
Abstract
Anti-fungal therapies remain sub-optimal, and resistant pathogens are increasing. New therapies are desperately needed, especially options that are less toxic than most of the currently available selection. In this review, I will discuss anti-fungal therapies that are in at least phase I human trials. These include VT-1161 and VT-1598, modified azoles with a tetrazole metal-binding group; the echinocandin rezafugin; the novel β-1,3-d-glucan synthase inhibitor ibrexafungerp; fosmanogepix, a novel anti-fungal targeting Gwt1; the arylamidine T-2307; the dihydroorotate inhibitor olorofim; and the cyclic hexapeptide ASP2397. The available data including spectrum of activity, toxicity and stage of clinical development will be discussed for each of these so clinicians are aware of promising anti-fungal agents with a strong likelihood of clinical availability in the next 5–7 years.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
COVID-19 has exposed our paltry supply of effective anti-viral therapies. However, the truth is that anti-infectives generally have vastly increased in the past two decades, and this is equally and perhaps especially true of anti-fungal therapies.
Existing anti-fungal agents have a variety of limitations, from toxicity to rising levels of resistance in common fungal pathogens, significant drug interactions and sub-optimal efficacy. The rising proportion of the community in western countries with induced immune deficiencies from immunomodulatory therapies and their expanding indications is gradually increasing the burden of fungal disease. Equally, unlike anti-bacterial therapies which have exceptionally good response rates in sensitive pathogens, anti-fungal therapies are not as efficacious, probably due to a mixture of poor host immunity, innate fungal properties and dosing limitations due to toxicity. There is no question that new anti-fungal agents would be welcomed by clinicians and patients routinely dealing with these infections.
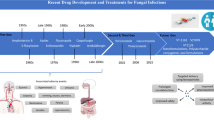
In this review, I will focus on promising anti-fungal therapies with at least phase I human data in the public domain (clinicaltrials.gov, PubMed and web search). While there are many agents not yet at this stage of development, their time frame for clinical use (if successful) is unlikely to be within the next decade.
VT-1161 and VT-1598
These two related compounds (and a third so far less advanced VT-1129) from the same company are modifications of the azoles with replacement of the triazole metal-binding group with a tetrazole and other structural changes to reduce the affinity for mammalian CYP 450 enzymes but maintain activity against fungal Cyp51 [1]. These changes are hoped will lead to less toxicity and less drug interactions.
VT-1161 has in vitro activity against C. glabrata and C. krusei isolates resistant to azoles and echinocandins [2], Trichophyton spp. [3], Coccidioides immitis and Coccidioides posadassii [4]. VT-1161 appears to have superior in vitro activity to VT-1120 for Candida and in interleukin-17 deficient mice was highly effective against fluconazole-susceptible and fluconazole-resistant oropharyngeal candidiasis [5]. Against C. posadassii in a murine model, VT-1161 had at least equivalent efficacy to fluconazole [4], and prophylactic administration was more effective than posaconazole in preventing Rhizopus arrhizus infection in neutropenic mice [6]. In an open-label study of naturally occurring coccidioidomycosis in dogs, VT-1161 was well tolerated and an apparent 83% successful response to therapy [7]. Other evidence of excellent anti-fungal activity of VT-1161 includes dermatophytosis in a guinea pig model [8],
In a phase II study of VT-1161 in recurrent vulvovaginal candidiasis, 215 women were randomized to one of five regimes i)150 mg once daily for 7 days followed by 150 mg once weekly for 11 weeks; ii)300 mg once daily for 7 days followed by 300 mg once weekly for 11 weeks; iii) 150 mg once daily for 7 days followed by 150 mg once weekly for 23 weeks; iv) 300 mg once daily for 7 days followed by 300 mg once weekly for 23 weeks; v) a matching placebo regime. On an intention to treat basis all VT-1161 arms substantially reduced the number of candidiasis episodes compared to placebo (0–7% vs 52%). No safety concerns arose during the trial with no effect on liver function observed. The basis for undertaking the phase II study was earlier work showing efficacy of VT-1161 in a murine model of vaginal candidiasis [9]. A second phase 2 study in vulvovaginal candidiasis compared 300 mg once daily for 3 days, 600 mg once daily for 3 days, 600 mg twice daily for 3 days or a single dose of 150 mg of fluconazole. No significant differences were observed between the VT-1161 and fluconazole treated groups, nor were there significant differences in response rates between the VT-1161 groups. A trend to higher recurrence rates at 3 and 6 months was observed in the fluconazole arm (0% at 3 and 6 months for VT-1161 groups vs 28.5% and 46.1% for fluconazole) [10]. Two identical phase 3 studies (as required by the Federal Drug Administration for registration) of VT-1161 in recurrent vaginal candidiasis are currently listed as active on clinicaltrials.gov (NCT03562156 and NCT03561701) with estimated study completion dates in mid-2021.
