
Abstract
Targeted protein degradation (TPD) technology has gradually become widespread in the past 20 years, which greatly boosts the development of disease treatment. Contrary to small inhibitors that act on protein kinases, transcription factors, ion channels, and other targets they can bind to, targeted protein degraders could target “undruggable targets” and overcome drug resistance through ubiquitin–proteasome pathway (UPP) and lysosome pathway. Nowadays, some bivalent degraders such as proteolysis-targeting chimeras (PROTACs) have aroused great interest in drug discovery, and some of them have successfully advanced into clinical trials. In this review, to better understand the mechanism of degraders, we elucidate the targeted protein degraders according to their action process, relying on the ubiquitin–proteasome system or lysosome pathway. Then, we briefly summarize the study of PROTACs employing different E3 ligases. Subsequently, the effect of protein of interest (POI) ligands, linker, and E3 ligands on PROTAC degradation activity is also discussed in detail. Other novel technologies based on UPP and lysosome pathway have been discussed in this paper such as in-cell click-formed proteolysis-targeting chimeras (CLIPTACs), molecular glues, Antibody-PROTACs (Ab-PROTACs), autophagy-targeting chimeras, and lysosome-targeting chimeras. Based on the introduction of these degradation technologies, we can clearly understand the action process and degradation mechanism of these approaches. From this perspective, it will be convenient to obtain the development status of these drugs, choose appropriate degradation methods to achieve better disease treatment and provide basis for future research and simultaneously distinguish the direction of future research efforts.
Graphical abstract
Protein is degraded by proteasome pathway and lysosome pathway.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Proteins are the main undertakers of life activities and the basis of all living things. They are constantly produced and constantly die in organisms [1]. Most diseases are related to abnormal protein expression or activity, and selective elimination of disease-causing proteins will contribute to cure diseases. Therefore, degradation or intervention of pathogenic proteins has become one of the most effective disease treatment strategies [2]. Currently, most drugs are protein inhibitors, which specifically bind to pathogenic proteins to inhibit their activity to obtain curative effects. However, the number of pathogenic proteins bound by available inhibitors is limited, less than 20% of the total number of disease-causing proteins, and 80% of them are undruggable targets [3, 4]. In addition, inhibition of traditional small inhibitors (SMIs) is an “occupancy-driven” event, which may eventually cause off-target side effects after medication. Also, drug resistance is a major problem due to overexpression or mutation of the target protein, which could not be completely avoided [3, 4]. Wang and his group have reported potent degraders targeting androgen receptor (AR) protein to address resistance to AR antagonists [5, 6]. Furthermore, Rao et al. published a series of proteolysis-targeting chimeras (PROTACs) to cure ibrutinib-resistant non-Hodgkin lymphomas [7].
Over the past 20 years, targeted protein degradation (TPD) has emerged as a novel chemical biology tool and potential therapeutic modality in several diseases, which can greatly expand the druggable space compared to SMIs [1, 8,9,10]. TPD drugs like PROTACs employ the “event-driven” approach, which means not “occupancy-driven,” so they can aim at undruggable targets. Moreover, the degraders deplete the target proteins quickly, in minutes to hours. Moreover, protein degrading methods are rapidly reversible and offer more fine-grained control [1].
In general, protein degradation is carried out by ubiquitin–proteasome pathway (UPP) and lysosome pathway, such as PROTAC technology and molecular glues hijack the ubiquitin–proteasome system, lysosomaltargeting chimeras (LYTACs) and macro-autophagy degradation target chimeras (MADTACs) co-opt lysosome pathway [10]. It is known that heterobifunctional PROTAC molecules consist of a ligand for a target protein of interest (POI), and a ligand for an E3 ubiquitin ligase, joined by a flexible linker. Herein, in this review, we divide the degraders into ubiquitin–proteasome system and lysosomal pathway-dependent degraders for a better understanding of TPD application and development.
Degraders based on the ubiquitin–proteasome pathway (UPP)
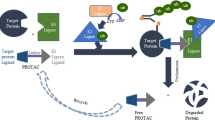
Proteolysis-targeting chimera (PROTAC) has been widely studied in the field of targeted protein degradation (TPD). Conventional PROTACs are heterobifunctional molecules designed to bind to an E3 ubiquitin ligase and a POI via two warheads [9]. The PROTAC molecule consists of three parts: an E3 ligase, a POI ligand, and a linker that connects the two parts. When the POI-PROTAC-E3 ligase forms the ternary complex, POI ubiquitination and degradation can be triggered by the ubiquitin–proteasome system (UPS), after which PROTAC is released and recycled to participate in a new round of degradation (Fig. 1). PROTAC employs a catalytic-type mechanism of action (MOA) and “event-driven” event, which is different from classical inhibitors [11]. Moreover, under the rapid development of TPD drugs, in addition to traditional PROTACs, other alternative degradation approaches based on UPP have also grown rapidly.

The process of PROTAC-mediated protein ubiquitination and degradation
Small-molecule proteolysis-targeting chimeras (PROTACs) based on different E3 ligases
There are more than 600 E3 ligases in eukaryotes. Different E3 ligases are specific because they can recognize different substrates due to their large differences in amino acid sequences [12]. Currently, however, only a few E3 ligases have been used in PROTAC studies, such as cereblon (CRBN), von Hipple-Lindau (VHL), cellular inhibitor of apoptosis protein-1 (cIAP1), mouse double minute 2 homologue (MDM2), DDB1-and-CUL4-associated factor 15 (DCAF15), and RING finger protein 4 (RNF4), which are mainly due to the fact that highly specific ligands of specific E3 ligases are few [13] (see Fig. 2).

The summary of E3 ligases described in this chapter
Mouse double-minute 2 homolog (MDM2) and cellular inhibitor of apoptosis protein-1(cIAP1)
In 2008, Crews’ group reported the first small-molecule PROTAC (1) (Fig. 3) which recruited the MDM2 E3 ligase to degrade the androgen receptor (AR) protein, but only produced protein degradation with micromolar concentration [14, 15]. Indeed, MDM2 is a primary cellular inhibitor of p53, the tumor suppressor, and plays a significant role in cancer therapy [16]. Most interestingly, Hines et al. published the first bromodomain-containing protein 4 (BRD4)-targeting MDM2-based PROTAC A1874 (2) (Fig. 3) triggering 98% POI degradation and achieving nanomolar degradation efficiency. Besides, this small-molecule PROTAC could mediate BRD4 degradation and p53 stabilization [15], thus, dually inhibiting BRD4 and MDM2 compared to BRD4 small inhibitors [17].

