
Abstract
Background
DNA barcoding have been considered as a tool to facilitate species identification based on its simplicity and high-level accuracy in compression to the complexity and subjective biases linked to morphological identification of taxa. MaturaseK gene (MatK gene) of the chloroplast is very vital in the plant system which is involved in the group II intron splicing. The main objective of this study is to determine the relative utility of the “MatK” chloroplast gene for barcoding in 15 legume as a tool to facilitate species identification based on their simplicity and high-level accuracy linked to morphological identification of taxa.
Methods and Results
MatK gene sequences were submitted to GenBank and the accession numbers were obtained with sequence length ranging from 730 to 1545 nucleotides. These DNA sequences were aligned with database sequence using PROMALS server, Clustal Omega server and Bioedit program. Maximum likelihood and neighbor-joining algorithms were employed for constructing phylogeny. Overall, these results indicated that the phylogenetic tree analysis and the evolutionary distances of an individual dataset of each species were agreed with a phylogenetic tree of all each other consisting of two clades, the first clade comprising (Enterolobium contortisiliquum, Albizia lebbek), Acacia saligna, Leucaena leucocephala, Dichrostachys Cinerea, (Delonix regia, Parkinsonia aculeata), (Senna surattensis, Cassia fistula, Cassia javanica) and Schotia brachypetala were more closely to each other, respectively. The remaining four species of Erythrina humeana, (Sophora secundiflora, Dalbergia Sissoo, Tipuana Tipu) constituted the second clade.
Conclusion
Moreover, their sequences could be successfully utilized in single nucleotide polymorphism or as part of the sequence as DNA fragment analysis utilizing polymerase chain reaction in plant systematic. Therefore, MatK gene is considered promising a candidate for DNA barcoding in the plant family Fabaceae and provides a clear relationship between the families.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fabaceae is considering a large and economically vital family of flowering plants which is usually known as the legume family [1,2,3,4,5]. The Fabaceae family, which has over 490 medicinal plant species 730 genera flowering plants and more than 19,400 species [5,6,7,8,9]. Documentation of the Mediterranean legume crops depending on morphological characteristics has shown tricky and much impossible [10,11,12,13]. So, using a DNA-based technique would offer accurate knowledge and facilitate the discrimination of the species. DNA barcoding is new, efficient, quick, low-cost, and standard technique for the fast identification and evaluation of plant and animal species based on DNA sequence from a small fragment of the whole genome in a rapid, accurate [14,15,16,17,18]. DNA barcoding can help to detect species, quick identification of any species that are possibly novel to science and to report the essential ecological and evolutionary questions as a biological instrument [19,20,21,22,23,24,25]. DNA barcoding are frequently promoted for their facility to enhance the accessibility of scientific information and new knowledge to the public and non-specialists [26, 27]. Short DNA sequences in DNA barcoding are used to identify the diversity between plant and animal species as molecular markers [28], also, it’s used in an assignment the unknown samples to a taxonomic group, and in-plant biodiversity documentation [29]. DNA barcoding is a potential tool to detect an error in identifying species because similarity-based approaches using DNA barcoding combined with morphology would solve the misidentification based on morphology [30,31,32]. DNA barcoding could help decrease the limitations of morphological characteristics and hurry up plant and animal species identification since it can detect the organisms at any stage of growth. DNA barcodes are designed to create a shared community resource of DNA sequence that is used in the identification or taxonomic classification of any organisms [33]. The usage of DNA barcodes as a tool for plant/ animal identification is based on the establishment of high-value reference databases of sequence [34] which cannot always distinguish between closely related species of land plants or fungi.
The MatK gene (1500 bp in length), located inside the intron of the mitochondrially encoded tRNA lysineprovided by HGNC (trnK) and codes for maturase as protein, which is involved in Group- II intron splicing, the trnK intron of plants encodes the MatK open reading frame (ORF). This gene has a high-level rate of substitution [3], a huge proportion of difference at nucleic acid levels at first and second codon place, and low transition and or/transversion ratio and the presence of mutationally conserved regions. Previous data were utilized to identify the molecular markers, which were used to identify the genus/species of these taxa, to provide valuable information for both conventional and molecular studies [11]. The current study, target to evaluate the capacity and the efficiency of MatK gene as normal plant barcode marker; documentation and identification of 45 plant specimens belonging to 15 species of Fabaceae plant species, and study the useful annotation, homology modeling and sequence analysis to permit an additional efficient use of these sequences between different plant species.
Materials and methods
Plant materials
Forty-five samples (three replicates for each species), which belonged to 15 species found in Fig. 1 were collected from Antoniadis Garden's (N 29" 56′ 55, E 18" 12′ 31), Alexandria, Egypt between July 2019 to January 2020.