In a similarly designed phase II study of VT-1161 in onychomycosis patients were randomized to 300 mg daily for 2 weeks followed by once weekly 300 mg dose for either 10 or 22 weeks or 600 mg daily for 2 weeks followed by 600 mg once weekly for 10 or 22 weeks or a matching placebo arm. Cure rates ranged from 32%-42% in the active arms compared to 0% in placebo. Again no significant adverse side effects were observed [11].
VT-1598 has been less well studied but also has activity against fluconazole-susceptible and fluconazole-resistant C.albicans [12], a variety of Candida and Cryptococcus spp., Aspergillus spp. and Rhizopus arrhizus [13]. VT-1598 also appears to have good efficacy in murine models of central nervous system coccidioidomycosis [14], cryptococcal meningitis [15] and invasive aspergillosis [16]. A phase I study of VT-1598 (NCT04208321) is currently listed to complete in mid-2021.
Although VT-1161 and VT-1598 show promise, some caution needs to be stated in that mutations of Erg3 have been described in C. albicans which confer resistance to both agents [17]. Similarly, the common mechanisms for azole resistance in C. glabrata, mutations within the zinc cluster transcription factor Pdr1 [18], can reduce susceptibility to both VT-1161 and VT-1598 [19]. Given the wide-spread problems that have arisen with azole-resistant C. glabrata [20], it is likely that resistance will rapidly become a problem with this pathogen.
Rezafungin (CD101)
Rezafungin is an echinocandin, targeting β1,3-D-glucan like caspofungin, micafungin and anidulafungin. The modifications incorporated into rezafungin were designed to give it a longer half-life and therefore potentially less frequent administration than daily dosing required with other echinocandins [21]. Pharmacokinetic studies in mice, rats, dogs, monkeys and chimpanzees confirmed that may be suitable for weekly administration. Indeed, in the chimpanzees, the t1/2 was 81 h compared to 30 h for anidulafungin [22].
Against a variety of fungal isolates in the SENTRY 2015 surveillance program rezafungin had equivalent efficacy to other echinocandins tested [23]. Similarly, rezafungin had equivalent efficacy to anidulafungin against a selection of Canadian clinical Candida spp. isolates, with both having potency against fluconazole-resistant isolates [24]. However, rezafungin did not maintain activity against a small number of anidulafungin-resistant isolates (5 C. glabrata and 2 C. tropicalis) [24]. The value of rezafungin is therefore in its longer half-life rather than increased spectrum of activity over existing echinocandins.
In a phase I study in 6 subjects, CD101 was tolerated at up to 400 mg as a weekly dose for 3 weeks. The t1/2 was estimated at greater than 81 h in the 400 mg and 200 mg doses with minimal urinary clearance and no change observed in renal, hepatic or hematological blood tests [25].
In a randomized, head to head trial with a single dose of 150 mg of oral fluconazole, topical CD101 6% ointment (one dose) or 3% gel (daily dose for 2 days) was studied in 126 women with vulvovaginal candidiasis. While both preparations of topical CD101 were well tolerated, clinical cure rates were lower in the gel (35%) and ointment (30%) groups compared to fluconazole (52.6%) [26].
Thompson et al. [27] recently published a phase 2, randomized, double-blind study comparing rezafungin in one of two dosing regimes (400 mg weekly or 400 mg in the first week then 200 mg weekly) or caspofungin 70 mg for one day followed by 50 mg daily, in the setting of candidemia in 207 patients. There were no statistically significant differences between therapy arms, although there was a trend to lowest response in the 400 mg rezafungin arm (cure rate 60.5% in the 400 mg weekly, 76.1% in the 200 mg weekly and 67.2% in the caspofungin arm. Side effects were no different between groups.
Two current rezafungin studies are listed on clinicaltrials.gov. A phase 3 trial comparing rezafungin to fluconazole for the prevention of invasive fungal disease in patients undergoing bone marrow transplantation is estimated to complete in 2022 (NCT04368559). A phase 3 comparison of rezafungin with caspofungin in the setting of candidemia is estimated to complete in mid-2021 (NCTNCT03667690). The dose chosen in the latter trial is the more efficacious arm of the rezafungin study by Thompson et al. of a 400 mg loading dose followed by 200 mg per week [27].
Ibrexafungerp (SCY-078)
Also targeting β-1,3-d-glucan synthase is a novel synthetic derivative of enfumafungin, ibrexafungerp that has the added advantage over the echinocandins of being orally bioavailable [28]. Initial studies in a murine model of candidiasis showed good activity against multiple Candida spp. including C.albicans, C.glabrata and C.parapsilosis [28]. In vitro data from clinical isolates [29] and animal models [30] also suggest reasonable activity against C.auris. However, like echinocandins, ibrezafungerp is poorly active against Mucorales and Fusarium, but was active against Scediosporium profliicans [31]. Against echinocandin-resistant strains of Candida including C.glabrata, ibrezafungerp generally maintained reasonable activity with MIC values increasing 2–eightfold compared to 2–133-fold for anidulafungin, 4–66-fold for caspofungin and 1–66 fold for micafungin [32].