Representative PROTACs based on MDM2 and cIAP1
It is known that cIAP1 may suppress apoptosis and promote cell-cycle progression [18]. Itoh et al. presented a degrader-targeting cellular retinoic acid-binding proteins (CRABPs) based on cIAP E3 ligase (3) (Fig. 3) in which a ligand for CRABPs was conjugated with Methyl bestatin (MeBS). This molecule eventually formed a ternary complex with cIAP1 and CRABPs, which degraded CRABPs by the ubiquitin–proteasome pathway [19]. In 2016, specific and Non-genetic inhibitors of apoptosis protein [IAP]-dependent Protein Erasers (SNIPER), BCR-ABL protein degradation inducers, were synthesized (4) (Fig. 3) [20]. Accordingly, SNIPER(ABL) induced the degradation of BCR-ABL protein and a subsequent reduction in cell growth to achieve the treatment of chronic myelogenous leukemia (CML) in this study. In addition, novel SNIPER capable of inducing protein degradation of AR has been shown (5) (Fig. 3). The SNIPER(AR) molecule with the AR ligand and IAP ligand produced effective protein knockdown activity against AR, suggesting that it could be a promising agent for prostate cancer (PC) [21]. In addition, 5 could effectively suppress AR-mediated gene expression and induce caspase activation and apoptosis in PC cells which did not emerge in cells treated with AR antagonists [22]. However, cIAP1-based PROTACs can also induce autoubiquitination and degradation of the E3 ligase itself [20, 21], thus, limiting the full potential of the technology [23].
Von Hipple-Lindau (VHL) and cereblon (CRBN)
Subsequently, it was still the Crews and co-workers who produced the first small-molecule ligand-targeting VHL ligase in 2012 [24]. From this study, the crystal structure of VHL bound to the most potent inhibitor was obtained, confirming the inhibitor binding at the hypoxia-inducible factor 1α (HIF-1α)-binding site [24] and providing a starting point for further ligand optimization [23]. In 2015, a VHL-based PROTAC was developed (6) (Fig. 4), which targeted estrogen receptor α (ERα) for degradation. The VHL-PROTACs developed from this study are highly specific for their targets and show efficient protein degradation of ERα in vivo in mice [25]. Later, Burslem et al. designed a series of small-molecule PROTACs based on VHL using specific lapatinib and gefitinib inhibitors as POI warheads to induce receptor tyrosine kinase (RTK) degradation. Compared with traditional RTK inhibitors, the corresponding PROTACs can not only effectively suppress downstream signaling and cell proliferation effectively but can also be amenable to solve the resistance of traditional inhibitors due to target protein mutation [26].

Representative PROTAC based on VHL
It is widely known that constraining a molecule’s bioactive conformation through macrocyclization represents an attractive strategy for rationally designing functional chemical compounds. Ciulli’s laboratory has made efforts to add a cyclic linker to the bromodomain and extra-terminal (BET) degrader MZ1 (7), a second linker to “close a circle” between the BRD4 ligand and the VHL ligand of MZ1 (8) (Fig. 5) [27]. Macrocyclization allows the constraint of a PROTAC molecule in its bioactive conformation, making it adopt or discriminate against a desired ternary complex, thus, achieving degradation potency and selectivity. Last year, this macrocyclization design strategy was also reported to develop anaplastic lymphoma kinase (ALK) PROTACs, which used VHL ligand to design compounds [28]. Importantly, this approach to promoting the small-molecule PROTAC process should be anticipated as an increasingly attractive and feasible method of drug design [27, 28].

The structure of MZ1 and representative macrocyclic PROTAC based on VHL
In 2010, cereblon (CRBN) was first exposed by Ito [29] from thalidomide, the immunomodulatory drugs (IMiDs). Prior to this, studies have revealed that other than thalidomide, its analogs pomalidomide, and lenalidomide degrad target proteins by binding to CRBN [30]. Cereblon ligands possess better drug-like properties-lower molecular weight (M.W.), fewer hydrogen donors, and fewer rotatable bonds, thus, holding excellent physicochemical properties [6] compared to VHL ligands.
In 2015, a small-molecule PROTAC recruited by CRBN E3 ligase targeting BRD4 was reported. The resulting compound, dBET1 (9) (Fig. 6), induced highly selective BET protein degradation in vitro and in vivo, and slowed leukemia progression in mice [31]. At almost the same time, similar to the design of dBET1, Crews’ laboratory chose BRD4 inhibitor JQ1 as a POI warhead and pomalidomide as an E3 ligand to synthesize ARV-825 (10) (Fig. 6) [32]. More interestingly, ARV-825 was 10 times more capable of degrading BRD4 than dBET1 and had a superior effect on suppression of Burkitt’s lymphoma cell proliferation compared to BRD4 inhibitors JQ1 and OTX-015.

Representative PROTACs based on CRBN
Surprisingly, in 2019, Arvinas discovered the first-inclass potent and orally active AR degrader-ARV-110 (11) (Fig. 6) and it advanced into clinical development to cure AR-positive metastatic castration-resistant prostate cancer (mCRPC) [6]. Concurrently, ARV-471 (12) (Fig. 6), an ER-targeting degrader, has entered the Phase II clinical trial focused on the treatment of metastatic ER-positive/human epidermal growth factor receptor 2 (HER2)-negative breast cancer [33]. Both ARV-110 and ARV-471 are CRBN-based small-molecule PROTACs, displaying low nanomolar concentrations of protein degradation and potential cell inhibitory activity [6, 33].
In the early days, PROTAC was generally applied to knock down single POI. At present, the discovery and development of drugs with multiple targets have opened up new possibilities for the treatment of diseases. In contrast to traditional combination therapies involving the use of two or more drugs acting on different processes, multi-target drug design aims to integrate function and structure against two or more targets in the same molecule [34]. Herein, based on similar structures, pharmacophores of two or more ligands can be connected, superimposed, or fused to obtain a ligand that can act on two or more targets [34]. In 2018, Zoppi et al. employed CRBN and VHL ligands to design and synthesize degraders to target BRD9 and BRD7. Particularly, potent and selective inhibitors that bind to BRD7/9 bromodomains have recently regarded as POI ligands. VZ185 (13) (Fig. 7) was filtered out as a highly selective, potent, and rapid dual degrader based on the VHL ligand [35]. Furthermore, a series of compounds have been published to achieve potent and rapid degradation of Cyclin-dependent kinase 2 (CDK2) and CDK9 [36]. Next, small-molecule degraders with dual targets were applied to other CDK families. Niu et al. employed the purine-based noncovalent CDK12/13 dual inhibitor as the target protein-binding ligand to develop CDK12-specific PROTAC degraders, such as PP-C8 which based on CRBN (14) (Fig. 7) [37]. Last but not least, dual-target histone deacetylase (HDAC) inhibitors became warheads to fulfill the degradation of related proteins [38]. Similarly, the dual BCL-xL and BCL-2 degraders based on CRBN or VHL have been developed in cancer therapy [39, 40].

Representative dual-target PROTACs based on VHL and CRBN
As mentioned above, classical small-molecule PROTACs usually combine with POIs and hijack E3 ligases to trigger degradation of non-native neosubstrates [41]. Then, homo-PROTACs, a small-molecule approach to effectively dimerize an E3 ubiquitin ligase to induce its own self-destruction, were described. Contrary to classical PROTACs, the POI of homo-PROTACs is E3 ligase. Ciulli’s group focused on VHL E3 ubiquitin ligase to achieve self-degradation and designed CM11 (15) (Fig. 8) which rapidly achieved protein knockdown [42]. In 2019, it was still Ciulli and colleagues published dually targeting CRBN-VHL-PROTACs that aimed to analyze the relative ability of CRBN and VHL E3 ligase to induce degradation of one another. In this study, among hetero-PROTACs, compound 16 (Fig. 8) achieved CRBN ligase degradation with high potency and up to deep levels [43]. Moreover, in addition to the above cases for CRBN and VHL, homo-PROTACs studied on MDM2 and hetero-PROTACs for MDM2 and CRBN have recently been published [41]. From this event, this approach opened powerful new avenues for drugging E3 ligases.

Representative homo-PROTACs and hetero-PROTACs based on VHL and CRBN
More interestingly, current studies use VHL and CRBN ligands to exploit trivalent degraders that can be narrowly defined as a molecule combining three ligands, one E3 ligand and two POI ligands. From our point of view, the higher strengthen degradation will be achieved owing to the development of trivalent degraders. The majority of PROTAC molecules reported only connect one inhibitor with one E3 ligand, often degrade only one target protein, and do not exceed the limit of two or more similar proteins. In this respect, Zheng et al. inspired by dual-targets drugs have synthesized dual PROTACs to simultaneously degrade two different types of POIs. In this paper, a series of degraders were designed to degrade both epidermal growth factor receptor (EGFR) and poly(ADP-ribose) polymerase (PARP). Among the compounds produced, DP-V-4 (17) (Fig. 9) employed by VHL ligand obtained the best degradation for both EGFR and PARP [44]. However, due to the larger M.W. compared to monovalent and bivalent TPD agents, these degraders appeared poor antiproliferative activity [23, 44].