showing difference between leaves in fifteen fabaceae plants (1) Cassia fistula, (2) Cassia javinca, (3) Albizia lebbek, (4) Delonix regia, (5) Senna surattensis, (6) Parkinsonia aculeata, (7) Schotia brachypetala, (8) Tipuana tipu, (9) Erythrina humeana, (10) Sophora secundiflora, (11) Leucaena leucocephala, (12) Enterolobium contortisiliquum, (13) Dichrostachys cinerea, (14) Acacia saligna and (15) Dalbergia sissoo
DNA extraction and sequencing of specimens
Total genomic DNA was extracted from fresh leaves tissue by (i-genomic plant DNA extraction Mini kit @ iNtron biotechnology, Berlin, Germany) corresponding to the protocol linked to the Plant Genomic DNA Kit (iNtRON Bio Co., South Korea) as found in Fig. 2. PCR of the MatK regions were conducted out in Techne Flexigene PCR Thermal Cycler programmed for 30 cycles as follows: 94 °C/5 min (1 cycle); 94 °C/45 s, 50 °C/45 s, 72 °C/45 s (30 cycles); 72 °C/7 min (1 cycle); 4 °C (infinitive). The designed common primers and reaction conditions of the MatK region is F: 5'-CGTACAGTACTTTTGTGTTTACGAG-3' (Tm, 53.9 and GC%, 40), R: 5'-ACCCAGTCCATCTGGAAATCTTGGTTC-3' (Tm, 60.4 and GC%, 48). The PCR products were run on a 1.0% agarose gel utilizing 1X TAE buffer containing 0.5 µg/mL ethidium bromide for electrophoresis of PCR products as found in Fig. 3. PCR products were purified using Mini kit @ iNtron Biotechnology Purification kits before being sequenced exploitation the dideoxynucleotide chain termination method with a DNA sequencer (Applied Biosystems® 3500 and 3500xL Genetic Analyzers) and a BigDye Terminator version 3.1 Cycle Sequencing RR-100 Kit (Applied Biosystems). The sequences were submitted to DDBJ/EMBL/GenBank database. Generic and species data was achieved from the taxonomy database of the National Centre for Biotechnology Information (NCBI).

Agarose gel electrophoresis for extracted DNA from samples (1) Cassia fistula, (2) Cassia javinca, (3) Albizia lebbek, (4) Delonix regia, (5) Senna surattensis, (6) Parkinsonia aculeata, (7) Schotia brachypetala, (8) Tipuana tipu, (9) Erythrina humeana, (10) Sophora secundiflora, (11) Leucaena leucocephala, (12) Enterolobium contortisiliquum, (13) Dichrostachys cinerea, (14) Acacia saligna and (15) Dalbergia sissoo

Agarose gel electrophoresis for amplified samples by using the primer MatK for the samples (1) Cassia fistula, (2) Cassia javinca, (3) Albizia lebbek, (4) Delonix regia, (5) Senna surattensis, (6) Parkinsonia aculeata, (7) Schotia brachypetala, (8) Tipuana tipu, (9) Erythrina humeana, (10) Sophora secundiflora, (11) Leucaena leucocephala, (12) Enterolobium contortisiliquum, (13) Dichrostachys cinerea, (14) Acacia saligna and (15) Dalbergia sissoo
Sequence analysis
The sequences results analysis was completed for the one grouped dataset, this set contains all the plant species of Fabaceae for which the sequences are available in GenBank to find the inter-species and inter-generic variation. Fabaceae species sequences of MatK were retrieved from NCBI in Fasta format. Multiple sequence alignments of the MatK gene were conducted from different species applying the PROMALS server [35], Clustal Omega server [36], the BIOEDIT software [37] and MEGA-11 [38] which are offline software that conducts optimal sequence alignment to find the conserved area. The MEGA 11 software has matured to contain a large collection of methods and tools of computational molecular evolution for building timetrees of species, pathogens, and gene families using rapid relaxed-clock methods and estimating divergence times and confidence intervals for node-dating and sequence sampling dates for tip-dating analyses. Comparing to the greatest alignment methods with development for distantly related sequences the “PROMALS” is up to 30% more accurate. Clustal Omega server is a new multiple sequence alignment software that generates alignments between three or more sequences using seeded guide trees and HMM profile-profile methods. The “BIOEDIT” software is a user-friendly biological sequences alignment editor that aims to provide fundamental functions for editing, aligning, manipulating, and analyzing protein sequences.