Ibrexafungerp alone or in combination with isavuconazole was studied in a neutropenic rabbit model of invasive aspergillosis [33]. The combination of both agents was clearly superior to monotherapy with either, with the higher dose of ibrexafungerp (7.5 mg/kg/day) combined with isavuconazole having the best survival, lowest pulmonary infarct scores and lowest fungal burden.
An open-label phase 2 study looked at the efficacy of ibrexafungerp following echinocandin therapy in 27 patients with invasive candidiasis. Seven subjects received ibrexafungerp 500 mg, another 7 received 750 mg and the remainder received fluconazole(7) or micafungin(1) as standard of care [34]. Clinical success rates were similar across all groups at the end of treatment, 2-week post and 6-week post-time points with no safety concerns identified. These findings are consistent with in vitro data suggesting ibrexafungerp is fungostatic against A.fumigatus and that synergy exists with azoles and amphotericin B against Aspergillus spp [35].
A phase I study of ibrezafungerp in healthy males is listed as completed in clinicaltrials.gov (NCT04307082), but results are not yet published. A number of other currently active studies include a phase 3 study in recurrent vulvovaginal candidiasis (NCT04029116), an open-label phase 1 study in invasive candidiasis due to C.auris (CT03363841) and an open-label study in patients with fungal disease refractory to other options (NCT03059992). All of these studies are due to complete by end 2021.
Fosmanogepix (APX001)
Fosmanogepix is a novel anti-fungal targeting Gwt1, which is an early enzyme in the synthesis of glycosylphosphatidylinositol [36, 37]. Murine neutropenic models suggest fosmanogepix may be an effective treatment option for invasive Scediorporium and Fusarium [38], invasive candidiasis [39] and promisingly also for Rhizopus arrhizus [40]. Fosmanogepix also had efficacy in C.albicans endophthalmitis and meningoencephalitis in non-neutropenic rabbits [41] (Table 1)
Serial passage experiments in candida species demonstrated that mutations leading to resistance to fosmanogepix developed at about the same rate as with existing anti-fungal therapies [42].
Three of four phase I studies of fosmanogepix listed on clinicaltrials.gov have been completed but so far data have only been presented in poster format [37]. Based on the data presented fosmanogepix has high bioavailability and there were no barriers identified raising concerns about moving to phase 2 trials. Current phase 2 trials on clinicaltrials.gov are targeting invasive disease by Aspergillus or rare molds (NTC04240886) and invasive candidiasis due to C.auris (NTC04148287).
T-2307
T-2307 is an arylamidine similar to pentamidine and targets fungal mitochondria[43] through uptake via the high-affinity spermine and spermidine carrier [44]. In vitro T-2307 is active against Cryptococcus neoformans [45] and gattii [46], A. fumigatus [45] as well as most species of Candida [45], including echinocandin-resistant C. glabrata [47]. In a murine ocular model of C.albicans, T-2307 showed equivalent efficacy to amphotericin and fluconazole [48]. A successful phase I trial of T-2307 was completed in 2015 (clinicaltrials.gov NCT02289599), and a phase II study is currently proposed for 2022 [49].
Olorofim (F901318)
Olorofim is currently in clinical development through a phase IIa trial in patients with invasive fungal infection lacking other treatment options (Clinicaltrials.gov NCT03583164) with an estimated study completion date of February 2021. Olorofim inhibits the enzyme dihydroorotate (DHOD), a key step in the synthesis of pyrimidines [50]. In vitro F910318 is highly active against azole-resistant Aspergillus fumigatus, Scedosporium spp. and Fusarium spp., but has no activity against Mucorales and Candida [51,52,53,54,55]. In a neutropenic, A.terreus-infected murine model, twice daily olorofim administration at 10 mg/kg was more effective than amphotericin (0% 2-week survival vs 90% for oral F901318 and 100% for intravenous) [56]. In a phase I study of oral olorofim at 360 mg per day was well tolerated with no adverse events [57], which is not surprising given there is only approximately 20% homology between human and fungal DHOD [58].
ASP2397
ASP2397 is produced by Acremonium persicinum and is a cyclic hexapeptide similar to a number of characterized fungal siderophores although its exact target is unknown [59]. ASP2397 is transported into A.fumigatus by the Sit 1 siderophore transporter, inactivation of which conveys resistance to its actions [60]. In vitro ASP2397 has excellent activity against A.fumigatus, A.terreus and A.flavus, as well as Cryptococcus, but not Candida, Scedosporium or Rhizopus [61]. In vivo in a neutropenic mouse A.fumigatus model, those in the ASP2397 treatment group demonstrated greater survival than these treated with posaconazole, amphotericin or caspofungin [61]. There are currently no active trials for ASP2397 listed on clinicaltrials.gov with a phase II study and develop program terminated by the sponsor in 2019.