Representative trivalent PROTACs based on VHL
In the same year, Imaide et al. designed trivalent PROTACs consisting of bivalent BET inhibitors and an E3 ligand tethered via a branched linker, considering that increasing binding valency within a PROTAC could enhance degradation. The best degraders SIM1 (18) (Fig. 9) links to VHL ligand and binds BET protein intramolecularly in a cis fashion to both BD1 and BD2, inducing a conformational change to form a 1:1:1 complex [45]. As a result, SIM1 displayed higher efficacy and potency than bivalent PROTACs due to enhanced pharmacodynamic properties, including avidity, cooperativity, and prolonged residence time [9, 45]. Lately, Huang et al. designed and synthesized trivalent PROTACs that each had a tert-butyl ester unit on the benzene ring as a controlled orientation for further functionalization, which were based on the reported structure of MZ1 in a complex with human VHL and BRD4BD2 [46]. From these examples described above, the utilization of VHL ligands is more remarkable than CRBN probably due to the different levels of E3 ligase expression in cells and tissues of the target protein [47].
Altogether, most of small-molecule PROTACs reported since 2015 exploit VHL and CRBN E3 ligases [3]. Owing to the good binding affinity to their E3 ligase targets, CRBN and VHL ligands are pivotal elements for PROTAC design research [23]. Accordingly, it has become routine to generate a new degrader in cell culture experiments and animal models of disease, using either VHL or CRBN as recruitment ligases [8]. In fact, the preclinical and clinical development of the VHL- and CRBN-based PROTAC molecules still pose challenges, such as resistance mechanisms to therapeutic agents can arise rapidly [8], due to decreased or lost expression of E2 and E3 ubiquitin ligases at mRNA or protein level [23].
DDB1-and-CUL4-associated factor 15 (DCAF15), DCAF16 and RING finger protein-114 (RNF114)
Most importantly, it is necessary to expand the scope of E3 ligases and search for new ones owing to the degradation selectivity often related to the specific E3 ligases applied [47]. DDB1-and-CUL4-associated factor 15 (DCAF15), an E3 ligase, was initially discovered in a proteomic screen of co-immunoprecipitants of DDB1 and CUL4. Recently, some research has revealed that further structural and biochemical evaluation in determining the use of DCAF15 for PROTAC targeting is imperative [48]. Besides, Zhang et al. also identified DCAF16, a nuclear protein forming the substrate receptor subunit of cullin 4-RING ligase (CRL4), as a target for electrophilic PROTACs that promoted nuclear-restricted degradation of POI (19) (Fig. 10) [49]. In 2019, Spradlin et al. utilized Nimbolide as a RING finger protein-114 (RNF114) recruiter to other protein substrates for proteasomal degradation through the development of heterobifunctional degraders (20) (Fig. 10) [50]. Additionally, both DCAF16 and RNF114 bind covalently to ligands, the irreversible binding appears to be beneficial in dynamics.

Representative PROTACs based on DCAF16 and RNF114
RING finger protein 4 (RNF4) and kelch-like ECH-associated protein-1 (KEAP1)
Besides, some covalent ligands have also been recruited by E3 ligases to degrade POI. In 2019, Ward and his co-workers screened and optimized the RING finger protein 4 (RNF4) ligand to connect to JQ1. CCW 28–3 (21) (Fig. 11) is the RNF4-based PROTAC which has the ability to knock down BRD4 in breast cancer cells by inducing interaction, ubiquitination, and proteasomal degradation [51]. Interestingly, last year, a noncovalent kelch-like ECH-associated protein-1 (KEAP1) ligand was employed to design compounds targeting the BET family protein. For instance, MS83 (22) (Fig. 11), which links KEAP1 ligand and BRD4/3/2 binder, achieves effective degradation of BRD4/3 [52].

Representative PROTACs based on RNF4 and KEAP1
Peptide-based PROTACs
In 2001, the first bifunctional molecule PROTAC was created by Crews and Deshaies laboratory, which hijacked Skp1-Cullin-F boxβ-TRCP (SCFβ-TRCP) E3 ubiquitin ligase via IκBα phosphopeptides to induce methionine aminopeptidase-2 (MetAP-2) degradation [23, 53]. However, dependence on phosphorylation of IκBα phosphopeptide for E3 ligase recruitment was limited [23].
More recently, PROTACs composed of peptide ligands have been defined as bioPROTACs roughly [8]. Schneekloth et al. conferred cell permeability by fusing a polylysine-penetrating peptide at the connection of PROTAC, and verified that the molecule could self-introduce into cells and degrade target proteins (23) (Fig. 12) [54]. Notably, in this study, the shortest peptides from HIF1α were used to recruit the VHL ligase, boosting the development of peptide PROTACs to a large extent [23].

Representative peptide-based PROTACs
In addition, Wang and co-workers utilized a peptide combined with signal transducer and activator of transcription 3 (STAT3) protein to design a peptide-based PROTAC, SD-36 (24) (Fig. 12), recruiting the CRBN ligase [55]. SD-36 achieved potent STAT3 degradation and inhibited leukemia and lymphoma in vitro and in vivo [55, 56]. Although peptide-based PROTACs have advantages in binding affinity, target specificity, and chemical synthesis, they possess limited membrane permeability [56, 57].
Nucleotide-based PROTACs
Currently, there are public nucleotide-based PROTACs, using oligonucleotides as POI ligands [56]. Transcription factors and RNA-binding proteins (RBPs) are two kinds of proteins that are required for DNA repair, replication, transcription, and many RNA-dependent processes [8]. Next, some studies have published RNA-PROTACs that target RBPs, oligonucleotide-based PROTACs (O’PROTACs), and transcription factor-targeting chimeras (TRAFTACs) that target transcription factors [8, 19].
The first RNA-PROTAC (25) (Fig. 13) targets the Lin28 protein, a stem cell factor, and oncoprotein of high interest as a potential drug target for several diseases [58]. RNA-PROTACs are docked with the RNA-binding site of RBP via structurally modified oligoribonucleotides, which have the same sequence as the natural RNA-binding element of RBP. Then, this PROTAC accomplished remarkable Lin28 degradation with high selectivity and negligible toxicity [56, 58].

Representative nucleotide-based PROTACs
For TRAFTACs, double-stranded DNA (dsDNA) consensus sequences are used as transcription factor-binding warheads [8]. Samarasinghe et al. designed PROTAC-based DNA, which employed DNA sequence as POI ligand to recognize the transcription factor NF-κB for targeted degradation [56, 59]. At the same time, in 2021, an oligonucleotide-based PROTAC (26) (Fig. 13) was presented to potentially and efficiently induce in vivo and in vitro degradation by lymphoid enhancer-binding factor 1 (LEF1) and ETS-related gene (ERG) [60]. These nucleotide-based PROTACs expand the concept of bivalent PROTACs and provide a novel strategy for disease treatment.
Given that the factors affect the degradation activity of PROTAC, we could conclude from numerous research articles that PROTAC molecule design always employs different SMIs, linkers, and E3 ligands to make paired combinations and analyzed degradation activity with cell viability, protein degradation activity, and tumor suppression. When it comes to ligands selection for POIs, normally, researchers exploited representative SMIs against target proteins.
For example, Dong’s group used Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib as a POI warhead to study orally bioavailable BTK-PROTACs. From this article, compound 27 (Fig. 14), which appeared high BTK degradation potency and oral exposure and induced BTK degradation with high selectivity, was discovered [61]. Besides, Wang and co-workers employed the previously reported AR POI ligand, and then synthesized dozens of PROTACs targeted AR, achieving excellent degradation activity in VCap cells. However, in order to improve oral exposure, new POI ligands are urgently needed for further design. Subsequently, other ligands were predicted again through computational modeling to explore the binding model of ligand in a complex with AR, thus, publishing a series of compounds as well. Among these PROTACs, ARD-2585 (28) (Fig. 14) became an exceptionally potent and orally active AR degrader [6], which undoubtedly demonstrated that modifications to POI warheads were significant in further degrader exploration. The combination of proteins could affect the degradation activity of compounds to some extent.

