial pressures at the O/M interface for different oxide phases, through their standard Gibbs free energy of formation \(\Delta G^\circ _{\text {Me}_{\rm x}{\rm O}_{\rm y}}\) [8]. The equilibrium oxygen partial pressure in which the oxide \(\text {Me}_{\rm x}{\rm O}_{\rm y}\) is in local equilibrium with the metal at the interface can be calculated according to:
where \(a_{{Me_x}{O_y}}\) and \(a_{Me}\) are the thermodynamic activities of the oxide phase \(\text {Me}_{\rm x}{\rm O}_{\rm y}\) and the alloy constituent (Me) at the O/M interface, respectively.

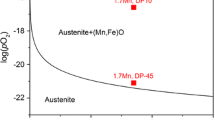
Stability regions of different iron and manganese oxide phases as a function of Mn weight fraction and oxygen partial pressure at 1000\({^{\circ }}\)C, obtained by using Factsage software [36]. The red marks show the experimental conditions of this work which are oxygen partial pressures of 10, 20, and 30kPa for three different alloys
A phase diagram that shows the iron and manganese oxides phases which can form at 1000\({^{\circ }}\)C is shown in Fig. 1. The mixture of MnO and FeO (wüstite) is the most stable oxide phase forming at very low oxygen pressure. Due to the higher diffusion coefficient of Fe and Mn in wüstite than in magnetite and hematite, wüstite grows much faster than the other oxides. This results for high-temperature oxidation of pure iron, in the formation of wüstite, magnetite, and hematite with thickness ratios of 95, 4, and 1, respectively [37]. In situ XRD results ([38]) also showed the formation of FeO and MnO together as a solid solution at oxidizing temperature. It is also known that during the linear growth of the oxide scale a local equilibrium between the oxide scale and the alloy is established, and thus the equilibrium oxygen partial pressure is fixed. The parabolic regime starts only after the formation of magnetite and hematite. Then the equilibrium oxygen partial pressures at the wüstite/magnetite and magnetite/hematite interfaces are also fixed. Although all the iron oxide phases can form in such conditions, it was seen that wüstite (FeO+MnO) was the main oxide phase that formed more than \(95\%\) of the oxide scale. Therefore, only FeO and MnO are the considered oxides in our simulations.
The simulation is based on a model which was developed and also validated previously [22, 24, 33]. A schematic illustration of the grid used for the model calculations at the start of the oxidation process (\(t=0\)) and also after some time (\(t=t\)) is shown in Fig. 2. At the beginning of oxidation, a number of slices L with an initial equal width of \(\Delta z^{i,0}\) are defined perpendicular to the surface of the alloy with total thickness Z. Z is considered thick enough to simulate a semi-finite diffusion case. The slices have a homogeneous and uniform composition \(C_{\text {Me}}^{i,0}\), where i and Me refer to the slice number and the constituent (Fe or Mn), respectively.

Schematic of the grid used for the calculations (a) at the start of oxidation (\(t=0\)) and (b) after a time (\(t=t\))
The compositional variations parallel to the metal surface are negligible, thus only diffusion perpendicular to the O/M interface within a limited depth of material is considered. The positions of the boundaries between slices are indicated with \(z^i\), and the concentration of each slice is assigned to the middle point of each slice which is located at \(l^i\). Therefore, the number of moles of each constituent Me at each slice at the beginning of oxidation is \(n^{i,0}_{\text {Me}}\), for which it holds that:
where \(N^{i,0}_{\text {Me}}\) and \(V_m\) are the initial mole fraction of the constituent Me and the molar volume of the alloy, respectively. After establishing the grid, the time is increased by a small increment \(\Delta t\). The explicit method for solving the above equation is limited by the stability criterion S [39] and as such the time steps \(\Delta t\) being used must be smaller than the stability limit.
where D is the diffusion coefficient of the diffusing element. After a certain amount of time t, a layer of oxide with the thickness \(d_{ox}\) is formed at the alloy’s surface; see Fig. 2b. The oxidation leads to an interface displacement \(\upxi\) with respect to the initial metal/gas interface.
At each time step, a number of calculations are performed which provide information such as oxide scale thickness, concentrations of the slices 2 to L, displacement of the O/M interface, and the interface concentrations for the next time step. It is assumed that local thermodynamic equilibrium at the O/M interface is established at each time step. The mass balance at the O/M interface plays an important role in the calculations. It is assumed that no material is lost, implying that all material that diffuses out of the alloy is present in the oxide scale.