Molecular evolution and phylogenetic analysis
The Neighbor Joining method was used to deduce the evolutionary narrative. Finding the topology and branch length of the tree that will offer the best chance of detecting amino acid sequence in current data is the approach for constructing the phylogenetic tree using maximum likelihood. So, for phylogenetic evaluation Mafft server [39], Clustal Omega server and “MEGA-11” software were applied. MEGA was used to analyze the sequencing data using the neighbor-joining technique and Unweighted Pair Group Mean Average "UPGMA." The “DNADIST” software of “PHYLIP” was used to calculate distances. NJ plot was used to do bootstrapping and decay analysis. MEGA determined parsimony analyses and different clades.
Results
DNA extraction and PCR amplification
The quality of the obtained DNA was detected 1% agarose gel electrophoresis. The results indicated that there is no fragmentation was observed in extracted DNA. The quantity of extracted DNA samples was determined by using Nanodrop Spectrophotometer and the concentration ranged from 30 to 50 ng/μl. The extracted DNA was directly used in PCR amplification for the MatK gene recorded on fragment in molecular weight (900 bp).
Development in DNA sequencing methods has allowed us to describe the genomes of numerous organisms quickly. Evaluations of the DNA sequences of several species are providing useful knowledge about their taxonomy, gene makeup, and utilization. In the current study using DNA sequence polymorphisms of the chloroplast, MatK gene is much more variable than many other genes. From Fifteen plant species belong to different genera of the same family Fabaceae as found in Table 1. In this data we organized a study to contribute to the knowledge of the major evolutionary relationship between the studied plant genus and species (clades) and discussed the application of MatK for molecular evolution. The chloroplast MatK marker was more useful as DNA markers. The present study included fifteen species from fifteen genera are deposited in GenBank; accession numbers were obtained for the respective plant species with different numbers of conserved domains, segment length and average entropy (Hx) (Table 1).
Phylogenetic analysis of collected plants
Numerous sequence alignments showed that there are varying numbers of “Indels” in the gene MatK. Using the neighbor-joining method, UPGMA and maximum likelihood, the evolutionary distances for the 15 plant species were recognized into individual clades. The alignment of MatK gene of Acacia saligna nucleotide sequences showed 15 conserved regions, 769 variable sites and 571 parsimony sites, the overall mean distance is 2.85 (Table 2). The combined tree showed two groups or cladograms and they are represented as follows: Group I include Acacia saligna was closely related to different species belonging to other genera of the same family (Fabaceae) such as Enterolobium, Pararchidendron, Archidendron, Samanea, Hydrochorea, Balizia and Abarema (Fig. 4). Also, Acacia comprising other species were closely arranged but distinguished into different genera such as Falcataria, Pararchidendron and Lacacia. In addition, the aligned MatK dataset was 793 nucleotide sites long, of which 102 sites were potentially parsimony informative. Consequently, Enterolobium contortisiliquum is more closely related to different species of genus Acacia according to phylogenetic analysis using maximum likelihood (Fig. 4). The length of MatK varies from 750 bp in Albizia lebbek (the smaller length of MatK gene for these species is due to incomplete sequencing, which was retrieved from GenBank) to 813 in different genera (Enterolobium, Acacia, Senegalia, Cojoba, Samanea, Hydrochorea, Balizia and Abarema). Maximum likelihood and Neighbor-joining analysis of the dataset resulted in tree with two groups. The clades established in the trees were mainly mixtures of numerous species. Consequently, creating a local barcode database will be useful for a broad range of potential ecological purposes, involving the building of community phylogenies [40]. Group I have three clusters comprising several genera (Albizia, Enterolobium, Mariosousa, Archidendron, Samanea, Balizia, and Abarema). Otherwise, group II has one genera acacia which is the most closely related to our plant Albizia lebbeck according to MatK gene partial cds (Fig. 4). The arrangement of MatK gene of Albizia lebbeck nucleotide sequence revealed 649 varying sites and 359 parsimony sites, the overall mean distance is 2.37 and the estimated Transition/Transversion bias (R) is 0.52 (Table 2, 3).

Phylogenetic tree analysis and the evolutionary distances of Acacia saligna, Albizia lebbeck and Enterolobium contortisiliquum were computed using the Maximum Likelihood technique using nucleotide sequences of the MatK gene. This analysis involved 49 nucleotide sequences. Codon positions included were 1st + 2nd + 3rd + Noncoding. All ambiguous positions were removed for each sequence pair (pairwise deletion option). There were a total of 856 positions in the final dataset
Furthermore, depending on the phylogenetic analysis, the two genera Cassia and Senna with different species are closely related and more highly similar than any other studies species (Fig. 5). The phylogeny tree was created using the neighbor-joining approach and the evolutionary distances were calculated employing the maximum composite likelihood approach. The combined trees showed that there are two groups, and they are as follows: Group I consisted of five clades representing different genera with different species such as (Chamaecrista, Senna, Erytherophleum, Arapatiell and Dinizia). Group II showed two branches: each one with many sub-branches containing five clades with different species of the genus Senna. According to MatK gene sequence, the collected plants (Cassia fistula and Cassia javanica) revealed a high percentage of identity with different 7 species of genus senna having the same clade (Fig. 5). Also, they are closely related to other different species of Erytherophleum, Arapatiell. On another hand, the sequence of MatK gene of collected Senna surattensis species has a high degree of similarity with many species in different genera in Fabaceae (Fig. 5), and consequently, this species is used as a template to estimate the similarity between different species in Fabaceae family.