Representative PROTACs target BTK, AR, BRD4, EGFR, and HER2
It is known that the linker is an important part of PROTAC’s design, and the length and flexibility of the linker between the two warheads bound to the POI and E3 ligase can dramatically affect potency and selectivity [4]. Moreover, the linkers do not simply connect two ligands but can also affect the E3 ligases-POI interactions and presentation of the target protein, thus, presupposing the basis for yielding the ternary complex with sufficient stability [8, 62]. Nowadays, some linker motifs have already appeared in a number of PROTAC designs, such as flexible linkers PEG, alkyl, amide acid and alkyne motifs, rigid linkers saturated heterocycles, and aromatic rings [63]. And then, linker as a crucial part of PROTAC molecule can influence target selectivity such as length and tethering orientation [23]. In addition, notably, if the linker length is too short, the formation of ternary complex will be unfavorable and longer linker cannot effectively recruit E3 ligase to remove POI.
In 2018, Burslem et al. employed a SMI-lapatinib which targeted EGFR and could also bind to human epidermal growth factor receptor 2 (HER2) as the POI warhead and used VHL ligand to synthesize PROTACs. The PROTAC (29) (Fig. 14) which has 2 PEG units, can induce EGFR and HER2, but increasing the length to 3 PEG units, the compound 30 (Fig. 14) selectively degrades EGFR rather than HER2 [23, 26]. From this study, the conclusion that linker length could define target selectivity were elucidated. Furthermore, as mentioned above, dBET1 and ARV-825 (Fig. 6) both possess the same POI and E3 ligands while the length and the composition of linker are different. In this case, ARV-825 was 10 times more capable of degrading BRD4 than dBET1. It has been proved that the length and the composition of linker are key variables determining the activity of degraders [31, 32].
Besides, conformational restriction has often been used as a strategy to improve oral bioavailability of small-molecule drugs [6]. Therefore, researchers used rigid linkers to develop and exploit more excellent PROTACs. As we know, the AR-targeting degrader, ARV-110 (Fig. 6), which entered clinical trials utilized rigid piperazine and piperidine rings and achieved great AR degradation in cells and tumors. Interestingly, Wang’s laboratory adopted the same composition linker of ARV-110 and reported ARD-2128 (31) (Fig. 14) which achieved 67% oral bioavailability in mice, effectively removed AR protein and suppressed AR-regulated genes in tumor tissues with oral administration [5]. It was hypothesized that increased conformational restriction of the linker in degrader may lead to a more stable and productive ternary complex [6]. In other words, linker rigidity could increase cooperativity of the ternary complex through improved protein degradation [23].
As mentioned above, the expression of different E3 ligases is specific in various tissues. For this reason, during the design of PROTACs, different E3 ligases were utilized to compare the degradation activity. Additionally, Steinebach et al. addressed VHL and cIAP1 ligases to explore CDK4/6 degraders, where VHL-based compounds were either specific for CDK6 or exhibited dual activity against CDK4 and CDK6, while IAP-based PROTACs produced a combined degradation of CDK4/6 [64]. More interestingly, different chiral configurations of E3 ligands could also cause different degradation effects. For example, the BET PROTAC ARV-771 (32) (Fig. 14) is a potent BET degrader in cell models of castration-resistant prostate cancer (CRPC) and has a high affinity with VHL, but ARV-766 (33) (Fig. 14), a diastereomer of ARV-771 with the opposite configuration at the hydroxyproline, has no affinity for VHL, leading to little degradation to BET [65].
In general, POI warhead, linker, and E3 ligand are indispensable for PROTAC development, and the selection of three parts is equally important. First, pairing the E3 ligase with POI is one of the most critical factors involved in the generation of potent and selective PROTACs [47]. However, there is no correlation between the affinity of the PROTAC molecule for kinases and the extent of induced degradation [47, 62]. Second, PROTACs usually yield novel protein–protein interactions (PPIs) between E3 ligases and target POIs that can vary significantly depending on the ligands used and the length and composition of linker [3, 63]. Thereby, linkerology can be used to adjust physicochemical properties, such as metabolic stability, membrane permeability, and aqueous solubility of PROTACs [23]. Ultimately, the formation of a stable, cooperative POI-PROTAC-E3 ligase ternary complex is a crucial step in achieving potent and selective degradation. PROTACs can stabilize PPIs between the POI and E3 ligase, thus, promoting positive cooperativity in ternary complex formation [23, 47]. In other words, the rate of formation of the ternary complex determines the degradation efficiency of PROTAC. It is believed that the further development and discovery of linker, POI, and E3 ligands will achieve better and deeper success in this field under constant pursuit.
Molecular glues, SERDs, and SARDs: based on UPP
The discovery and development of bivalent targeted protein degraders seem to have become a major focus in the field of TPD. However, unlike PROTACs, which recruit an E3 ligase to degrade a POI, monovalent protein degraders simply bind to POI through a direct or induced mechanism [13]. Some monovalent degraders can induce the POI degradation by producing conformational or other changes that make the protein susceptible to detection by cellular quality control and ubiquitin-degradation machinery, such as molecular glues [66]. It is not surprising that the successful application of this strategy will pave the way for the advancement of TPD development.
Molecular glues
Molecular glues, which have a lower molecular weight than PROTAC molecules, can recognize and promote ubiquitination to degrade target proteins by remodeling the surface of E3 ligases and inducing or enhancing protein–protein interactions (PPIs) between E3 ligases and target proteins, and are classified as monovalent degraders [67] (Fig. 15). The physicochemical properties of molecular glues are similar to those of traditional small-molecule drugs, and most of them fall into the range of the “rule of five.” Therefore, molecular glues are theoretically more drug-like than PROTACs in terms of lower M.W., increased oral bioavailability, and improved pharmacokinetic (PK)/pharmacodynamic (PD) profiles [68]. At present, there are mainly two kinds of mature molecular glues: one is IMiDs such as thalidomide and its derivatives, and the other is aryl sulfonamide drugs.

Mode of action and structural features of PROTAC and molecular glue
For example, transcription factors Ikaros family zinc finger protein-1 (IKZF1) and IKZF3 have been reported to be ubiquitinated by IMiD-induced CUL4 (cullin 4)-RBX1-DDB1-CRBN (CRL4CRBN) and degraded by proteasome [68, 69]. 34 (Fig. 16) is an IMiD analogs. For the aryl sulfonamide derivative, molecular glue 35 (Fig. 16) is a representative drug. DCAF15, the CRL4 substrate receptor, was used to degrade splicing factor RNA-Binding Motif Protein 39 (RBM39) [48, 67, 70], which could also be employed in the design of PROTAC molecules [48].