PC945
PC945 is a novel triazole targeting CYP51, inhibiting ergosterol synthesis, and was designed for inhalation rather than systemic administration [62]. In vitro the inhibition of ergosterol by PC945 appears to be significantly greater than that of voriconazole or posaconazole [63]. Against 96 clinical isolates of A.fumigatus the potency of PC945 was assessed as superior to voriconazole and comparable to posaconazole [63]. As well as Aspergillus spp., PC945 has activity against a broad range of fungi including Candida spp., Cryptococcus spp. and Rhizopus oryzae [63].
In a phase I trial, PC945 was well tolerated in healthy controls and subjects with mild asthma [64]. No current trials for PC945 are listed on clincaltrials.gov although a phase III trial is slated for 2021 in patients with refractory or resistant pulmonary fungal disease [65].
Inhaled itraconazole
Although itraconazole has been available for over two decades, a novel inhaled formulation is in clinical trials. For airways disease, such as allergic bronchopulmonary aspergillosis and for prophylaxis in immunocompromised patients an inhaled route may offer improved efficacy at much lower toxicity than systemic therapy. A phase 1 study demonstrated good tolerability of an inhaled itraconazole formulation, PUR1900, with 70-fold higher sputum concentrations than with systemic therapy and 66-fold lower systemic concentrations. A randomized, double-blind study of PUR1900 is, however, listed as terminated on clinicaltrials.gov, and no further trials are listed although the parent company lists a planned phase IIb study in development.
Summary
A number of anti-fungals are in or approaching phase III trials that promise to significantly increase the therapeutic options available to clinicians and patients. That these agents have collectively a wide range of activity including drug-resistant Candida, Aspergillus and Mucorales will be particularly welcome.
None of these new agents will be immune to the development of fungal resistance, particularly given the steadily increasing number of patients requiring anti-fungal therapy and/or prophylaxis. However, with greater bioavailability, less toxicity and different modes of action promised by the emerging anti-fungal therapies discussed above, the possibility of combination therapy to reduce the likelihood of resistance developing, as well as potentially increasing the efficacy of therapy, becomes a real possibility. While it is highly unlikely all the candidates discussed will pass the phase 3 testing ground and get to market, a significant number is likely to be our new tools in the decade ahead.
References
Warrilow AG, Hull CM, Parker JE, Garvey EP, Hoekstra WJ, Moore WR, et al. The clinical candidate VT-1161 is a highly potent inhibitor of Candida albicans CYP51 but fails to bind the human enzyme. Antimicrob Agents Chemother. 2014;58(12):7121–7. https://doi.org/10.1128/AAC.03707-14.
Schell WA, Jones AM, Garvey EP, Hoekstra WJ, Schotzinger RJ, Alexander BD. Fungal CYP51 Inhibitors VT-1161 and VT-1129 Exhibit Strong In Vitro Activity against Candida glabrata and C. krusei Isolates Clinically Resistant to Azole and Echinocandin Antifungal Compounds. Antimicrob Agents Chemother. 2017;61(3). https://doi.org/10.1128/AAC.01817-16.
Warrilow AGS, Parker JE, Price CL, Garvey EP, Hoekstra WJ, Schotzinger RJ et al. The Tetrazole VT-1161 Is a Potent Inhibitor of Trichophyton rubrum through Its Inhibition of T. rubrum CYP51. Antimicrob Agents Chemother. 2017; https://doi.org/10.1128/AAC.00333-17.
Shubitz LF, Trinh HT, Galgiani JN, Lewis ML, Fothergill AW, Wiederhold NP, et al. Evaluation of VT-1161 for Treatment of Coccidioidomycosis in Murine Infection Models. Antimicrob Agents Chemother. 2015;59(12):7249–54. https://doi.org/10.1128/AAC.00593-15.
Break TJ, Desai JV, Natarajan M, Ferre EMN, Henderson C, Zelazny AM, et al. VT-1161 protects mice against oropharyngeal candidiasis caused by fluconazole-susceptible and -resistant Candida albicans. J Antimicrob Chemother. 2018;73(1):151–5. https://doi.org/10.1093/jac/dkx352.
Gebremariam T, Alkhazraji S, Lin L, Wiederhold NP, Garvey EP, Hoekstra WJ et al. Prophylactic Treatment with VT-1161 Protects Immunosuppressed Mice from Rhizopus arrhizus var. arrhizus Infection. Antimicrob Agents Chemother. 2017; https://doi.org/10.1128/AAC.00390-17.
Shubitz LF, Roy ME, Trinh HT, Hoekstra WJ, Schotzinger RJ, Garvey EP. Efficacy of the Investigational Antifungal VT-1161 in Treating Naturally Occurring Coccidioidomycosis in Dogs. Antimicrob Agents Chemother. 2017; https://doi.org/10.1128/AAC.00111-17.
Garvey EP, Hoekstra WJ, Moore WR, Schotzinger RJ, Long L, Ghannoum MA. VT-1161 dosed once daily or once weekly exhibits potent efficacy in treatment of dermatophytosis in a guinea pig model. Antimicrob Agents Chemother. 2015;59(4):1992–7. https://doi.org/10.1128/AAC.04902-14.