Oxide Scale Thickness
In order to calculate the total oxide scale thickness at time t, the scale growth kinetic data such as linear and parabolic constants are needed. The fast initial stage of oxidation is linear which changes into a parabolic regime with a slower growth rate at \(t=\,t_\text{trans}\). Having the required kinetic data, the calculations are as follows:
where \(k_\text{lin}\) and \(k_\text{par}\) are the kinetic constants for the linear and parabolic regimes, respectively.
The total oxide scale thickness \(d_\text{ox}\) formed at time t can be related to the total amount of oxides \({\text {Me}_{\rm x}{\rm O}_{\rm y}}\) that is formed at the O/M interface, which are FeO and MnO based on the assumption of this work. It is expressed in terms of the volume of each oxide per unit interface area of the alloy, \(\upvarphi _{\text {Me}_{\rm x}{\rm O}_{\rm y}}\). Then, it holds that:
Consumption of the material from the alloy also results in the displacement of the O/M interface [20]. Compared to its original location at \(t=0\), the displacement of the OM interface can be calculated by considering the mass balance, as:
where \(V_{MeO}\) is the molar volume of FeO and MnO, and \(V_{Me}\) is the partial molar volume of each component at the OM interface in the alloy.
Concentrations of the Second to Last Slices
Due to the consumption of the constituents from the metal substrate by the oxide formation, a concentration difference establishes between neighboring slices, which drives diffusion of the constituting elements. For calculating the concentration profile underneath the alloy-surface, a modified flux equation was used [33,34,35]. The flux of manganese at time t from slice i to \(i+1\) is given by:
where R is the gas constant (\(J mol^{-1} K^{-1}\) ), T is the temperature (K), \(M_{Mn}^{i,t}\) and \(\mu _{Mn}^{i,t}\) are the mobility and chemical potential of manganese at time t (s) in slice i, respectively. The value for the mobility of iron and manganese is obtained from the tracer diffusion coefficient of each constituent Me in the phase \(\phi\) (here austenite), which is included in [40]:
where \(D*\) is the tracer diffusion coefficient of Me in \(\phi\). Calculations conducted via DICTRA [40] show that the diffusion coefficient of Mn is only weakly dependent on concentration; if the Mn concentration in the alloy increases from 0.5 to 8%, the diffusion coefficient of Mn in the matrix would increase only around 10%. This ideal behavior of Fe-Mn alloys has also been reported before in previous works [41]. Assuming ideal behavior for low concentration of the alloying elements, i.e., negligible interaction between the elements, the chemical potential of each constituent can be simply related to its mole fraction, according to:
A zero-flux slice is considered at depth Z in the bulk, which is far enough from the interface to be unaffected by the oxidation:
The value for the flux of both iron and manganese between all the slices at each time t allows calculating the number of moles of the constituents in the slices 2 up to the last one for the next time step \(t+\Delta t\) by:
Therefore, at each time step, a series of calculations through Eq. (8) to Eq. (12) results in the concentration for the second to the last slice for the next time step to be calculated as shown in the flowchart; see Fig. 3. However, for calculating the composition of the alloy at the O/M interface another method, described in Sect. 2.3, has been implemented.

Flowchart of the model’s operations
The Interface Composition and Displacement
The interface concentration of manganese is constantly changing during oxidation because of the consumption of manganese. To determine whether oxidation occurs of only manganese or also iron, a series of calculations was performed [8]. Since Mn forms a more stable oxide than Fe (i.e., \(p_{O_{2_{\text {MnO}}}} < p_{O_{2_{\text {FeO}}}}\); cf. Equation (2)), oxidation of Mn at the O/M interface occurs first, and the interface concentration of manganese would decrease. Due to the limited amount of Mn present at the interface (which is related to the concentration and diffusion coefficient of manganese in the alloy), all the Mn at the interface is consumed at each time step. Moreover, the mass balance at the O/M interface must be fulfilled. This implies that the total amount of iron and manganese incorporated into the oxide layer at time t must be equal to the same amount consumed from the alloy until time t. So at time t, the mass balance can be written as:
where \(C_{Me}^0\) is the initial concentration of the constituents, and \(\xi\) is the interface recession. Therefore at each time step, the total amount of Mn consumed from the substrate metal, which is denoted as the so-called accessible amount \(\Gamma ^t\), can be calculated by Eq. (14). As explained before, all the Mn at the interface is considered to be consumed, so \(C_{\text {Mn}}^{1, t+\Delta t}\) is virtually zero. It is shown as the green area in Fig. 4.
Where the difference between \(\xi ^{t+\Delta t}\) and \(\xi ^{t}\) is the O/M interface displacement due to consumption of \(\Gamma _{Mn}^t\) during \(\Delta t\), which can be calculated with Eq. (7).

