
Abstract
Material selection for thermal cracking processes of anthropogenic resources is challenging. H2S and HCl are formed, and corrosion occurs at high temperatures. Several steels were investigated in the past. In all cases, corrosion layers were characteristically structured. On top, large chromium sulfide crystals are located, followed by a Cr2O3 layer, followed by a layer enriched in chlorine and nickel. In this paper, we propose a model on the course of the corrosion reactions. By considering the water–gas shift reaction as well as the influence of H2S, the layer formation can be explained. As an example S31400, 240 h corrosion time at 680 °C was chosen.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In several industrial processes, construction materials suffer from chlorine and/or sulfide high-temperature corrosion, e.g., ethylendichloride production, gasification of biomass, coal or waste. Most processes work at higher partial pressures of oxygen, where metallic materials are able to form protective oxide layers. However, if chlorine is present, porous and non-protective corrosion layers are formed [1,2,3,4,5].
The mechanism of chlorine-induced high-temperature corrosion in oxidizing atmosphere is well established and was investigated by numerous authors [7,8,9,10,11,12,13,14,15,16,17,18,19,20]. At temperatures above 400 °C, chlorine or low molecular chlorine-containing compounds, like HCl, are able to move through the initial, protective oxide layer. The mechanisms how chlorine or HCl is able to do so are still part of investigations and not clear yet. Below the oxide layer, metal chlorides are formed, which are volatile and evaporate. The gaseous metal chlorides diffuse out of the oxide layer until they reach areas with a higher oxygen partial pressure. There they react to metal oxides and HCl. The formed HCl is able to reenter the reaction cycle and an autocatalytic corrosion process is initiated, which leads to rapid material failures. Generally, chromium forms its oxide in most environments due to its high affinity to oxygen [7,8,9,10,11,12,13,14,15,16,17,18,19,20].
Stability diagrams help to estimate the thermodynamic stability of a distinct phase when a metal is subjected to corrosion by two or more oxidants. These diagrams were often used by authors to explain the corrosion of metals in mixed oxidant atmospheres [7, 8]. In order to obtain more realistic results, in case of involvement of volatile compounds (e.g., FeCl2), the concept of quasi-stability diagrams was introduced by Schwalm et al. [1,2,3]. These quasi-stability diagrams are based on a limiting vapor pressure of the volatile species of 10−4 bar which, in this case, is the metal chloride. Quasi-stability diagrams reach their limits when a third oxidant is added to the gas mixture.
In coal gasification or thermal cracking of municipal solid waste processes, H2S is additionally present beside HCl and CO2 [21, 22]. The influence of H2S on the chlorine-induced high-temperature corrosion is not well investigated. Only Bakker et al. [21, 22] and Pan et al. [23] dealt with this topic and even found contradictory results in some cases.
During earlier experiments, corrosion resistance of several steels and nickel-based alloys was investigated in H2S- and HCl-containing atmosphere at high temperatures [4,5,6]. The potential for oxidation in the gas mixture was low because CO2 and no O2 were present. Because the interpretation of the observed corrosion layers was non-trivial and was only suggested in the previous publications; in this paper, a model about the course of the corrosion reaction is proposed based on additionally performed thermodynamic calculations. As an example alloy the austenitic stainless steel S31400 (1.4841), which was tested at 680 °C for 240 h in the corrosive gas atmosphere was chosen.
Materials and Methods
All corrosion experiments were performed in an inert silica tube, which was heated with a tube furnace. In order to reach a well-defined substrate surface, all specimens were polished with 1000 grit SiC paper, washed, degreased and gauged before they were placed on the specimen holder in the silica tube. The whole system was purged with nitrogen for 30 min before the experiment was started and during the heating process. Until the furnace reached the required temperature, the testing gas mixture was added with 120 ml/min. By purging the system with nitrogen for 30 min before the furnace was turned off, the experiment was stopped.