Representative molecular glues
Selective estrogen receptor degraders (SERDs) and selective androgen receptor degraders (SARDs)
Recently, some monomeric degraders which target such as ERɑ, AR, HER2, phosphatidylinositol 3-kinase ɑ (PI3Kɑ), BTK, and cIAP have been studied and exploited [71]. Among them, the most representative and notable is the publication of monovalent degraders targeting ERɑ and AR. Previously, selective estrogen receptor modulators (SERMs) and aromatase inhibitors (AIs) that could directly antagonize ERα transcriptional activity or block estrogen biosynthesis were employed in the cure of ERα-positive breast cancer [72]. Lately, fulvestrant (36) (Fig. 17), a selective estrogen receptor degrader, which binds and downregulates ER by inducing conformational instability could induce ERα degradation and overcome resistance to AIs and SERMs [13, 72]. Until recently, Lu et al. discovered a thieno[2,3‑e] indazole derivative as novel oral selective estrogen receptor degraders (SERDs) (37) (Fig. 17) for the treatment of ER-positive breast cancer, which has been evaluated in preclinical trials [72].

Representative SERDs and SARDs
Encouraged by the application of SERDs, selective androgen receptor degraders (SARDs) have been investigated in the clinic to block androgen receptor function in patients with mCRPC, owing to primary and acquired resistance to AR modulators [13, 66]. In 2013, an orally bioavailable SARD-AZD3514 (38) (Fig. 17), advanced into Phase I clinical trials, was disclosed by Loddick and co-workers [71, 73]. From his study, AZD3514 could obviously suppress androgen-dependent and -independent AR signaling. AZD3514 modulates AR signaling through two distinct mechanisms, inhibition of ligand-driven nuclear translocation of AR, and downregulation of receptor levels [73]. In addition, Miller’s group reported a series of degraders applying to the treatment of enzalutamide-resistant prostate cancer, which is increasingly prevalent in the clinic [74].
Other novel degradation technologies based on UPP
In general, in addition to the above-mentioned PROTACs that induce degradation by UPS, there are other novel PROTACs that work through this system (Table 1). The ubiquitin proteasome pathway (UPP) (Fig. 18) is an important approach for selective protein degradation. In this system, the detached PROTAC enters the next degradation cycle, after the modified polyubiquitin POI is recognized and degraded by proteasome.

The ubiquitin proteasome pathway
Hypoxia-activated PRTOACs (Ha-PROTACs)
Regardless of important milestones established, small-molecule PROTACs still have some concerns [75]. To date, the most important issue is how to precisely control their functions to reduce potential toxicity from systemic degradation [75, 76]. As TH-302, a prodrug containing a nitroimidazole unit that can be recognized with nitroreductase (NTR), entered Phase III clinical trials [75, 77], the nitroimidazole unit was selected to explore the application of PROTAC. NTR is conventionally overexpressed in the hypoxic region of solid tumors, while very low or no expression in normal tissues or cells [78]. Zhang’s group and Xu’s group also introduced nitroimidazole unit as a hypoxia-activated leaving group (HALG) on POI or E3 ligands to develop hypoxia-activated PRTOACs (ha-PROTACs), such as compounds 39 and 40 (Fig. 19), demonstrating the feasibility of the strategy with precisely releasing PROTAC in a tumor hypoxia microenvironment [75, 79] (Fig. 20).

Representative ha-PROTACs

Mechanism of action of ha-PROTACs. The PROTAC can be released under a tumor hypoxia microenvironment
Photosensitive-caged PROTACs (Pc-PROTACs) and azobenzene-PROTACs (Azo-PROTACs)
To effectively improve the targeting of PROTAC technology and reduce potential toxicity, some research groups have independently developed a general strategy to spatiotemporally regulate protein degradation using light, named as photo-PROTACs [75] (Fig. 21). For instance, Xue et al. have developed a kind of light-induced PROTACs, called photosensitive caged PROTACs (pc-PROTACs). The pc-PROTAC (41) (Fig. 22), based on a ligand of CRBN and JQ1, applied to aim at BRD4 [80]. Based on this, photoswitchable azobenzene-PROTACs (Azo-PROTACs) (42) (Fig. 22) by incorporating azobenzene moieties between ligands for the E3 ligase and POI have been found to knock down protein through light control [81]. Unfortunately, some photo-PROTACs only work after UV irradiation [75]. Worse still, prolonged exposure to UV radiation can cause DNA damage, threatening human skin health [11, 75].

Mechanism of action of pc-PROTACs. The PROTAC can be released upon light irradiation

Representative pc-PROTAC and Azo-PROTAC
In-cell click-formed proteolysis-targeting chimeras (CLIPTACs)
As we know, due to the high molecular weight (M.W.), PROTACs own poor solubility and cellular permeability in vivo, preventing these agents from entering cells to induce protein degradation. It is quite clear that reducing the M.W. is essential [11, 56]. In 2016, Lebraud et al. developed a novel PROTAC technology called in-cell click-formed proteolysis-targeting chimeras (CLIPTACs) to overcome limitations [82] (Fig. 23). A tetrazine-tagged thalidomide derivative and a trans-cyclo-octene (TCO)-tagged ligand in cells to form a cereblon E3 ligase recruiting PROTAC molecules are introduced into the design, which have lower M.W. and better permeability, thus successfully degrading oncogenic BRD4 (43) or extracellular-regulated protein kinase 1/2 (ERK1/2) (44) (Fig. 24) [11, 56, 82].

Mechanism of action of CLIPTACs. Click reaction is applied in cells to form CLIPTAC to facilitate E3 ligase degrading protein of interest

Representative CLIPTACs
Chloroalkane-containing PROTACs (HaloPROTACs) and degradation tag (dTAG) molecules
Next, Crews and co-workers established chloroalkane-containing PROTACs (HaloPROTACs) using HaloTag (HT) technology, which incorporates VHL ligands to effectively degrade HaloTag7 fusion proteins (45) (Fig. 25). HaloPROTAC utilizes E3 ligands to combine with chloroalkanes for the degradation of HT fusion proteins as HaloTag can covalently bind to chloroalkanes [8, 83]. Similar to the working principle of HaloPROTAC, the degradation tag (dTAG) system has been developed to rapidly deplete POI (46) (Fig. 25) [84, 85]. From this system, the engineered variant of FK506-binding protein (FKBP12)-FKBP12F36V was used as fusion label, and then the dTAG molecule recruited VHL ligase to induce rapid degradation of the tagged protein, compared to previously reported CRBN-recruiting dTAG molecules [84].

Representative HaloPROTAC and dTAG molecule
Auxin-inducible degron (AID) and Trim-Away
Lately, other bivalent TPD tools, like the auxin-inducible degron (AID) system, have appeared in human vision. In this pathway, there are three essential components: the E3 ubiquitin ligase, target proteins, and phytohormone, similar to small-molecule PRTOACs, where the E3 ligase is the SCF (Skp1, Cullin 1, and F-box) complex [85]. Auxin such as Indole-3-acetic acid (IAA) and Naphthylacetic acid (NAA) can interact with F-box Transport inhibitor response protein-1 (TIR1) and promote the interaction of the E3 ubiquitin ligase SCF-TIR1 with AID, ultimately leading to the ubiquitination and degradation of AID [85] (Fig. 26).

Mechanism of AID. The auxin can be degraded under the ubiquitin–proteasome pathway
Clift et al. reported a post-translational protein depletion method called ‘Trim-Away’ (Fig. 27) to degrade endogenous protein. Trim-Away is based on the ubiquitin ligase and Fc receptor tripartite-motif protein 21 (TRIM21), which recognizes antibody-bound proteins and targets them for proteasome degradation [86, 87]. It may become possible in the future to adapt the Trim-Away method to develop novel therapeutics that target disease-causing proteins for degradation.