Schematic of accessible amount, i.e., the total amount of Mn from the substrate metal consumed by the oxide within the time step \(\Delta t\)
Now \(\Delta \upvarphi _{\text {MnO}}\) can be used as a criterion to determine if only the most stable oxide is present or that simultaneously the other constituent’s oxide form. So at each time step, \(\Delta \upvarphi _{\text {MnO}}\) is compared with \(\Delta d_\text{ox}\) (the thickness of the oxide scale formed during \(\Delta t\)) to establish the oxides formed.
-
1.
If \(\Delta \upvarphi _{\text {MnO}} \ge \Delta d_\text{ox}\), only MnO is formed during the time step \(\Delta t\). Then the amount of MnO formed during this time step, \(\upvarphi ^{\Delta t} _{\text {MnO}}\), is equal to \(\Delta d_\text{ox}\) (see Eq. (6)). Therefore, the displacement of the O/M interface can be obtained by Eq. (7), with \(\Delta \upvarphi _{\text {FeO}}\) equal with zero.
-
2.
If \(\Delta \upvarphi _{\text {MnO}} < \Delta d_\text{ox}\), simultaneous oxidation of Mn and Fe occurs during \(\Delta t\). Then the amount of FeO formed (\(\Delta \upvarphi _{\text {FeO}}\)) is equal with \(\Delta d_\text{ox} - \Delta \upvarphi _{\text {MnO}}\). Accordingly, the interface displacement can be calculated by Eq. (7).
Grid Adjustment
After each series of calculations for a time step, the interface displacement requires a rearrangement of the grid system in order to obtain a new set of semi-distance grid points (grid points are at the middle point of each slice). To achieve this, the size \(\Delta z\) and the number L of the slices are kept constant, as well as the total studied thickness of the alloy at each time step. Therefore at each time step, the starting point of the grid is moved inward by the value of \(\Delta \xi ^{\Delta t}\). The position of slices’ walls \(z^i\) can be calculated with:
Eventually, by having the oxide scale growth kinetics of the alloy, the concentration profile for both iron and manganese inside the alloy as well as the amount of each oxide phase formed can be obtained as a function of time by numerically solving Eqs. (2) to (15). To this end, a numerical model, based on finite difference technique [42], was developed in MATLAB [43].
Experiments
The kinetic data needed as an input for the simulations were obtained from oxidation experiments conducted using TGA.
The Fe-Mn alloys were provided by ChemPur (Karlsruhe, Germany). The chemical composition of the alloys with different manganese contents as obtained with inductive coupled plasma-optical emission spectrometry (ICP-OES) is presented in Table 1. The alloys were cut into 2 \(\times\) 8 \(\times\) 15 mm pieces with a hole of 2.2 mm diameter by electric discharge machining (EDM). Then, they were ground using SiC emery paper and cleaned ultrasonically in isopropanol. Finally, the specimens were stored in airtight membrane boxes (Agar Scientific, G3319, Essex, UK) after drying with a flow of pure nitrogen gas.
The alloys were oxidized in a symmetrical thermogravimetric analyzer (TGA, Setaram TAG 16/18, Caluire, France) in order to calculate the kinetic constants from the mass gain data. An alumina pin with a diameter of 2.2mm was inserted into the hole in the sample and placed onto a sapphire rod. The initial mass of the sample was measured by a Mettler Toledo balance (accuracy ±1\({\upmu }g\)). To eliminate any buoyancy effect, a dummy sample of alumina of the same size was mounted onto a sapphire rod the counterpart of the balance. The furnace chambers were two identical tubes with 280mm length and an inner diameter of 15mm. First, the TGA system was pumped to vacuum (\(<{50}{Pa}\)). Then the gas lines, balance, and furnaces were flushed with pure nitrogen three times. The purity of nitrogen was 5N vol.%, and it was filtered additionally to remove any residual hydrocarbons, moisture, and oxygen, with Accosorb (\(<10\) ppb hydrocarbons), Hydrosorb (\(<10\) ppb \(\text {H}_{2}\text {O}\)) and Oxysorb (\(<5\) ppb \(\text {O}_{2}\)) filters (Messer Griesheim, Germany), respectively. Next, both furnaces were heated up with a rate of 10\({^{\circ}}C/min\) with a flow of pure nitrogen. Temperatures were chosen between 950\({^{\circ }}\)C and 1150\({^{\circ }}\)C. After reaching the target temperature, the chambers’ atmospheres were switched to oxidizing by introducing a gas mixture of oxygen, and nitrogen. Different flow rates (26.6, 53.3, 100, 150, 200, and 250\(\text {mL}{\text {min}^{-1}}\), corresponding to an average gas velocities of around 1 to 10cm\(s^{-1}\) at 1000\({^{\circ }}\)C) and oxygen partial pressures (10 to 30kPa) were applied by changing the ratio between \(\text {O}_{2}\) and \(\text {N}_{2}\) gases, while the total chamber pressure was kept at 101kPa (1 atm). After oxidation, the furnace tube was cooled to room temperature with a rate of 10\({^{\circ}}C/min\) while flushing with pure \(\text {N}_{2}\).