The gas for the tests consisted of 3.8 vol.% HCl, 1.9 vol.% CO2, 0.3 vol.% CO, 2.8 vol.% H2, 0.02 vol.% H2S, bal. N2 and represents the corrosive gas atmosphere in a thermal cracking process for anthropogenic resources. It was provided in two separated bottles by Linde gas (Eggendorf, AUT) as HCl cannot be stored together with H2S. Stainless steel tubes were used to transport the gas from the bottles to the silica tube. The detailed experimental procedure and a scheme of the testing equipment can be found in 4 and 6; the chemical composition of steel S31400 is shown in Table 1. The corrosion experiment discussed here was performed at 680 °C in a silica glass tube for ten days.
Thermodynamic Calculations
Thermodynamic calculations were performed with the software package FactSage 7.2 (© 1976–2018 Thermfact and GTT-Technologies). The Gibbs free energies of given chemical reactions were obtained with the FactSage module Reaction using the FactPS compound database (FACT pure substances database). The built-in calculation routine is based on Gibbs energy minimization and accesses only compound databases. Further, the calculation assumes all gas phase species to be ideal and neglects expansivity and compressibility of solid and liquid phases.
Metallographic Preparation of the Samples After the Corrosion Experiments
In order to prepare cross sections of the corroded samples, they were cold-mounted in epoxy resin (Struers® Epo Fix). A vacuum guaranteed the absence of gas inclusions. Polishing was performed water-free to avoid the dissolution of the water-soluble metal chlorides. With the exception of the 3-µm polishing, SiC paper was used. The bounded grains maintained corrosion products way better than loose grains. Isopropyl alcohol was used as lubricant.
Results
The general appearance of the glass tube and the samples after a 240-h corrosion experiment at 680 °C is illustrated in Fig. 1. Samples show a black layer of corrosion products (Fig. 1b) with black crystals grown on top. Colorless crystals are visible in gas flow direction, outside the furnace (Fig. 1c). EDX and XRD analysis identified these crystals as pure FeCl2 [4, 5]. A corrosion mechanism was developed to explain the results obtained during metallographic preparation.

Samples and silica glass tube after corrosion experiment: a silica tube with samples (in the furnace) and FeCl2 deposition (outside the furnace), b samples in the sample holder, c deposited FeCl2
The Corrosion Experiment at 680 °C
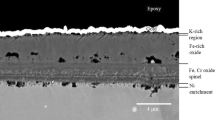
Figure 2 shows a SEM image of a cross section of steel S31400 after 240-h experiment at 680 °C. The big, black crystals of Fig. 1b are also visible in the cross section. They grew on top of a porous structure, which is filled with corrosion products. Thickness of the corrosion products is about 100 µm. The crack through the porous layer in Fig. 2 (right) illustrates the poor adhesion of the corrosion products on the sample. An EDX mapping of the same region as shown in the SEM image is displayed in Fig. 3. The big crystals on top of the corrosion products consist of chromium and sulfur, with minor amounts of oxygen. Below these crystals, a chromium- and oxygen-containing layer had formed, while a high amount of chlorine was detected in the lowest layer of corrosion products.

SEM image of a cross section of S31400, after 240-h experiment at 680 °C

EDX mapping of cross section of S31400, after the 240-h experiment at 680 °C
An XRD analysis of the corrosion products revealed the phases Cr3S4, Cr2S3 and Cr2O3. No metal chlorides were found using XRD. This is most probably due to the very hygroscopic character of metal chlorides, which may react with atmospheric water and therefore are not crystalline. Furthermore, there was an enrichment of nickel in the border area of the base metal, next to the corrosion products. Chromium and iron were depleted in this area and were found in the corrosion products as Cr3S4, Cr2S3, Cr2O3. The volatile FeCl2 re-sublimated in the colder parts of the silica tube. The enrichment of nickel reveals that there is no reaction between this metal and the gas atmosphere. A possible course of corrosion reactions was developed to explain this setup of corrosion products.