Garvey EP, Hoekstra WJ, Schotzinger RJ, Sobel JD, Lilly EA, Fidel PL Jr. Efficacy of the clinical agent VT-1161 against fluconazole-sensitive and -resistant Candida albicans in a murine model of vaginal candidiasis. Antimicrob Agents Chemother. 2015;59(9):5567–73. https://doi.org/10.1128/AAC.00185-15.
Brand SR, Sobel JD, Nyirjesy P, Ghannoum MA, Schotzinger RJ, Degenhardt TP. Randomized Phase 2 Study of VT-1161 for the Treatment of Acute Vulvovaginal Candidiasis. Clin Infect Dis. 2020. https://doi.org/10.1093/cid/ciaa1204.
Elewski B, Brand S, Degenhardt T, Curelop S, Pollak R, Schotzinger R, et al. A phase II, randomized, double-blind, placebo-controlled, dose-ranging study to evaluate the efficacy and safety of VT-1161 oral tablets in the treatment of patients with distal and lateral subungual onychomycosis of the toenail. Br J Dermatol. 2020. https://doi.org/10.1111/bjd.19224.
Break TJ, Desai JV, Healey KR, Natarajan M, Ferre EMN, Henderson C, et al. VT-1598 inhibits the in vitro growth of mucosal Candida strains and protects against fluconazole-susceptible and -resistant oral candidiasis in IL-17 signalling-deficient mice. J Antimicrob Chemother. 2018;73(8):2089–94. https://doi.org/10.1093/jac/dky170.
Wiederhold NP, Patterson HP, Tran BH, Yates CM, Schotzinger RJ, Garvey EP. Fungal-specific Cyp51 inhibitor VT-1598 demonstrates in vitro activity against Candida and Cryptococcus species, endemic fungi, including Coccidioides species, Aspergillus species and Rhizopus arrhizus. J Antimicrob Chemother. 2018;73(2):404–8. https://doi.org/10.1093/jac/dkx410.
Wiederhold NP, Shubitz LF, Najvar LK, Jaramillo R, Olivo M, Catano G et al. The Novel Fungal Cyp51 Inhibitor VT-1598 Is Efficacious in Experimental Models of Central Nervous System Coccidioidomycosis Caused by Coccidioides posadasii and Coccidioides immitis. Antimicrob Agents Chemother. 2018;https://doi.org/10.1128/AAC.02258-17.
Garvey EP, Sharp AD, Warn PA, Yates CM, Schotzinger RJ. The novel fungal CYP51 inhibitor VT-1598 is efficacious alone and in combination with liposomal amphotericin B in a murine model of cryptococcal meningitis. J Antimicrob Chemother. 2018;73(10):2815–22. https://doi.org/10.1093/jac/dky242.
Garvey EP, Sharp AD, Warn PA, Yates CM, Atari M, Thomas S, et al. The novel fungal CYP51 inhibitor VT-1598 displays classic dose-dependent antifungal activity in murine models of invasive aspergillosis. Med Mycol. 2020;58(4):505–13. https://doi.org/10.1093/mmy/myz092.
Nishimoto AT, Wiederhold NP, Flowers SA, Zhang Q, Kelly SL, Morschhauser J et al. In Vitro Activities of the Novel Investigational Tetrazoles VT-1161 and VT-1598 Compared to the Triazole Antifungals against Azole-Resistant Strains and Clinical Isolates of Candida albicans. Antimicrob Agents Chemother. 2019;https://doi.org/10.1128/AAC.00341-19.
Vermitsky JP, Edlind TD. Azole resistance in Candida glabrata: coordinate upregulation of multidrug transporters and evidence for a Pdr1-like transcription factor. Antimicrob Agents Chemother. 2004;48(10):3773–81. https://doi.org/10.1128/AAC.48.10.3773-3781.2004.
Nishimoto AT, Whaley SG, Wiederhold NP, Zhang Q, Yates CM, Hoekstra WJ et al. Impact of the Major Candida glabrata Triazole Resistance Determinants on the Activity of the Novel Investigational Tetrazoles VT-1598 and VT-1161. Antimicrob Agents Chemother. 2019; https://doi.org/10.1128/AAC.01304-19.
Bassetti M, Righi E, Montravers P, Cornely OA. What has changed in the treatment of invasive candidiasis? A look at the past 10 years and ahead. J Antimicrob Chemother. 2018;73(suppl_1):i14-i25. https://doi.org/10.1093/jac/dkx445.
Sofjan AK, Mitchell A, Shah DN, Nguyen T, Sim M, Trojcak A, et al. Rezafungin (CD101), a next-generation echinocandin: A systematic literature review and assessment of possible place in therapy. J Glob Antimicrob Resist. 2018;14:58–64. https://doi.org/10.1016/j.jgar.2018.02.013.