The mass gain per unit area for an oxidation experiment in the TGA system is shown in Fig. 5. The mass gain data correspond with the total weight of oxygen consumed by the metal to form the oxide scale. The internal oxidation zone (IOZ) was negligible compared to the scale’s thickness. The mass gain data show the initial linear growth of the oxide layer, which is followed by parabolic growth. The linear and parabolic rate constants are, respectively, the slope of the lines in mass gain and squared mass gain divided by area versus time. To obtain the slope of the line within the linear regime, the very first part of it is not considered due to the lack of oxygen in the beginning.

a The mass gain per unit surface area and b the squared mass gain per unit surface area data for 20 minutes of oxidation of Fe-8Mn binary alloys at 1000\({^{\circ }}\)C in 20kPa oxygen partial pressure with a total gas flow rate of 100\(\text {mL}{\text {min}^{-1}}\). The slopes of the lines represent the kinetic constants
The details about the mechanism of high-temperature oxidation and characterization of the oxidized samples are described in our previous work [38]. It was concluded that for our experimental conditions, the linear growth of the oxide layer is controlled by the diffusion of the oxidizing gas through the boundary layer.
Different parameters which could influence the oxidation kinetics were studied. The effect of flow rate on the mass gain data is shown in Fig. 6. It can be seen that an increase in the gas flow rate increases the linear constant (\(k_l\)) and total mass gain (Fig. 6a), while it has almost no effect on the parabolic growth constant (Fig. 6b). The linear constant increases from \(1.4\times 10^{-5}\;\pm 3.1\times 10^{-7}\;g\,cm^{-2}\,s^{-1}\) for oxidation with gas flow rate of \(26.6\;mL\,min^{-1}\) to \(1.0\times 10^{-4}\;\pm 4.1\times 10^{-7} g\,cm^{-2}\,s^{-1}\) for oxidation with gas flow rate of \(250\;mL\,min^{-1}\). The parabolic constant only varies between \(5.6\times 10^{-7}\;\pm 1.9\times 10^{-9}\) to \(5.7\times 10^{-7}\;\pm 3.0\times 10^{-9}\;g^2\,cm^{-4}\,s^{-1}\) for oxidation with flow rates of 100 to \(250\;mL\,min^{-1}\).

The experimental results for oxidation of Fe-8Mn binary alloy in 20kPa oxygen partial pressure at 1000\({^{\circ }}\)C for 20 minutes with a total gas flow rate between 26.6 and 250\(\text {mL}{\text {min}^{-1}}\): (a) mass gain per area, and (b) squared mass gain per area as a function of time
The effect of increasing the oxygen partial pressure of the oxidizing gas mixture on the kinetics of the oxidation reaction is shown in Fig. 7, and it was almost the same as rising the flow rate. It increased the linear constant from \(4.3\times 10^{-5}\;\pm 2.5\times 10^{-7}\) to \(1.5\times 10^{-4}\;\pm 3.3\times 10^{-6}\;g\,cm^{-2}\,s^{-1}\), respectively, for 10 and 30kPa, while the parabolic constant varied between \(6.0\times 10^{-7}\;\pm 5.6\times 10^{-9}\) and \(6.6\times 10^{-7}\;\pm 6.4\times 10^{-9}\;g^2\,cm^{-4}\,s^{-1}\) in the same range of oxygen partial pressure.

The experimental results for oxidation of Fe-8Mn binary alloy in 10, 20, and 30kPa oxygen partial pressure at 1000\({^{\circ }}\)C for 20 minutes with a total gas flow rate of 250\(\text {mL}{\text {min}^{-1}}\): a mass gain per area, and b squared mass gain per area as a function of time
The effect of temperature on high-temperature oxidation of Fe-8Mn binary alloys oxidized in an atmosphere with 20kPa oxygen with a flow rate of 200\(\text {mL}{\text {min}^{-1}}\) is shown in Fig. 8. Increasing the temperature from 950 to 1150\({^{\circ }}\)C does not affect the slope of the linear part of the mass gain data, but only leads to a longer linear-to-parabolic transition time; see Fig. 8a. However, oxidation at higher temperatures increases the parabolic kinetic constant; see Fig. 8b. This is expected because of the higher diffusion rate through the oxide scale at higher temperatures. This also results in a longer linear regime because it takes a longer time before diffusion through the oxide layer becomes the rate-determining step. The parabolic constant increases from \(5.90\times 10^{-7}\;{\pm 1.59\times 10^{-8}}\) to \(1.12\times 10^{-6}\;{\pm 3.47\times 10^{-8}}\;g^2\,cm^{-4}\,s^{-1}\), for oxidation at 950 and 1150\({^{\circ }}\)C, respectively.