Discussion
Suggested Corrosion Mechanism
Based on the experimental results, considerations about possible chemical reactions, as well as some thermodynamic considerations and a sequence of these chemical reactions, can be proposed. Figure 4 shows a schematic illustration of the proposed corrosion mechanism.

Illustration of the proposed corrosion mechanism for austenitic steels in H2S-, HCl- and CO2-containing atmosphere
Early Stage of Corrosion
High-alloyed steels form an initial, thin and protective oxide layer. The most aggressive compound in the gas mixture is HCl, which is able to penetrate through this initial oxide layer. However, the initial oxide layer itself will not react with HCl as Cr2O3 is thermodynamically too stable [10]. Below this layer, it reacts with the metals to form metal chlorides. The various alloying elements show different reactivities with HCl (reaction 1).
Thermodynamic calculations (Fig. 5a) illustrate the Gibbs free energies for the reactions of iron, chromium and nickel with HCl between 480 and 680 °C. The reactions of iron and chromium show negative Gibbs free energies in contrast to nickel with a positive Gibbs free energy. From this point of view, FeCl2 and CrCl2 will be formed and nickel remains metallic. Additionally, FeCl2 is volatile at 680 °C and evaporates, and it seems that there is no further reaction in the gas phase, as pure FeCl2 was observed after resublimation in the cold parts of the testing equipment. The evaporating FeCl2 results in a growing porosity with increasing corrosion times.

Gibbs free energies of some reactions: a metallic Fe, Cr, Ni with HCl, b possible metal chlorides with H2O, c chromium chloride with H2S
The vapor pressure of CrCl2 is about 10−2 mbar at 680 °C, which causes slow evaporation rates.
The explanation for the chromium sulfide crystals on the surface and Cr2O3 layer formation can be found in the water–gas shift reaction, which forms H2O from CO2 and H2.
The Water–Gas Shift Reaction
The water–gas shift reaction is an equilibrium reaction which can be catalyzed on surfaces, thus increasing its reaction rate there. Bustamante et al. [24] found a difference in conversion rate of two orders of magnitude for the reversed water–gas shift reaction on an Inconel surface compared to a quartz surface. However, impurities like sulfur can restrain the catalytic effect. The position of the equilibrium is defined by the reaction temperature. At lower temperatures of about 200 °C (reaction 2), it is used to generate H2 from CO and H2O.
At higher temperatures (about 1000 °C), the equilibrium reaction (reaction 3) is used to produce H2O very slowly for depositing Al2O3 coatings by chemical vapor deposition (CVD) [25,26,27].
The presence of H2S influences the catalytic reaction at low as well as high temperatures. At low temperatures, the decrease in reaction rate is negative for H2 production. In CVD processes, H2S also reduces the reaction rate of H2O formation, which reduce Al2O3 powder formation in the gas phase [28, 29].
Steady-State Stage of Corrosion
After the first attack of HCl and the formation of a porous layer of corrosion products, diffusion processes determine the further development of the corrosion process. HCl will still diffuse to the metal to form metal chlorides and hydrogen. The amount of hydrogen will increase in the layer, but it will also diffuse outwards. As shown above, due to the presence of H2S and missing of a catalytic surface, the water–gas shift reaction is negligible in the main gas flow.
Because of the diffusion gradient, CO2 diffuses into the corrosion layer, and the water–gas shift reaction can take place on these surfaces. Formed H2O will react with the CrCl2 already present (reaction 4) (Fig. 5b). This explains the thick oxide layer observed in the EDX mapping.
From a kinetic point of view, it is unlikely that this reaction takes place in one step. Possibly, CrO is formed first as an intermediate (reaction 5), followed by further oxidation of this thermodynamically unstable molecule (reaction 6).