Ong V, James KD, Smith S, Krishnan BR. Pharmacokinetics of the Novel Echinocandin CD101 in Multiple Animal Species. Antimicrob Agents Chemother. 2017; https://doi.org/10.1128/AAC.01626-16.
Pfaller MA, Messer SA, Rhomberg PR, Castanheira M. CD101, a long-acting echinocandin, and comparator antifungal agents tested against a global collection of invasive fungal isolates in the SENTRY 2015 Antifungal Surveillance Program. Int J Antimicrob Agents. 2017;50(3):352–8. https://doi.org/10.1016/j.ijantimicag.2017.03.028.
Hall D, Bonifas R, Stapert L, Thwaites M, Shinabarger DL, Pillar CM. In vitro potency and fungicidal activity of CD101, a novel echinocandin, against recent clinical isolates of Candida spp. Diagn Microbiol Infect Dis. 2017;89(3):205–11. https://doi.org/10.1016/j.diagmicrobio.2017.07.007.
Sandison T, Ong V, Lee J, Thye D. Safety and Pharmacokinetics of CD101 IV, a Novel Echinocandin, in Healthy Adults. Antimicrob Agents Chemother. 2017;https://doi.org/10.1128/AAC.01627-16.
Nyirjesy P, Alessio C, Jandourek A, Lee JD, Sandison T, Sobel JD. CD101 Topical Compared With Oral Fluconazole for Acute Vulvovaginal Candidiasis: A Randomized Controlled Trial. J Low Genit Tract Dis. 2019;23(3):226–9. https://doi.org/10.1097/LGT.0000000000000473.
Thompson GR, Soriano A, Skoutelis A, Vazquez JA, Honore PM, Horcajada JP, et al. Rezafungin versus Caspofungin in a Phase 2, Randomized, Double-Blind Study for the Treatment of Candidemia and Invasive Candidiasis- The STRIVE Trial. Clin Infect Dis. 2020. https://doi.org/10.1093/cid/ciaa1380.
Lepak AJ, Marchillo K, Andes DR. Pharmacodynamic target evaluation of a novel oral glucan synthase inhibitor, SCY-078 (MK-3118), using an in vivo murine invasive candidiasis model. Antimicrob Agents Chemother. 2015;59(2):1265–72. https://doi.org/10.1128/AAC.04445-14.
Berkow EL, Angulo D, Lockhart SR. In Vitro Activity of a Novel Glucan Synthase Inhibitor, SCY-078, against Clinical Isolates of Candida auris. Antimicrob Agents Chemother. 2017; https://doi.org/10.1128/AAC.00435-17.
Ghannoum M, Isham N, Angulo D, Borroto-Esoda K, Barat S, Long L. Efficacy of Ibrexafungerp (SCY-078) against Candida auris in an In Vivo Guinea Pig Cutaneous Infection Model. Antimicrob Agents Chemother. 2020; https://doi.org/10.1128/AAC.00854-20.
Lamoth F, Alexander BD. Antifungal activities of SCY-078 (MK-3118) and standard antifungal agents against clinical non-Aspergillus mold isolates. Antimicrob Agents Chemother. 2015;59(7):4308–11. https://doi.org/10.1128/AAC.00234-15.
Pfaller MA, Messer SA, Rhomberg PR, Borroto-Esoda K, Castanheira M. Differential Activity of the Oral Glucan Synthase Inhibitor SCY-078 against Wild-Type and Echinocandin-Resistant Strains of Candida Species. Antimicrob Agents Chemother. 2017; https://doi.org/10.1128/AAC.00161-17.
Petraitis V, Petraitiene R, Katragkou A, Maung BBW, Naing E, Kavaliauskas P et al. Combination Therapy with Ibrexafungerp (Formerly SCY-078), a First-in-Class Triterpenoid Inhibitor of (1-->3)-beta-d-Glucan Synthesis, and Isavuconazole for Treatment of Experimental Invasive Pulmonary Aspergillosis. Antimicrob Agents Chemother. 2020; https://doi.org/10.1128/AAC.02429-19.
Spec A, Pullman J, Thompson GR, Powderly WG, Tobin EH, Vazquez J, et al. MSG-10: a Phase 2 study of oral ibrexafungerp (SCY-078) following initial echinocandin therapy in non-neutropenic patients with invasive candidiasis. J Antimicrob Chemother. 2019;74(10):3056–62. https://doi.org/10.1093/jac/dkz277.
Ghannoum M, Long L, Larkin EL, Isham N, Sherif R, Borroto-Esoda K et al. Evaluation of the Antifungal Activity of the Novel Oral Glucan Synthase Inhibitor SCY-078, Singly and in Combination, for the Treatment of Invasive Aspergillosis. Antimicrob Agents Chemother. 2018; https://doi.org/10.1128/AAC.00244-18.