The experimental results for oxidation of Fe-8Mn binary alloy in 20kPa oxygen partial pressure for 20 minutes with a total gas flow rate of 200\(\text {mL}{\text {min}^{-1}}\): a mass gain per area, and b squared mass gain per area as a function of time
Simulation Results and Discussion
It is practically impossible to observe the O/M interface of the alloy, while oxidizing at very high temperatures. Only after the sample has cooled to room temperature the oxide scale can be analyzed. Within the cooling process, which is slow with TGA (10\({^{\circ}}C/min\)), phase transformations and homogenization within the oxide scale and the alloy substrate will occur. Simulating the high-temperature oxidation allows the prediction of the changes in the alloy during oxidation. As mentioned earlier, this method for simulation was applied for MCrAlY coatings and alloys before and it has been validated by experiments [8, 23, 33].
The simulations were performed based on the model described in Sect. 2. The following data were used as input. The molar volume of the alloys was calculated with Thermo-Calc using the TCFE 5 database [40]; the molar volumes of MnO and FeO are taken to be 13.21 and \(12.24\;cm^3\;mol^{-1}\), respectively [44]. The diffusion coefficients are taken from [45], and following Ref. [46] the mobilities of the alloy constituents in the alloy are calculated using Eq. (9). The oxidation experiments with the Fe-Mn alloys (see Table 1) were conducted with TGA at 950 to 1150\({^{\circ }}\)C, with 10 to 30kPa oxygen partial pressure, and flow rates of 26.6 to 250\(\text {mL}{\text {min}^{-1}}\). The oxide scale’s growth kinetic constants for the linear and parabolic part were obtained from the mass gain data; see Sect. 3. As shown in Fig. 9, the oxide’s growth kinetics in both linear and parabolic regimes could be described adequately with a combined linear and parabolic law (R-Squared=0.98). Therefore, the developed model can predict the behavior of Fe-Mn alloys, which are oxidized within the mentioned range of oxidizing conditions.

Experimental data and linear-parabolic calculation for oxidation of Fe-8Mn alloy at 1000\({^{\circ }}\)C in 20kPa oxygen partial pressure with a gas flow rate of 250\(\text {mL}{\text {min}^{-1}}\), with linear and parabolic kinetic constants of \(1.0\times 10^{-4}\;{\pm 2.3\times 10^{-6}}\,g\,cm^{-2}\,s^{-1}\) and \(5.7\times 10^{-7}\;{\pm 1.9\times 10^{-8}}\,g^2\,cm^{-4}\,s^{-1}\), respectively
The simulated concentration profiles of Mn in a Fe-8Mn alloy when it is oxidized in an environment with 20kPa oxygen partial pressure at different temperatures are demonstrated in Fig. 10. It is observed that the depleted zone increased with increasing temperature, due to the faster diffusion of Mn within the alloy. Hence, the Mn depletion layer of less than 2 microns at 950\({^{\circ }}\)C increases to more than 20 microns at 1150\({^{\circ }}\)C.

The calculated Mn concentration profiles within the alloy, when Fe-8Mn oxidized in the temperature range of 950 to 1150\({^{\circ }}\)C in 20kPa oxygen partial pressure for 20 minutes with a total gas flow rate of 53.3\(\text {mL}{\text {min}^{-1}}\)
The effect of the oxygen content in the oxidizing gas on the composition depth profile is calculated as well. In Fig. 11 the simulated concentration profiles of Mn for Fe-8Mn when oxidized at 1000\({^{\circ }}\)C for 20 minutes are demonstrated. It shows that reducing the oxygen partial pressure in the gas mixture from 30 to 10kPa expands the manganese depleted zone in the substrate alloy from 2 to more than 4 microns.

The calculated Mn concentration profiles within the Fe-8Mn substrate alloy when oxidized at the 1000\({^{\circ }}\)C with oxygen partial pressure of 10 to 30kPa for 20 minutes and a total gas flow rate of 53.3\(\text {mL}{\text {min}^{-1}}\)
The effect of the composition of the alloy on concentration profiles is illustrated in Fig. 12. The Mn depleted zone is calculated to be less than 2 microns for all three alloy compositions. The Mn content has no clear effect on Mn depleted zone width. Also, a negligible effect of Mn content on the mass gain data (i.e., the kinetics of oxidation) was seen; see [38].