The proteins degradation by Trim-Away to degrade endogenous protein based on TRIM21 and antibody
Folate-PROTACs and Antibody-PROTACs (Ab-PROTACs)
More interestingly, in 2021, Liu et al. employed the folate-conjugating strategy (Fig. 28) to link the conventional PROTAC molecule (47) (Fig. 29) to achieve controllable targeted degradation of POI in tumor cells with high folate receptor ɑ (FOLR1) expression [88]. However, this approach is restricted in that these PROTACs can simply be applied to some diseases with FOLR1 overexpression.

Mechanism of action of Folate-PROTACs. The PROTAC can be released in FOLR1 overexpression tumor cell

Representative Folate-PROTACs and Ab-PROTACs
Additionally, antibody–drug conjugate (ADC), whose structure is remarkably similar to classical PROTACs, has also been developed recently. Consequently, an antibody-PROTAC conjugate can be considered as an alternative approach for selective delivery of broad-spectrum PROTAC into specific cell types [89]. Dragovich et al. employed this method to explore the first phase, the development of ADCs derived from BRD4-targeting chimeric degrader entities (48) (Fig. 29) [90, 91]. Significantly, Antibody-PROTAC (Ab-PROTAC) exhibits potent and antigen-dependent BRD4 degradation and antiproliferation activities in cells, and antitumor efficacy in mouse xenograft models [23, 90, 91], which once again provides an alternative option for TPD drug research. Taken together, there is no doubt that the exploitation of trivalent degraders will greatly expand the application of TPD technology and open up a new field of drug discovery.
Degraders based on the lysosome pathway
As we all know, most intracellular protein degradation is conducted by the UPP and lysosome pathway [92]. Furthermore, the lysosome degradation system is divided into autophagy-lysosome system and endosome-lysosome system, according to different principles of protein depletion [9]. More importantly, lysosome pathway-based degraders have greatly broadened the spectrum of degradable targets. Autophagy-targeting chimeras (AUTACs) can deplete organelle such as mitochondria and lysosome-targeting chimeras (LYTACs) can degrade membrane protein and extracellular protein [93], which shows unprecedented research compared to other TPD agents based on the ubiquitin–proteasome pathway.
Autophagy-targeting chimeras (AUTACs) and autophagosome-tethering compounds (ATTECs)
Autophagy-targeting chimeras (AUTACs) link a warhead for POI to a guanine derivative that tags the protein for degradation by the autophagy machinery [8, 94] (Fig. 30). Takahashi et al. developed AUTACs that apply proteins and dysfunctional mitochondria to selective autophagy. A mitochondria-targeted AUTAC accelerates mitochondrial turnover and inhibits apoptosis against mitochondrial injury (49) (Fig. 31) [94]. Besides, another subclass belonging to this machinery known as autophagosome-tethering compounds (ATTECs) directly links a POI warhead to a ligand without a linker, thus, being a part of monovalent degraders (Fig. 30). It is worth noting that ATTEC works like molecular glues: ATTECs do not require the intervention of linkers, simply linking a POI warhead to a ligand that binds to the autophagy protein microtubule-associated protein-1 light chain 3α (LC3), and bind POIs to autophagosomes, causing autophagic degradation [8]. In 2019, Li et al. identified mHTT (mutant huntingtin)-LC3 linker compounds (50) (Fig. 31) that were capable of lowering mHTT levels in vivo and demonstrated the possibility of targeting proteins for degradation using autophagosome-tethering compounds, which provided new entry points for drug discovery [95].

Mechanism of AUTACs and ATTECs. The AUTAC and ATTEC enter into the cell and bind to protein of interest degradation by autophagosome

Representative AUTAC and LC3 linker compound
Lysosome-targeting chimeras (LYTACs)
In 2020, Bertozzi laboratory studied lysosome-targeting chimeras (LYTACs) technology (Fig. 32) that could target extracellular and membrane-associated proteins [9, 96]. LYTACs have two binding domains: oligoglycopeptide groups and antibodies or small molecules that can bind to target proteins, which are connected by a linker. To date, LYTACs have been exploited to selectively degrade apolipoprotein E4 (ApoE), EGFR, and programmed death ligand 1 (PD-L1) [9, 96]. Notably, TPD agents based on a lysosome system have made superior progress in the field of drug design, presenting a promising prospect in preclinical and clinical processes.

The proteins degradation of LYTACs. The membrane protein and extracellular protein can be bound to antibody with oligoglycopeptide group and be degraded by intracellular lysosome
Degraders for the treatment of others diseases
With the development of TPD technology, a large number of degraders are used in cancer treatment and numerous targets are exploited to develop PROTAC molecules, including “undruggable targets,” which, to be honest, have dramatically promoted the study of cancer therapy. In the new era of TPD, it is important to extend the clinical reach of modality beyond oncology [8]. Nowadays, ALK, AR, ER, BCR-ABL, BET, BRD9 and BRD7, BTK, CDK4/6 and so on, these targets have been employed into oncology [4, 33, 56]. Moreover, the disease spectrum includes prostate cancer (PC), breast cancer (BC), colorectal cancer (CRC), non-small cell lung cancer (NSCLC), acute myeloid leukemia (AML), and so on [97].
Treatment of rheumatoid arthritis and neurodegenerative diseases
Given the potential to degrade any target of choice, the field of TPD is extending beyond oncology. For example, a BTK degrader (NX-5948) and an interleukin-1 receptor-associated kinase 4 (IRAK4) degrader (KT-474) in Phase I clinical trials could treat patients with various immuno-inflammatory diseases, such as rheumatoid arthritis [4, 8]. Currently, measures are being taken to develop degraders to target Tau. Tau plays an essential role in neuronal cells stabilizing microtubules (MTs), providing tracks in the transport of cargo proteins and maintaining cell shape [4]. Lu et al. disclosed a Keap1-dependent peptide PROTAC to knock down Tau by UPP, which showed promising character in the treatment of neurodegenerative disease [98] and realized further progress in the field of “undruggable proteins.”
Treatment of viral infections
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has rapidly spread around the world and resulted in the scale Corona Virus Disease 2019 (COVID-19) pandemic, which has destroyed healthcare systems and our daily lives [23]. In 2020, Chatterjee et al. reported their development of the first anti-COVID (peptide-based) PROTAC by computational design [99], which targeted the viral spike protein receptor-binding domain (RBD) with TRIM21 [87]. Interestingly, some scientists have recently developed ribonuclease-targeting chimeras (RIBOTACs) concept by connecting an RNA-binding molecule to a latent ribonuclease (RNase L) ligand to induce RNA degradation [11]. And then, a RIBOTAC molecule (51) (Fig. 33) was discovered pointing to the degradation of SARS-CoV-2 FSE (frameshifting element) RNA for SARS-CoV-2 therapy [100].