The absence of oxygen in deeper parts of the corrosion layer can be explained by the fact that CO2 is converted completely into H2O and subsequently to Cr2O3 near the surface.
Excessive CrCl2 in the corrosion layer, which is not converted to Cr2O3, will diffuse out of the corrosion layers. In contact with the main gas flow containing 200 ppm H2S, the CrCl2 will form the detected Cr2S3 and Cr3S4 sulfides via the reactions in Fig. 5c (reaction 7). On top of the corrosion surface, large crystals of Cr2S3 or Cr3S4, respectively, were observed (Fig. 3). As mentioned above for the oxides, the formation of an intermediate CrS is plausible as well (reaction 8), followed by further oxidation (reaction 9).
The fact that H2S suppresses the water–gas shift reaction, and that there is no H2O in the main gas flow, explains the absence of oxygen in the chromium sulfide crystals.
Thermodynamically, the formation of iron sulfide from FeCl2 is possible as well. However, no sulfides of iron were detected at 680 °C, and only FeCl2 crystals were present in the cold parts of the testing equipment. Kinetic reasons seem plausible for the suppression of the conversion of FeCl2 to FeS with H2S.
The enrichment of nickel in relation to the steel matrix (Fig. 3) can be explained by its low reactivity with HCl (Fig. 5a) and the fact that iron and chromium are lost due to the evaporating FeCl2 and CrCl2. The chlorine content below the corrosion layers is caused by remaining metal chlorides, which could not be evaporated until the experiment was completed and the furnace was turned off.
Conclusions
For the corrosion of austenitic steels like S31400 in a H2S-, HCl- and CO2-containing environment at 680 °C, a characteristic setup of the corrosion products was observed. On top of the substrate, large chromium sulfide crystals were detected, followed by a porous layer containing Cr2O3, and in contact with the metallic substrate, a porous area was found containing chlorine and an enrichment of nickel with respect to the metal matrix.
The phenomenon of separated regions with Cr2O3 and chromium sulfides can be explained by the peculiarity of the water–gas shift reactions, which forms H2O from CO2. Inside the corrosion layers, CO2 can react with already present hydrogen to H2O, which immediately forms Cr2O3 via the reaction with CrCl2. Excessive CrCl2 diffuses out of the corrosion layers and reacts on the surface with H2S to Cr3S4 and Cr2S3. The reason that oxides and sulfides are not mixed can be explained by the influences of H2S on the water–gas shift reaction.
References
C. Schwalm and M. Schütze, Materials and Corrosion, 51, 34–49 (2000).
R. Bender and M. Schütze, Materials and Corrosion, 54, 567–586 (2003).
H. Latreche, S. Doublet and M. Schütze, Oxidation of Metals, 72, 1–30 (2009).
A. Schmid, G. Mori, R. Haubner, M. Weil and S. Hönig, Materials and Corrosion, 69, 1328–1337 (2018).
A. Schmid, G. Mori, S. Strobl, R. Haubner and S. Hönig, Corrosion of various Fe and Ni based alloys in HCl, H2S containing environments, with low oxygen partial pressure, at 680 °C, Presentation: EUROCORR 2017, 20th International Corrosion Congress & Process Safety Congress 2017, Prague, Czech Republic; 03.09.–07.09. 2017, in Conference Proceedings EUROCORR 2017, Paper-Nr. 79016, 10 S (2017).
A. Schmid, G. Mori, S. Hönig, M. Weil, S. Strobl nad R. Haubner, Corrosion Science, 139, 76–82 (2018).
A. Zahs, M. Spiegel and H.J. Grabke, Materials and Corrosion, 50, 561–578 (1999).
D. Bramhoff, H.J. Grabke and E. Reese, Werkstoffe und Korrosion, 41, 303–307 (1990).
Y. Ihara, H. Ohgame and K. Sakiyama, Corrosion Science, 21, 805–817 (1981).
Y. Ihara, H. Ohgame and K. Sakiyama, Corrosion Science, 23, 167–181 (1983).
Y. Ihara, H. Ohgame and K. Sakiyama, Corrosion Science, 22, 901–912 (1982).
H. Asteman and M. Spiegel, Corrosion Science, 49, 3626–3637 (2007).
D. Bramhoff, H.J. Grabke and H.P. Schmidt, in The role of active elements, ed. E. Lang, (Elsevier, 1989), pp 335–349.
W.T. Bakker, Materials at high temperatures, 14, 197–206 (1997).