The calculated Mn concentration profiles within the substrate alloy for samples with different Mn contents when oxidized at the 1000\({^{\circ }}\)C in 20kPa oxygen partial pressure for 20 minutes with a total gas flow rate of 53.3\(\text {mL}{\text {min}^{-1}}\)
Generally, the oxide scale growth kinetics, and consequently the resulting compositional changes in the alloy, are significantly influenced by the oxidizing condition (e.g., T and \(p_{\text {O}_{2}}\)), microstructure, composition, and surface condition of the alloy [47]. The oxidation model is applied to study the effect of the scale’s growth kinetics on the composition near the surface of the alloy as well as the oxide phase constitution of the developing oxide layer, for the oxidation of Fe-Mn alloys at high temperatures. The effect of initial linear growth rate on the relative amount of Mn oxide (\(\upvarphi _{\text {MnO}}/d_{\text{ox}}\)) is shown in Fig. 13.

Calculated values for the ratio of MnO thickness to oxide scale thickness (\(\upvarphi _{\text {MnO}}/d_{\text{ox}}\)) as a function of linear oxidation growth rate for Fe-8Mn alloy oxidized at 1000\({^{\circ }}\)C for 20 minutes
Since the dissociation oxygen partial pressure of MnO is less than that of FeO, the onset of oxidation starts with the preferential oxidation of Mn from the alloy. However, due to low Mn content and the rapid scale growth, the Mn interfacial mole fraction instantly (within the first time steps) drops. The early stage of oxidation then begins with the formation and development of FeO and MnO at the same time. The total amounts of MnO and FeO forming within the 20 minutes of oxidation are shown in Fig. 14. Within the linear regime, only the most stable oxide phases of iron and manganese can form (i.e., FeO and MnO). It is known that when the growth regime is changed to parabolic, the other oxide phases (magnetite and hematite) are stable to form as well [41]. However, even during the parabolic growth most of the oxide scale consists of wüstite.

Calculated amount of each oxide as a function of time, when Fe-8Mn alloy is oxidized at 1000\({^{\circ }}\)C in 20kPa oxygen atmosphere with a gas flow rate of 250\(\text {mL}{\text {min}^{-1}}\); the transition time is indicated by the vertical line
Conclusions
The in situ methods for investigating the high-temperature oxidation behavior of alloys are limited, and it is practically impossible to experimentally access the alloy’s microstructure and composition in that condition. Measurements at room temperature require fast cooling to avoid homogenization; however, fast cooling can lead to phase transformations which also change the material. Therefore, to study such a process, a numerical model was developed. The methodology of such a model is developed and verified by experiments before in several works [8, 22, 33]. The kinetic data necessary as input for the simulations were obtained from a series of TGA experiments. Iron with 1, 3, and 8 wt% Mn were oxidized at 950-1150\({^{\circ }}\)C in gas mixtures with 10-30kPa oxygen partial pressure and linear gas flow rates of 26.6 to 250\(\text {mL}{\text {min}^{-1}}\) in a furnace tube of 15mm inner diameter. The following conclusions can be drawn:
-
1.
Considering the composition profile of Mn within the alloys and the depleted zone after the oxidation, temperature change plays an important role. Increasing the temperature increases the depleted zone due to the faster diffusion of Mn within the alloy.
-
2.
Increasing the oxygen partial pressure of the oxidizing gas mixture leads to faster linear growth of the oxide scale when only FeO and MnO form. Faster oxidation results in higher O/M recession and a thinner Mn depleted zone.
-
3.
The ratio between Mn and Fe within the oxide layer can be predicted via the introduced simulations. Therefore, the amount of Mn that the alloy has lost during the high-temperature oxidation can be calculated.
The ability to predict the initial high-temperature oxidation of alloys in various oxidizing conditions can help to control the surface behavior of steels at high-temperature processing. Further developments on other iron binary or ternary alloys will be necessary, in order to give more insights regarding the role of different alloying elements on the oxidizing behavior of the alloys.
Data availability
The data that support the findings of this study and the code are available from the corresponding author upon reasonable request.
References
Melfo, W. M. C. Analysis of hot rolling events that lead to ridge-buckle defect in steel strips. Ph.D. thesis, University of Wollongong (2006).
Chen, R. & Yuen, W. Oxidation of low-carbon, low-silicon mild steel at 450-\(900^\circ\)C under conditions relevant to hot-strip processing. Oxidation of Metals 57 (1–2), 53–79 (2002).
Lashgari, V., Zimbitas, G., Kwakernaak, C. & Sloof, W. G. Kinetics of internal oxidation of Mn-steel alloys. Oxidation of Metals 82 (3–4), 249–269 (2014).