Phylogenetic tree analysis and the evolutionary distances of Cassia fistula, Cassia javanica and Senna surattensis using the Neighbor-Joining technique using nucleotide sequences of the MatK gene. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site with the highest log likelihood (− 16,447.32). This analysis involved 55 nucleotide sequences and there were a total of 854 positions in the final dataset
Nevertheless, the Delonix regia is the more studied species having a good similarity to different species of different genera of the Fabaceae family. Polymorphism obtained from the DNA sequence indels or replacements of the MatK gene indicated that Delonix regia, Umtiza listeriana, Diptychandra aurantiaca, Moldenhawera blanchetiana, Schizolobium parahyba, Tachigali costaricensis, Arapatiella psilophylla and Parkinsonia Africana were evolved from a Common ancestor (Fig. 6). In addition, Dichrostachys cinerea is closely related to different species of genera Leucaena, Senegalia, Falcataria and prosopis (Fig. 6). Furthermore, applying the same incremental method of informative sites starting at the 5/-end of the MatK gene, completely different results were found. The consensus tree of 15 most parsimonious trees demonstrated unresolved clades until 250 informative sites. At that point, 1 highly parsimonious tree was created, which was congruent with the topology of the stable tree achieved from the 3/-end. To recognize the greatest DNA barcode marker for species documentation and traceability, the value of genetic divergence for all the confirmed loci were calculated in each analyzed group at dissimilar taxonomic level and by considering only fresh morphologically identified samples. Results indicated the species of Delonix regia, Parkinsonia aculeata and Leucaena leucocephala are more like other species of the same genus and less similar to species of other genera of the Fabaceae family (Fig. 6). This reflected that the Parkinsonia aculeata was closely related and in the same clade with Schizolobium parahyba, Diptychandra aurantiaca, Delonix regia, Conzattia multiflora and Colvillea racemose (Fig. 6).

Phylogenetic tree analysis and the evolutionary distances of Leucaena leucocephala, Dichrostachys Cinerea, Delonix regia and parkinsonia aculeata using the Neighbor-Joining technique using nucleotide sequences of the MatK gene. This analysis involved 73 nucleotide sequences. All ambiguous positions were removed for each sequence pair (pairwise deletion option). There were a total of 924 positions in the final dataset
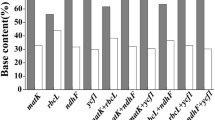
The current sequences showed little variations in the percentage of guanine plus cytosine content (% G + C) related to that in the sequences of MatK. In case of MatK, the nucleotide structure was biased toward the guanine and cytosine content (G + C) with frequencies were 30.4 to 34.8%, respectively. The NJ, ML, and MP analyses all resulted in comparable trees in each of the data sets. There are often variations between the trees from the various analyses involving non-resolution (polytomies). Analyses carried out on samples belonging to Parkinsonia aculeata, Schotia brachypetala, Sophora secundiflora and Tipuana Tipu indicated that the sequences divergences of marker MatK were clearly distinguished from other species of Fabaceae. Figures 7 showed phylogenetic clusters constructed using ML and NJ; The difference observed in MatK does separate several species; however, there is a wide range of intra- specific and inter-specific variation. Furthermore, On the Neighbor-Joining Phylogram, the Schotia group is a sister taxon to the Macrolobium group and this observation is found in 50% of the most clade in this cladistic analysis (Fig. 7).

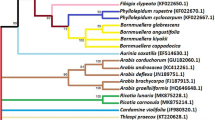
Phylogenetic tree analysis and the evolutionary distances of Dalbergia Sissoo, Erythrina humeana, Schotia brachypetala, Sophora secundiflora and Tipuana Tipu using using the Maximum Likelihood method. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site with the highest log likelihood (− 4725.80). This analysis involved 55 nucleotide sequences. There were a total of 902 positions in the final dataset
The last two members i.e., Sophora secundiflora and Tipuana Tipu produced an independent clade and confirmed the ambiguous position relative to the other genera of Fabaceae based on the combined cladistic analysis data from chloroplast DNA restriction sites and morphology. Sophora secundiflora shared a common ancestor with Angylocalyx braunii, Zollernia splendens, Ormosia xylocarpa and Dermatophyllum secundiflorum. Also, Tipuana Tipu is in the same clade with different species of two genera Centrolobium and Pterocarpus (Fig. 7). Additionally, highly Fabaceae species in the current research were detected to have a unique sequence in the MatK gene. These results will offer a valuable way to authenticate various MatK species. MatK sequence created in this analysis will be applied to construct reference sequence libraries, and the sequences extracted from samples with particular identity classifications will be utilized to search the database.