Representative RIBOTAC
Conclusion and perspective
As mentioned above, PROTACs based on different E3 ligases, molecular glues, novel technologies for TPD, AUTACs, LYTACs, etc. have been exhibited in this review. TPD drugs have generated many new strategies that have a more promising future among targeted protein degraders, but also face some challenges (Table 2). Such as small-molecule PROTACs, their toxicology is uncertain and side effects are largely unknown although they have a clear mechanism of action. Besides, other degraders based on UPP can be employed efficiently in their specific areas, which similarly limits their effectiveness. Nucleotide-based PROTACs like O’PROTACs have no “hook effect” and are convenient to apply to “undruggable targets” such as TF of interest, which may be an effective treatment of diseases such as cancer. However, the time of protein degradation is longer than conventional PROTACs. Furthermore, photo-PROTACs, which take advantage of light to regulate protein degradation, can cause damage to human. Folate-PROTACs and CLIPTACs are controllable targets for protein degradation, achieving low side effects but are also limited in application. In this regard, HaloPROTACs and degraders from the dTAG system only elicit fusion proteins. Moreover, although trivalent degraders obtain stronger degradation to target proteins, the difficulty of synthesis also interferes with their further development.
In addition, monovalent degraders have lower molecular weight than bivalent and trivalent degraders, thus, possessing better physicochemical properties in vivo, which could be used for the treatment of diseases of the central nervous system. However, monovalent drugs like molecular glues cannot target specific proteins and do not have systematic design strategies, in this regard, limiting the efficiency and applicability of molecular glue discovery. ATTECs, which could target intracellular proteins while binding only to LC3 fragments, narrow the scope of development. Taken together, the benefits outweigh the disadvantages. Owing to the specificity of different targeted protein degradation technologies, further development and application should be directed to drugs required according to current demand.
For the above problems, the vast majority of efforts are considered urgent. Exploitation of PROTACs should focus on broadening the scope of E3 ligases through screening techniques, developing undruggable targets and finding the optimal linker. Of course, from our point of view, we need to consider these three parts in a comprehensive manner. There is no doubt that small-molecule PROTACs still have broad and promising development prospects in the future. In addition, although peptide-based PROTACs have poor membrane permeability, the high specificity and low toxicity have driven researchers to explore novel strategy. peptide PROTACs combined with cell-penetrating peptides, constrained conformation technique, and targeted delivery systems could be the future efforts. Based on a paper published in 2020, the CDK inhibitor, CR8-a, acted as a molecular glue to degradation protein via UPS according to its solvent-exposed pyridyl moiety [101]. This strategy can be further studied in the research progress of molecular glues. The molecular glue is simple to make since it has a small molecular weight. In addition, clinical trials have begun on current molecular glue agents. Furthermore, SERDs and SARDs due to their high degradation and binding specificity are expected to operate on the mechanism exploration. And then, other new technologies can be applied in a specific area. CLIPTAC technology is expected to be applied in other inhibitor-protein systems because successful use of linkers that can occur “click reaction” may effectively deplete proteins. We have received great achievements from TPD technologies, such as benefiting from the ADC drugs, antibody-PROTACs are designed, achieving in vivo and in vitro antitumor activity. Then, we all know, controlling the activity of small-molecule probes with higher accuracy has always been the goal of drug discovery and development. The caged PROTACs such as ha-PROTACs, photo-PROTACs, and folate-PROTACs can be incorporated to treat localized diseases.. In addition, AUTACs, ATTECs, and LYTACs have emerged in recent years, which can deplete intracellular proteins including organelle, transmembrane protein, etc. [94, 96]. Since the ubiquitination-proteasome process of PROTAC requires substrates and ubiquitin molecules to be in the proper position and distance, exploiting degradation technologies based on the lysosome pathway may be less complicated [93]. These technologies are developing vigorously and will bring us more fruits in the future. More studies and efforts should be carried out to explore degradation model, clarify structure–activity relationship and so on.
Subsequently, TPD technology not only provides new insights for drug research and development but also provides new choices for the treatment of diseases. At present, it has been successfully used in the therapeutic research of many diseases, including cancer, immune diseases, neurodegenerative diseases, and viral diseases. Also, studies should be further explored on the premise of existing research.
TPD agents have made some achievements that we cannot ignore. Above all, these approaches have the potential to greatly promote the development of targeted therapy drugs. It is of great importance to continuously develop TPD technology with the joint efforts of researchers.
References
Wu T, Yoon H, Xiong Y, Dixon-Clarke SE, Nowak RP, Fischer ES (2020) Targeted protein degradation as a powerful research tool in basic biology and drug target discovery. Nat Struct Mol Biol 27:605–614. https://doi.org/10.1038/s41594-020-0438-0
Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, Sabatini DM (2015) Identification and characterization of essential genes in the human genome. Science (New York) 350:1096–1101. https://doi.org/10.1126/science.aac7041
An S, Fu L (2018) Small-molecule PROTACs: an emerging and promising approach for the development of targeted therapy drugs. EBioMedicine 36:553–562. https://doi.org/10.1016/j.ebiom.2018.09.005
Sun X, Gao H, Yang Y, He M, Wu Y, Song Y, Tong Y, Rao Y (2019) PROTACs: great opportunities for academia and industry. Signal Transduct Target Ther 4:64. https://doi.org/10.1038/s41392-019-0101-6
Han X, Zhao L, Xiang W, Qin C, Miao B, McEachern D, Wang Y, Metwally H, Wang L, Matvekas A, Wen B, Sun D, Wang S (2021) Strategies toward discovery of potent and orally bioavailable proteolysis targeting chimera degraders of androgen receptor for the treatment of prostate cancer. J Med Chem 64:12831–12854. https://doi.org/10.1021/acs.jmedchem.1c00882
Xiang W, Zhao L, Han X, Qin C, Miao B, McEachern D, Wang Y, Metwally H, Kirchhoff PD, Wang L, Matvekas A, He M, Wen B, Sun D, Wang S (2021) Discovery of ARD-2585 as an exceptionally potent and orally active PROTAC degrader of androgen receptor for the treatment of advanced prostate cancer. J Med Chem 64:13487–13509. https://doi.org/10.1021/acs.jmedchem.1c00900
Bueno C, Velasco-Hernandez T, Gutierrez-Aguera F, Zanetti SR, Baroni ML, Sanchez-Martinez D, Molina O, Closa A, Agraz-Doblas A, Marin P, Eyras E, Varela I, Menendez P (2019) CD133-directed CAR T-cells for MLL leukemia: on-target, off-tumor myeloablative toxicity. Leukemia 33:2090–2125. https://doi.org/10.1038/s41375-019-0418-8
Bekes M, Langley DR, Crews CM (2022) PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov 21:181–200. https://doi.org/10.1038/s41573-021-00371-6
Barghout SH (2022) New frontiers in the discovery and development of PROTACs. Anticancer Agents Med Chem. https://doi.org/10.2174/1871520622666220412132759