Bastow, B., Whittle, D. & Wood, G. C. Alloy depletion profiles resulting from the preferential removal of the less noble metal during alloy oxidation. Oxidation of Metals 12 (5), 413–438 (1978).
Gesmundo, F. & Niu, Y. The criteria for the transitions between the various oxidation modes of binary solid-solution alloys forming immiscible oxides at high oxidant pressures. Oxidation of Metals 50 (1–2), 1–26 (1998).
Guan, S., Yi, H. & Smeltzer, W. Internal oxidation of ternary alloys Part II: Kinetics in the presence of an external scale. Oxidation of Metals 41 (5-6), 389–400 (1994) .
Gesmundo, F. & Niu, Y. The formation of two layers in the internal oxidation of binary alloys by two oxidants in the absence of external scales. Oxidation of Metals 51 (1–2), 129–158 (1999).
Nijdam, T., Jeurgens, L. & Sloof, W. G. Modelling the thermal oxidation of ternary alloys-compositional changes in the alloy and the development of oxide phases. Acta Materialia 51 (18), 5295–5307 (2003).
Chatterjee, A. et al. Kinetic modeling of high-temperature oxidation of Ni-base alloys. Computational Materials Science 50 (3), 811–819 (2011).
Carter, P., Gleeson, B. & Young, D. Calculation of precipitate dissolution zone kinetics in oxidising binary two-phase alloys. Acta Materialia 44 (10), 4033–4038 (1996).
Nesbitt, J. & Heckel, R. Modeling degradation and failure of Ni-Cr-Al overlay coatings. Thin Solid Films 119 (3), 281–290 (1984).
Niu, Y. & Gesmundo, F. An approximate analysis of the external oxidation of ternary alloys forming insoluble oxides I: High oxidant pressures. Oxidation of Metals 56 (5), 517–536 (2001) .
Kvernes, I. A. & Kofstad, P. The oxidation behavior of some Ni-Cr-Al alloys at high temperatures. Metallurgical Transactions 3 (6), 1511–1519 (1972).
Young, D. J. High temperature oxidation and corrosion of metals Vol. 1. Elsevier, London. (2008).
Mao, W., Ma, Y. & Sloof, W. G. Internal oxidation of Fe-Mn-Cr steels, simulations and experiments. Oxidation of Metals 90 (1), 237–253 (2018).
Gong, L., Ruscassier, N., Ayouz, M., Haghi-Ashtiani, P. & Giorgi, M.-L. Analytical model of selective external oxidation of Fe-Mn binary alloys during isothermal annealing treatment. Corrosion Science 166, 108454 (2020).
Gong, L., Jiang, W., Balloy, D. & Giorgi, M.-L. Numerical model of selective external oxidation of Fe-Mn binary alloys during non-isothermal annealing treatment. Corrosion Science 178, 108921 (2021).
Wagner, C. Theoretical analysis of the diffusion processes determining the oxidation rate of alloys. Journal of The Electrochemical Society 99 (10), 369 (1952).
Wang, G., Gleeson, B. & Douglass, D. An extension of Wagner’s analysis of competing scale formation. Oxidation of Metals 35 (3–4), 317–332 (1991).
Whittle, D., Evans, D., Scully, D. & Wood, G. Compositional changes in the underlying alloy during the protective oxidation of alloys. Acta Metallurgica 15 (9), 1421–1430 (1967).
Yuan, K. et al. MCrAlY coating design based on oxidation-diffusion modelling part I: Microstructural evolution. Surface and Coatings Technology 254, 79–96 (2014) .
Pillai, R., Sloof, W. G., Chyrkin, A., Singheiser, L. & Quadakkers, W. J. A new computational approach for modelling the microstructural evolution and residual lifetime assessment of MCrAlY coatings. Materials at High Temperatures 32 (1–2), 57–67 (2015).
Pillai, R., Kane, K., Lance, M. & Pint, B. A. Computational methods to accelerate development of corrosion resistant coatings for industrial gas turbines. Superalloys 2020 824–833 (2020).
Pillai, R., Chyrkin, A. & Quadakkers, W. J. Modeling in high-temperature corrosion: A review and outlook. Oxidation of Metals 96 (5), 385–436 (2021).
Swaminathan, S. & Spiegel, M. Thermodynamic and kinetic aspects on the selective surface oxidation of binary, ternary and quarternary model alloys. Applied Surface Science 253 (10), 4607–4619 (2007).
Vanden Eynde, X., Servais, J.-P. & Lamberigts, M. Thermochemical surface treatment of iron-silicon and iron-manganese alloys. Surface and Interface Analysis 33 (4), 322–329 (2002).