Lastly, utilizing BLAST1 and the closest genetic distance approach, we will be able to define the species identities of the query sequences based on these data. In the dataset of MatK, the nearest genetic distance approach achieved 99.68% to 96.45% identification accomplishment rates at the species level for “BLAST1” and distance discrimination methodology, respectively, with no equivocal identification at the genus level. The planned barcoding portion of MatK is about 760 base pairs in Fabaceae. The phylogenetic tree (Fig. 8) consists of two clades, the first clade comprising (Enterolobium contortisiliquum, Albizia lebbek), Acacia saligna, Leucaena leucocephala, Dichrostachys Cinerea, (Delonix regia, Parkinsonia aculeata), (Senna surattensis, Cassia fistula, Cassia javanica) and Schotia brachypetala were more closely to each other, respectively. The other four species of Erythrina humeana with Sophora secundiflora and (Dalbergia Sissoo, Tipuana Tipu) constituted the second clade.

Evolutionary analysis of legume tree species grown in Egypt in this study using MatK gene by Maximum Likelihood method
Discussion
Because plant genomes include several copies of MatK sequences, it's unclear if the sequence obtained by PCR will be balanced and representative [41]. As a result, we suggest MatK as a potential barcode sequence in the Fabaceae family, as well as a wider range of plant species. Utilizing MatK as a DNA barcode would extend our knowledge of phylogenetics and population genetics in Fabaceae species as reviewed by [42,43,44,45]. We also recommend that MatK be used as a DNA barcode sequence to overcome difficulties in Fabaceae genus and species categorization [44, 46]. MatK might serve as a starting point for quality control and assurance of plant materials utilized in research, manufacturing, customs, and forensics.
The MatK was discovered to be a necessarily variable DNA region between Fabaceae species as determined by genetic divergences, and it demonstrated a greater potential of effective discrimination. MatK can be a powerful taxonomic marker for identifying species and resolving taxonomic issues [30, 32, 41]. For instance, the MatK sequence of Enterolobium contortisiliquum is highly like Albizia lebbek, so our results indicate that in the genus Cassia, in which the species were poorly graded, MatK was still able to distinguish among some confusing species[47]. The evolutionary distances for the 15 plant species that were separated into distinct clades were analyzed using the maximum likelihood and neighbor-joining methods, which discriminated most of the species better than previous techniques [48].
The identification by MatK region paired with morphological recognition 100% to species (Fig. 1) level; for the set of plants studied, it appears to be an accurate approximation of species identification using this one locus. Short sequence, universality, and unique identifiers are three features of a common barcode [48, 49]. According to our results of sequence length and composition of MatK barcode gene for the 15 plant species, MatK regions have high rate of nucleotide substitutions as showed by [50] or the locus remodeling ring [51]. Alternate primer sequences may increase the success rate of MatK amplification for some of the current taxa, making it a barcoding locus. The species in which the MatK region is amplified, however, had wide taxonomic coverage in the Fabaceae family, indicating that the locus' conserved sequence is notable.
Consequently, the partial amplification sequence of MatK was further utilized to investigate the evolutionary linkage of the selected plants. The evolutionary distances between the 15 plant species were divided into two clades using the neighbor-joining approach., the first clade comprising (Enterolobium contortisiliquum, Albizia lebbek), Acacia saligna, Leucaena leucocephala, Dichrostachys Cinerea, (Delonix regia, Parkinsonia aculeata), (Senna surattensis, Cassia fistula, Cassia javanica) and Schotia brachypetala which were more closely to each other, respectively. The remaining four species of Erythrina humeana, (Sophora secundiflora, Dalbergia Sissoo, Tipuana Tipu) constituted the second clade. The results are encouraging, which give a backbone of knowledge in the data set. As additional species become accessible, more research for species resolution of a genus may be undertaken.