Trzoss M, Covel JA, Kapoor M, Moloney MK, Soltow QA, Webb PJ, et al. Synthesis of analogs of the Gwt1 inhibitor manogepix (APX001A) and in vitro evaluation against Cryptococcus spp. Bioorg Med Chem Lett. 2019; https://doi.org/10.1016/j.bmcl.2019.126713.
Shaw KJ, Ibrahim AS. Fosmanogepix: A Review of the First-in-Class Broad Spectrum Agent for the Treatment of Invasive Fungal Infections. J Fungi (Basel). 2020;. https://doi.org/10.3390/jof6040239.
Alkhazraji S, Gebremariam T, Alqarihi A, Gu Y, Mamouei Z, Singh S et al. Fosmanogepix (APX001) Is Effective in the Treatment of Immunocompromised Mice Infected with Invasive Pulmonary Scedosporiosis or Disseminated Fusariosis. Antimicrob Agents Chemother. 2020; https://doi.org/10.1128/AAC.01735-19.
Wiederhold NP, Najvar LK, Shaw KJ, Jaramillo R, Patterson H, Olivo M et al. Efficacy of Delayed Therapy with Fosmanogepix (APX001) in a Murine Model of Candida auris Invasive Candidiasis. Antimicrob Agents Chemother. 2019; https://doi.org/10.1128/AAC.01120-19.
Gebremariam T, Alkhazraji S, Alqarihi A, Wiederhold NP, Shaw KJ, Patterson TF et al. Fosmanogepix (APX001) Is Effective in the Treatment of Pulmonary Murine Mucormycosis Due to Rhizopus arrhizus. Antimicrob Agents Chemother. 2020; https://doi.org/10.1128/AAC.00178-20.
Petraitiene R, Petraitis V, Maung BBW, Mansbach RS, Hodges MR, Finkelman MA, et al. Efficacy and Pharmacokinetics of Fosmanogepix (APX001) in the Treatment of Candida Endophthalmitis and Hematogenous Meningoencephalitis in Non-neutropenic Rabbits. Antimicrob Agents Chemother. 2020. https://doi.org/10.1128/AAC.01795-20.
Kapoor M, Moloney M, Soltow QA, Pillar CM, Shaw KJ. Evaluation of Resistance Development to the Gwt1 Inhibitor Manogepix (APX001A) in Candida Species. Antimicrob Agents Chemother. 2019; https://doi.org/10.1128/AAC.01387-19.
Yamashita K, Miyazaki T, Fukuda Y, Mitsuyama J, Saijo T, Shimamura S et al. The Novel Arylamidine T-2307 Selectively Disrupts Yeast Mitochondrial Function by Inhibiting Respiratory Chain Complexes. Antimicrob Agents Chemother. 2019; https://doi.org/10.1128/AAC.00374-19.
Nishikawa H, Sakagami T, Yamada E, Fukuda Y, Hayakawa H, Nomura N, et al. T-2307, a novel arylamidine, is transported into Candida albicans by a high-affinity spermine and spermidine carrier regulated by Agp2. J Antimicrob Chemother. 2016;71(7):1845–55. https://doi.org/10.1093/jac/dkw095.
Mitsuyama J, Nomura N, Hashimoto K, Yamada E, Nishikawa H, Kaeriyama M, et al. In vitro and in vivo antifungal activities of T-2307, a novel arylamidine. Antimicrob Agents Chemother. 2008;52(4):1318–24. https://doi.org/10.1128/AAC.01159-07.
Nishikawa H, Fukuda Y, Mitsuyama J, Tashiro M, Tanaka A, Takazono T, et al. In vitro and in vivo antifungal activities of T-2307, a novel arylamidine, against Cryptococcus gattii: an emerging fungal pathogen. J Antimicrob Chemother. 2017;72(6):1709–13. https://doi.org/10.1093/jac/dkx020.
Wiederhold NP, Najvar LK, Fothergill AW, Bocanegra R, Olivo M, McCarthy DI, et al. The novel arylamidine T-2307 demonstrates in vitro and in vivo activity against echinocandin-resistant Candida glabrata. J Antimicrob Chemother. 2016;71(3):692–5. https://doi.org/10.1093/jac/dkv398.
Abe M, Nakamura S, Kinjo Y, Masuyama Y, Mitsuyama J, Kaku M, et al. Efficacy of T-2307, a novel arylamidine, against ocular complications of disseminated candidiasis in mice. J Antimicrob Chemother. 2019;74(5):1327–32. https://doi.org/10.1093/jac/dkz020.
Therapeutics A. A novel, broad spectrum, clinical stage antifungal to address severe and difficult-to-treat invasive fungal infections. 2020. https://www.appilitherapeutics.com/ati-2307. Accessed December 18 2020.
Oliver JD, Sibley GEM, Beckmann N, Dobb KS, Slater MJ, McEntee L, et al. F901318 represents a novel class of antifungal drug that inhibits dihydroorotate dehydrogenase. Proc Natl Acad Sci USA. 2016;113(45):12809–14. https://doi.org/10.1073/pnas.1608304113.