Martinez, C., Cremer, R., Neuschütz, D. & Von Richthofen, A. In-situ surface analysis of annealed Fe-1.5% Mn and Fe-0.6% Mn low alloy steels. Analytical and Bioanalytical Chemistry 374 (4), 742–745 (2002).
Martínez, C., Cremer, R., Loison, D. & Servais, J. P. In-situ investigation on the oxidation behaviour of low alloyed steels annealed under N\(_{2}\)-5% H2 protective atmospheres. Steel Research 72 (11–12), 508–511 (2001).
Shinoda, K., Yamamoto, T. & Suzuki, S. Characterization of selective oxidation of manganese in surface layers of Fe-Mn alloys by different analytical methods. ISIJ international 53 (11), 2000–2006 (2013).
Mayer, P. & Smeltzer, W. Kinetics of manganeseo-wustite scale formation on iron-manganese alloys. Journal of the Electrochemical Society 119 (5), 626 (1972).
Jackson, P. & Wallwork, G. The oxidation of binary iron-manganese alloys. Oxidation of Metals 20 (1–2), 1–17 (1983).
Wilson, P. & Chen, Z. The effect of manganese and chromium on surface oxidation products formed during batch annealing of low carbon steel strip. Corrosion Science 49 (3), 1305–1320 (2007).
Nijdam, T. & Sloof, W. G. Modelling of composition and phase changes in multiphase alloys due to growth of an oxide layer. Acta Materialia 56 (18), 4972–4983 (2008).
Larsson, H. & Engström, A. A homogenization approach to diffusion simulations applied to \(\alpha\)+\(\gamma\) Fe-Cr-Ni diffusion couples. Acta Materialia 54 (9), 2431–2439 (2006).
Larsson, H., Strandlund, H. & Hillert, M. Unified treatment of kirkendall shift and migration of phase interfaces. Acta Materialia 54 (4), 945–951 (2006).
Bale, C. W. et al. Factsage thermochemical software and databases. Calphad 26 (2), 189–228 (2002).
Goursat, A. & Smeltzer, W. Kinetics and morphological development of the oxide scale on iron at high temperatures in oxygen at low pressure. Oxidation of Metals 6 (2), 101–116 (1973).
Aghaeian, S., Sloof, W. G., Mol, J. M. C. & Böttger, A. J. Initial high-temperature oxidation behavior of Fe-Mn binaries in air: The kinetics and mechanism of oxidation. Oxidation of Metals 98, 217–237 (2022).
Feulvarch, E., Bergheau, J. & Leblond, J. An implicit finite element algorithm for the simulation of diffusion with phase changes in solids. International Journal for Numerical Methods in Engineering 78 (12), 1492–1512 (2009).
Andersson, J.-O., Helander, T., Höglund, L., Shi, P. & Sundman, B. Thermo-Calc & DICTRA, computational tools for materials science. Calphad 26 (2), 273–312 (2002).
Mao, W. Oxidation phenomena in advanced high strength steels: Modelling and experiment. Ph.D. thesis, Delft University of Technology (2018).
Ames, W. & Brezinski, C. Handbook of differential equations (North-Holland, 1992).
MATLAB. version 9.8.0.1721703 (R2020a) (The MathWorks Inc., Natick, Massachusetts, 2020).
Haynes, W. M., Lide, D. R. & Bruno, T. J. CRC handbook of chemistry and physics CRC press, Boca Raton (2016).
Auinger, M., Müller-Lorenz, E.-M. & Rohwerder, M. Modelling and experiment of selective oxidation and nitridation of binary model alloys at \(700^\circ\)C-the systems Fe, 1 wt%\(\{\)Al, Cr, Mn, Si\(\}\). Corrosion Science 90, 503–510 (2015) .
Andersson, J.-O. & Ågren, J. Models for numerical treatment of multicomponent diffusion in simple phases. Journal of applied physics 72 (4), 1350–1355 (1992).
Stott, F., Wood, G. & Stringer, J. The influence of alloying elements on the development and maintenance of protective scales. Oxidation of Metals 44 (1), 113–145 (1995).
Acknowledgements
This research was carried out under project number T17019p in the framework of the research program of the Materials innovation institute (M2i, www.m2i.nl) supported by the Dutch government. The authors are indebted to Dr. W. Melfo of Tata Steel (IJmuiden, The Netherlands) for the valuable discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aghaeian, S., Brouwer, J.C., Sloof, W.G. et al. Numerical Model For Short-Time High-Temperature Isothermal Oxidation of Fe–Mn Binaries at High Oxygen Partial Pressure. High Temperature Corrosion of mater. 99, 201–218 (2023). https://doi.org/10.1007/s11085-023-10153-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-023-10153-7