Conclusion
During the current study, DNA barcoding using MatK chloroplast gene was applied in fifteen legume trees by both single region and multiregional approaches. The obtained chloroplast gene sequences were submitted to GenBank, and fifteen accession numbers were recorded as LC602060, LC602154, LC602263, LC603347, LC603655, LC603845, LC603846, LC603847, LC604717, LC604718, LC605994, LC604799, LC605995, LC606468, LC606469) with length ranging from 730 to 1545 nucleotides. The current results indicated that the phylogenetic tree analysis and the evolutionary distances of an individual dataset of each studied species were agreed with a phylogenetic tree of all each other consisting of two clades, the first clade comprising (Enterolobium contortisiliquum, Albizia lebbek), A. saligna, Leucaena leucocephala, Dichrostachys Cinerea, (Delonix regia, Parkinsonia aculeata), (Senna surattensis, C. fistula, C. javanica) and Schotia brachypetala were more closely to each other, respectively. The remaining four species of Erythrina humeana, (Sophora secundiflora, Dalbergia Sissoo, Tipuana Tipu) constituted the second clade. Finally, it could be concluded that, MatK gene is considered promising a candidate for DNA barcoding in plant family Fabaceae and providing a clear relationship between the families.
References
Gepts P, Beavis WD, Brummer EC, Shoemaker RC, Stalker HT, Weeden NF, Young ND (2005) Legumes as a model plant family. Genomics for food and feed report of the cross-legume advances through genomics conference. Plant Physiol 137:1228. https://doi.org/10.1104/pp.105.060871
Antunes A, Nunes R, Novaes E, Coelho A, Soares T, Telles M (2020) Large number of repetitive elements in the draft genome assembly of Dipteryx alata (Fabaceae). Genet Mol Res 19:GMR18463
Xiong Y, Xiong Y, He J, Yu Q, Zhao J, Lei X, Dong Z, Yang J, Peng Y, Zhang X et al (2020) The complete chloroplast genome of two important annual clover species, Trifolium alexandrinum and T. resupinatum: genome structure, comparative analyses and phylogenetic relationships with relatives in Leguminosae. Plants. https://doi.org/10.3390/plants9040478
Tungmunnithum D, Drouet S, Lorenzo JM, Hano C (2021) Effect of traditional cooking and in vitro gastrointestinal digestion of the ten most consumed beans from the fabaceae family in Thailand on their phytochemicals, antioxidant and anti-diabetic potentials. Plants 11:67
Janarny G, Ranaweera KKDS, Gunathilake KDPP (2022) Digestive recovery of polyphenols, antioxidant activity, and anti-inflammatory activity of selected edible flowers from the family Fabaceae. J Food Biochem 46:e14052
Gao T, Yao H, Song J, Liu C, Zhu Y, Ma X, Pang X, Xu H, Chen S (2010) Identification of medicinal plants in the family Fabaceae using a potential DNA barcode ITS2. J Ethnopharmacol 130:116–121. https://doi.org/10.1016/j.jep.2010.04.026
Van Wyk BE (2020) A family-level floristic inventory and analysis of medicinal plants used in Traditional African Medicine. J Ethnopharmacol 249:112351. https://doi.org/10.1016/j.jep.2019.112351
Christenhusz MJ, Byng JW (2016) The number of known plants species in the world and its annual increase. Phytotaxa 261:201–217. https://doi.org/10.11646/phytotaxa.261.3.1
Palmer CM, Wershoven NL, Martinson SJ, ter Hofstede HM, Kress WJ, Symes LB (2022) Patterns of herbivory in neotropical forest katydids as revealed by DNA barcoding of digestive tract contents. Diversity. https://doi.org/10.3390/d14020152
Smartt J (1980) Evolution and evolutionary problems in food legumes. Econ Bot 34:219–235
Enriquez-Hidalgo D, Cruz T, Teixeira DL, Steinfort U (2020) Phenological stages of mediterranean forage legumes, based on the BBCH scale. Ann Appl Biol 176:357–368. https://doi.org/10.1111/aab.12578
Lombardo E, Bancheva S, Domina G, Venturella G (2020) Distribution, ecological role and symbioses of selected shrubby species in the Mediterranean Basin: a review. Plant Biosystems - An International Journal Dealing with all Aspects of Plant Biology 154:438–454. https://doi.org/10.1080/11263504.2020.1727988
Zsögön A, Peres LE, Xiao Y, Yan J, Fernie AR (2022) Enhancing crop diversity for food security in the face of climate uncertainty. Plant J 109:402–414