S.H. Lee and M.J. Castaldi, The effects of varied hydrogen chloride gas concentrations on corrosion rates of commercial tube alloys under simulated environment of WTE facilities, Waste-to-Energy conference, Pennsylvania (2008).
R. Prescott, F.H. Stott and P. Elliott, Corrosion Science, 29, 465–475 (1989).
D. Bramhoff, H.J. Grabke and H.P. Schmidt, Werkstoffe und Korrosion, 40, 642–650 (1989).
V.A.C. Haanappel, T. Fransen and P.J. Gellings, High Temperature Materials and Processes, 10, 67–89 (1992).
Zahs, M. Spiegel nad H.J. Grabke, Corrosion Science, 42, 1093–1122 (2000).
F.H. Stott, R. Prescott and P. Elliott, Materials Science and Technology, 6, 364–370 (1990).
W.T. Bakker and R.A. Perkins, in Materials for coal gasification, eds. W.T. Bakker, S. Dapkunas and V. Hill, (ASM International, 1988), pp 85–96.
W.T. Bakker and R.A. Perkins, Beyond mixed oxidant corrosion-corrosion Phenomena in gasifiers, in EPRI Gasification Conference (1990).
T.J. Pan et.al., Corrosion Science, 49, 1362–1377 (2007).
F. Bustamante, R. Enick, K. Rothenberger, B. Howard, A. Cugini, M. Ciocco and B. Morreale, Fuel Chemistry Division Reprints, 47, 663–664 (2002).
M. Kornmann, H. Schachner, R. Funk and B. Lux, Journal of Crystal Growth, 28, 259–262 (1975).
R. Funk, H. Schachner, C. Triquet, M. Kornmann and B. Lux, Journal of the Electrochemical Society, 123, 285–289 (1976).
R.J. Byron Smith, L. Muruganandam and S.S. Murthy, International Journal of Chemical Reactor Engineering, 8, 1–32 (2010).
S. Ruppi and A. Larsson, Thin Solid Films, 388, 50–61 (2001).
A. Blomqvist, C. Århammar, H. Pedersen, F. Silvearv, S. Norgren and R. Ahuja, Surface and Coatings Technology, 206, 1771–1779 (2011).
Acknowledgments
Open access funding provided by Montanuniversität Leoben. The material was provided by Salzgitter AG, which is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
Alexander Schmid and Gregor Mori contributed to conceptualization; Alexander Schmid contributed to methodology; Edith Bucher contributed to software; Alexander Schmid, Gregor Mori and Roland Haubner contributed to validation; Alexander Schmid contributed to formal analysis; Alexander Schmid contributed to investigation; Alexander Schmid and Gregor Mori contributed to resources; Alexander Schmid and Edith Bucher contributed to data curation; Alexander Schmid contributed to writing—original draft preparation; Alexander Schmid, Gregor Mori and Roland Haubner contributed to writing—review and editing; Alexander Schmid contributed to visualization; Gregor Mori and Roland Haubner supervised the study; and Gregor Mori contributed to project administration.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Schmid, A., Mori, G., Bucher, E. et al. Model About the Course of Corrosion Reactions of Austenitic Steels in H2S-, HCl- and CO2-Containing Atmospheres at 680 °C. Oxid Met 91, 1–10 (2019). https://doi.org/10.1007/s11085-018-9876-z
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-018-9876-z