Jorgensen KM, Astvad KMT, Hare RK, Arendrup MC. EUCAST Determination of Olorofim (F901318) Susceptibility of Mold Species, Method Validation, and MICs. Antimicrob Agents Chemother. 2018; https://doi.org/10.1128/AAC.00487-18.
Rivero-Menendez O, Cuenca-Estrella M, Alastruey-Izquierdo A. In vitro activity of olorofim (F901318) against clinical isolates of cryptic species of Aspergillus by EUCAST and CLSI methodologies. J Antimicrob Chemother. 2019;74(6):1586–90. https://doi.org/10.1093/jac/dkz078.
Biswas C, Law D, Birch M, Halliday C, Sorrell TC, Rex J, et al. In vitro activity of the novel antifungal compound F901318 against Australian Scedosporium and Lomentospora fungi. Med Mycol. 2018;56(8):1050–4. https://doi.org/10.1093/mmy/myx161.
Buil JB, Rijs A, Meis JF, Birch M, Law D, Melchers WJG, et al. In vitro activity of the novel antifungal compound F901318 against difficult-to-treat Aspergillus isolates. J Antimicrob Chemother. 2017;72(9):2548–52. https://doi.org/10.1093/jac/dkx177.
Wiederhold NP, Law D, Birch M. Dihydroorotate dehydrogenase inhibitor F901318 has potent in vitro activity against Scedosporium species and Lomentospora prolificans. J Antimicrob Chemother. 2017;72(7):1977–80. https://doi.org/10.1093/jac/dkx065.
Lackner M, Birch M, Naschberger V, Grassle D, Beckmann N, Warn P, et al. Dihydroorotate dehydrogenase inhibitor olorofim exhibits promising activity against all clinically relevant species within Aspergillus section Terrei. J Antimicrob Chemother. 2018;73(11):3068–73. https://doi.org/10.1093/jac/dky329.
Kennedy T, Allen G, Steiner J, Heep M, Oliver J, Sibley G et al. Multiple dose pharmacokinetics of an immediate-release tablet formaulation of F901318 in health male and female subjects. ECCMID 20172017.
du Pre S, Birch M, Law D, Beckmann N, Sibley GEM, Bromley MJ et al. The Dynamic Influence of Olorofim (F901318) on the Cell Morphology and Organization of Living Cells of Aspergillus fumigatus. J Fungi (Basel). 2020; https://doi.org/10.3390/jof6020047.
Nakamura I, Yoshimura S, Masaki T, Takase S, Ohsumi K, Hashimoto M, et al. ASP2397: a novel antifungal agent produced by Acremonium persicinum MF-347833. J Antibiot (Tokyo). 2017;70(1):45–51. https://doi.org/10.1038/ja.2016.107.
Dietl AM, Misslinger M, Aguiar MM, Ivashov V, Teis D, Pfister J et al. The Siderophore Transporter Sit1 Determines Susceptibility to the Antifungal VL-2397. Antimicrob Agents Chemother. 2019; https://doi.org/10.1128/AAC.00807-19.
Nakamura I, Ohsumi K, Takeda S, Katsumata K, Matsumoto S, Akamatsu S et al. ASP2397 Is a Novel Natural Compound That Exhibits Rapid and Potent Fungicidal Activity against Aspergillus Species through a Specific Transporter. Antimicrob Agents Chemother. 2019; https://doi.org/10.1128/AAC.02689-18.
Murray A, Cass L, Ito K, Pagani N, Armstrong-James D, Dalal P et al. PC945, a Novel Inhaled Antifungal Agent, for the Treatment of Respiratory Fungal Infections. J Fungi (Basel). 2020; https://doi.org/10.3390/jof6040373.
Colley T, Alanio A, Kelly SL, Sehra G, Kizawa Y, Warrilow AGS et al. In Vitro and In Vivo Antifungal Profile of a Novel and Long-Acting Inhaled Azole, PC945, on Aspergillus fumigatus Infection. Antimicrob Agents Chemother. 2017; https://doi.org/10.1128/AAC.02280-16.
Cass L, Murray A, Davis A, Woodward K, Albayaty M, Ito K, et al. Safety and nonclinical and clinical pharmacokinetics of PC945, a novel inhaled triazole antifungal agent. Pharmacol Res Perspect. 2021; https://doi.org/10.1002/prp2.690.
Hava DL, Tan L, Johnson P, Curran AK, Perry J, Kramer S, et al. A phase 1/1b study of PUR1900, an inhaled formulation of itraconazole, in healthy volunteers and asthmatics to study safety, tolerability and pharmacokinetics. Br J Clin Pharmacol. 2020;86(4):723–33. https://doi.org/10.1111/bcp.14166.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
I have no conflicts of interest to declare.
Additional information
Handling Editor: Sanjay Haresh Chotirmall.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Waterer, G. Advances in anti-fungal therapies. Mycopathologia 186, 665–672 (2021). https://doi.org/10.1007/s11046-021-00560-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11046-021-00560-2