Hajibabaei M, de Waard JR, Ivanova NV, Ratnasingham S, Dooh RT, Kirk SL, Mackie PM, Hebert PDN (2005) Critical factors for assembling a high volume of DNA barcodes. Philosoph Trans R Soc B 360:1959–1967. https://doi.org/10.1098/rstb.2005.1727
Baldi P, La Porta N (2020) Molecular approaches for low-cost point-of-care pathogen detection in agriculture and forestry. Front Plant Sci 11:570862–570862. https://doi.org/10.3389/fpls.2020.570862
Jalali SK, Ojha R, Venkatesan T (2015) DNA barcoding for identification of agriculturally important insects. In: Chakravarthy AK (ed) New horizons in insect science: towards sustainable pest management. New Delhi, Springer India, pp 13–23
Khan MQ, Khalil AT, Shinwari ZK (2015) Searching for DNA barcodes in plants. Am Eurasian J Agric Environ Sci 15:504–513
Zhao J, Abdelsalam NR, Khalaf L, Chuang W-P, Zhao L, Smith CM, Carver B, Bai G (2019) Development of single nucleotide polymorphism markers for the wheat curl mite resistance gene Cmc4. Crop Sci 59:1567–1575. https://doi.org/10.2135/cropsci2018.11.0695
Kress WJ, Erickson DL (2012) DNA barcodes: methods and protocols. In: Kress WJ, Erickson DL (Eds) DNA barcodes: methods and protocols. Humana Press, Totowa, NJ, pp 3–8
Lahaye R, van der Bank M, Bogarin D, Warner J, Pupulin F, Gigot G, Maurin O, Duthoit S, Barraclough TG, Savolainen V (2008) DNA barcoding the floras of biodiversity hotspots. Proc Natl Acad Sci 105:2923–2928. https://doi.org/10.1073/pnas.0709936105
Park H-S, Jayakodi M, Lee SH, Jeon J-H, Lee H-O, Park JY, Moon BC, Kim C-K, Wing RA, Newmaster SG et al (2020) Mitochondrial plastid DNA can cause DNA barcoding paradox in plants. Sci Rep 10:6112. https://doi.org/10.1038/s41598-020-63233-y
Abdelsalam NR, Salem MZ, Ali HM, Mackled MI, Mervat E-H, Elshikh MS, Hatamleh AA (2019) Morphological, biochemical, molecular, and oil toxicity properties of Taxodium trees from different locations. Ind Crops Prod 139:111515
Abdelsalam NR, Ali HM, Salem MZ, Ibrahem EG, Elshikh MS (2018) Genetic and morphological characterization of Mangifera indica L. growing in Egypt. HortScience 53:1266–1270
Abdelsalam NR, Awad RM, Ali HM, Salem MZ, Abdellatif KF, Elshikh MS (2019) Morphological, pomological, and specific molecular marker resources for genetic diversity analyses in fig (Ficus carica l). HortScience 54:1299–1309
Abdelsalam NR, Ali HM, Salem MZ, El-Wakil HE (2020) Quantitative and qualitative genetic studies of some Acacia species grown in Egypt. Plants 9:243
Kress WJ, Erickson DL (2008) DNA barcoding: a windfall for tropical biology? Biotropica 40:405–408
Li Q, Wu J, Wang Y, Lian X, Wu F, Zhou L, Huang Z, Zhu S (2017) The phylogenetic analysis of Dalbergia (Fabaceae: Papilionaceae) based on different DNA barcodes. Holzforschung 71:939–949. https://doi.org/10.1515/hf-2017-0052
Selvaraj D, Sarma RK, Sathishkumar R (2008) Phylogenetic analysis of chloroplast MatK gene from Zingiberaceae for plant DNA barcoding. Bioinformation 3:24–27. https://doi.org/10.6026/97320630003024
Balachandran KRS, Mohanasundaram S, Ramalingam S (2015) DNA barcoding: a genomic-based tool for authentication of phytomedicinals and its products. Botanics 5:77–84. https://doi.org/10.2147/BTAT.S61121
Liu Z-F, Ci X-Q, Li L, Li H-W, Conran JG, Li J (2017) DNA barcoding evaluation and implications for phylogenetic relationships in Lauraceae from China. PLoS ONE 12:e0175788. https://doi.org/10.1371/journal.pone.0175788
Abdelsalam NR (2014) Polymorphism in some Egyptian wheat varieties based on SSR-markers. J Exp Agric Int. https://doi.org/10.9734/AJEA/2014/9235
Ahmed SS (2022) DNA barcoding in plants and animals: a critical review
Chakraborty C, Doss CGP, Patra BC, Bandyopadhyay S (2014) DNA barcoding to map the microbial communities: current advances and future directions. Appl Microbiol Biotechnol 98:3425–3436. https://doi.org/10.1007/s00253-014-5550-9
Katoh K, Misawa K, Kuma KI, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. https://doi.org/10.1093/nar/gkf436
Pei J, Grishin NV (2007) PROMALS: towards accurate multiple sequence alignments of distantly related proteins. Bioinformatics 23:802–808. https://doi.org/10.1093/bioinformatics/btm017
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J et al (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. https://doi.org/10.1038/msb.2011.75
Hall T (2001) BioEdit version 5.0. 6. Department of Microbiology, North Carolina State University
Stecher G, Tamura K, Kumar S (2020) Molecular evolutionary genetics analysis (MEGA) for macOS. Mol Biol Evol 37:1237–1239. https://doi.org/10.1093/molbev/msz312
Katoh Y, Nozaki S, Hartanto D, Miyano R, Nakayama K (2015) Architectures of multisubunit complexes revealed by a visible immunoprecipitation assay using fluorescent fusion proteins. J Cell Sci 128:2351–2362. https://doi.org/10.1242/jcs.168740
Kress WJ, Erickson DL, Jones FA, Swenson NG, Perez R, Sanjur O, Bermingham E (2009) Plant DNA barcodes and a community phylogeny of a tropical forest dynamics plot in Panama. Proc Natl Acad Sci 106:18621–18626. https://doi.org/10.1073/pnas.0909820106
De Mattia F, Bruni I, Galimberti A, Cattaneo F, Casiraghi M, Labra M (2011) A comparative study of different DNA barcoding markers for the identification of some members of Lamiacaea. Food Res Int 44:693–702. https://doi.org/10.1016/j.foodres.2010.12.032
Wattoo JI, Saleem MZ, Shahzad MS, Arif A, Hameed A, Saleem MA (2016) DNA Barcoding: Amplification and sequence analysis of rbcl and MatK genome regions in three divergent plant species. Adv Life Sci 4:03–07
Ali MA, Gyulai G, Hidvegi N, Kerti B, Al Hemaid FM, Pandey AK, Lee J (2014) The changing epitome of species identification–DNA barcoding. Saudi J Biol Sci 21:204–231
Adolfo LM, Rao X, Dixon RA (2022) Identification of Pueraria spp. through DNA barcoding and comparative transcriptomics. BMC Plant Biol 22:1–18
Kenfack D, Abiem I, Chapman H (2022) The efficiency of DNA barcoding in the identification of afromontane forest tree species. Diversity 14:233
Gholami A, Malik S, Dhabe AS, Pandey AK, Babbar SB (2020) DNA barcoding of Indian Alysicarpus (Fabaceae): ITS alone distinguishes species. Vegetos 33:592–600
Neugebauer K, El-Serehy HA, George TS, McNicol JW, Moraes MF, Sorreano MC, White PJ (2020) The influence of phylogeny and ecology on root, shoot and plant ionomes of 14 native Brazilian species. Physiol Plant 168:790–802
Presting GG (2006) Identification of conserved regions in the plastid genome: implications for DNA barcoding and biological function. Can J Bot 84:1434–1443. https://doi.org/10.1139/b06-117
Stoeckle M (2003) Taxonomy, DNA, and the Bar Code of Life. Bioscience 53:796–797. https://doi.org/10.1641/0006-3568(2003)053[0796:TDATBC]2.0.CO;2
Hilu KW, Borsch T, Müller K, Soltis DE, Soltis PS, Savolainen V, Chase MW, Powell MP, Alice LA, Evans R et al (2003) Angiosperm phylogeny based on <011>MatK sequence information. Am J Bot 90:1758–1776. https://doi.org/10.3732/ajb.90.12.1758
de Groot GA, During HJ, Maas JW, Schneider H, Vogel JC, Erkens RHJ (2011) Use of rbcL and trnL-F as a two-locus DNA barcode for identification of NW-European ferns: an ecological perspective. PLoS ONE 6:e16371. https://doi.org/10.1371/journal.pone.0016371
Acknowledgements
The authors gratefully acknowledge their respective universities for their support.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). The current research received no external funding.
Author information
Authors and Affiliations
Contributions
Data curation, AZA, SMAR, HEDMFE, AFZ, MEH & NRA; Formal analysis, SMAR, HEDMFE, AFZ, HMA and MEH & NRA; Funding acquisition, SMAR, HMA, EDMFE, AFZ and MEH, AAH & NRA; Methodology, SMAR, HEDMFE, AFZ, HMA and MEH, AAH & NRA; Resources, SMAR, HEDMFE, AFZ; Writing—original draft, MEH, SMAR, HEDMFE, AFZ, AAH and NRA; Writing—review & editing, TJ, RYG, ANS, AAH and NRA.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
Not applicable.
Informed consent
Not applicable.
Consent to Publish
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abdelsalam, N.R., Hasan, M.E., Javed, T. et al. Endorsement and phylogenetic analysis of some Fabaceae plants based on DNA barcoding. Mol Biol Rep 49, 5645–5657 (2022). https://doi.org/10.1007/s11033-022-07574-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-07574-z