Alabi SB, Crews CM (2021) Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. J Biol Chem 296:100647. https://doi.org/10.1016/j.jbc.2021.100647
Wang C, Zheng C, Wang H, Zhang L, Liu Z, Xu P (2022) The state of the art of PROTAC technologies for drug discovery. Eur J Med Chem 235:114290. https://doi.org/10.1016/j.ejmech.2022.114290
Pickart CM (2001) Mechanisms underlying ubiquitination. Annu Rev Biochem 70:503–533. https://doi.org/10.1146/annurev.biochem.70.1.503
Raina K, Crews CM (2017) Targeted protein knockdown using small molecule degraders. Curr Opin Chem Biol 39:46–53. https://doi.org/10.1016/j.cbpa.2017.05.016
Schneekloth AR, Pucheault M, Tae HS, Crews CM (2008) Targeted intracellular protein degradation induced by a small molecule: en route to chemical proteomics. Bioorg Med Chem Lett 18:5904–5908. https://doi.org/10.1016/j.bmcl.2008.07.114
Hines J, Lartigue S, Dong H, Qian Y, Crews CM (2019) MDM2-recruiting PROTAC offers superior, synergistic antiproliferative activity via simultaneous degradation of BRD4 and stabilization of p53. Cancer Res 79:251–262. https://doi.org/10.1158/0008-5472.CAN-18-2918
Wang S, Zhao Y, Aguilar A, Bernard D, Yang CY (2017) Targeting the MDM2-p53 protein-protein interaction for new cancer therapy: progress and challenges. Cold Spring Harb Perspect Med. https://doi.org/10.1101/cshperspect.a026245
Han X, Wei W, Sun Y (2022) PROTAC degraders with ligands recruiting MDM2 E3 ubiquitin ligase: an updated perspective. Acta Materia Medica. https://doi.org/10.15212/amm-2022-0010
Rathore R, McCallum JE, Varghese E, Florea AM, Busselberg D (2017) Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis 22:898–919. https://doi.org/10.1007/s10495-017-1375-1
Itoh Y, Ishikawa M, Naito M, Hashimoto Y (2010) Protein knockdown using methyl bestatin-ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J Am Chem Soc 132:5820–5826. https://doi.org/10.1021/ja100691p
Demizu Y, Shibata N, Hattori T, Ohoka N, Motoi H, Misawa T, Shoda T, Naito M, Kurihara M (2016) Development of BCR-ABL degradation inducers via the conjugation of an imatinib derivative and a cIAP1 ligand. Bioorg Med Chem Lett 26:4865–4869. https://doi.org/10.1016/j.bmcl.2016.09.041
Shibata N, Nagai K, Morita Y, Ujikawa O, Ohoka N, Hattori T, Koyama R, Sano O, Imaeda Y, Nara H, Cho N, Naito M (2018) Development of protein degradation inducers of androgen receptor by conjugation of androgen receptor ligands and inhibitor of apoptosis protein ligands. J Med Chem 61:543–575. https://doi.org/10.1021/acs.jmedchem.7b00168
Wang C, Zhang Y, Shi L, Yang S, Chang J, Zhong Y, Li Q, Xing D (2022) Recent advances in IAP-based PROTACs (SNIPERs) as potential therapeutic agents. J Enzyme Inhib Med Chem 37:1437–1453. https://doi.org/10.1080/14756366.2022.2074414
Li K, Crews CM (2022) PROTACs: past, present and future. Chem Soc Rev 51:5214–5236. https://doi.org/10.1039/d2cs00193d
Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, Noblin DJ, Jorgensen WL, Ciulli A, Crews CM (2012) Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. J Am Chem Soc 134:4465–4468. https://doi.org/10.1021/ja209924v
Bondeson DP, Mares A, Smith IE, Ko E, Campos S, Miah AH, Mulholland KE, Routly N, Buckley DL, Gustafson JL, Zinn N, Grandi P, Shimamura S, Bergamini G, Faelth-Savitski M, Bantscheff M, Cox C, Gordon DA, Willard RR, Flanagan JJ, Casillas LN, Votta BJ, den Besten W, Famm K, Kruidenier L, Carter PS, Harling JD, Churcher I, Crews CM (2015) Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol 11:611–617. https://doi.org/10.1038/nchembio.1858
Burslem GM, Smith BE, Lai AC, Jaime-Figueroa S, McQuaid DC, Bondeson DP, Toure M, Dong H, Qian Y, Wang J, Crew AP, Hines J, Crews CM (2018) The advantages of targeted protein degradation over inhibition: an RTK case study. Cell Chem Biol 25:67–7763. https://doi.org/10.1016/j.chembiol.2017.09.009
Testa A, Hughes SJ, Lucas X, Wright JE, Ciulli A (2020) Structure-based design of a macrocyclic PROTAC. Angew Chem Int Ed Engl 59:1727–1734. https://doi.org/10.1002/anie.201914396
Song X, Zhong H, Qu X, Yang L, Jiang B (2020) Two novel strategies to overcome the resistance to ALK tyrosine kinase inhibitor drugs: Macrocyclic inhibitors and proteolysis-targeting chimeras. MedComm 2(2021):341–350. https://doi.org/10.1002/mco2.42
Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, Yamaguchi Y, Handa H (2010) Identification of a primary target of thalidomide teratogenicity. Science (New York) 327:1345–1350. https://doi.org/10.1126/science.1177319
Sun Y, Zhao X, Ding N, Gao H, Wu Y, Yang Y, Zhao M, Hwang J, Song Y, Liu W, Rao Y (2018) PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res 28:779–781. https://doi.org/10.1038/s41422-018-0055-1
Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE (2015) DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science (New York) 348:1376–1381. https://doi.org/10.1126/science.aab1433
Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, Crews CM (2015) Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem Biol 22:755–763. https://doi.org/10.1016/j.chembiol.2015.05.009
Dale B, Cheng M, Park KS, Kaniskan HU, Xiong Y, Jin J (2021) Advancing targeted protein degradation for cancer therapy. Nat Rev Cancer 21:638–654. https://doi.org/10.1038/s41568-021-00365-x
Ma Y, Frutos-Beltran E, Kang D, Pannecouque C, De Clercq E, Menendez-Arias L, Liu X, Zhan P (2021) Medicinal chemistry strategies for discovering antivirals effective against drug-resistant viruses. Chem Soc Rev 50:4514–4540. https://doi.org/10.1039/d0cs01084g
Zoppi V, Hughes SJ, Maniaci C, Testa A, Gmaschitz T, Wieshofer C, Koegl M, Riching KM, Daniels DL, Spallarossa A, Ciulli A (2019) Iterative design and optimization of initially inactive proteolysis targeting chimeras (PROTACs) identify VZ185 as a potent, fast, and selective von hippel-lindau (VHL) based dual degrader probe of BRD9 and BRD7. J Med Chem 62:699–726. https://doi.org/10.1021/acs.jmedchem.8b01413
Zhou F, Chen L, Cao C, Yu J, Luo X, Zhou P, Zhao L, Du W, Cheng J, Xie Y, Chen Y (2020) Development of selective mono or dual PROTAC degrader probe of CDK isoforms. Eur J Med Chem 187:111952. https://doi.org/10.1016/j.ejmech.2019.111952
Niu T, Li K, Jiang L, Zhou Z, Hong J, Chen X, Dong X, He Q, Cao J, Yang B, Zhu CL (2022) Noncovalent CDK12/13 dual inhibitors-based PROTACs degrade CDK12-Cyclin K complex and induce synthetic lethality with PARP inhibitor. Eur J Med Chem 228:114012. https://doi.org/10.1016/j.ejmech.2021.114012
Sinatra L, Bandolik JJ, Roatsch M, Sonnichsen M, Schoeder CT, Hamacher A, Scholer A, Borkhardt A, Meiler J, Bhatia S, Kassack MU, Hansen FK (2020) Hydroxamic Acids Immobilized on Resins (HAIRs): synthesis of dual-targeting HDAC inhibitors and HDAC degraders (PROTACs). Angew Chem Int Ed Engl 59:22494–22499. https://doi.org/10.1002/anie.202006725
Zhang X, Thummuri D, Liu X, Hu W, Zhang P, Khan S, Yuan Y, Zhou D, Zheng G (2020) Discovery of PROTAC BCL-X(L) degraders as potent anticancer agents with low on-target platelet toxicity. Eur J Med Chem 192:112186. https://doi.org/10.1016/j.ejmech.2020.112186
Lv D, Pal P, Liu X, Jia Y, Thummuri D, Zhang P, Hu W, Pei J, Zhang Q, Zhou S, Khan S, Zhang X, Hua N, Yang Q, Arango S, Zhang W, Nayak D, Olsen SK, Weintraub ST, Hromas R, Konopleva M, Yuan Y, Zheng G, Zhou D (2021) Development of a BCL-xL and BCL-2 dual degrader with improved anti-leukemic activity. Nat Commun 12:6896. https://doi.org/10.1038/s41467-021-27210-x
Yan J, Li T, Miao Z, Wang P, Sheng C, Zhuang C (2022) Homobivalent, trivalent, and covalent PROTACs: emerging strategies for protein degradation. J Med Chem 65:8798–8827. https://doi.org/10.1021/acs.jmedchem.2c00728
Maniaci C, Hughes SJ, Testa A, Chen W, Lamont DJ, Rocha S, Alessi DR, Romeo R, Ciulli A (2017) Homo-PROTACs: bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat Commun 8:830. https://doi.org/10.1038/s41467-017-00954-1
Girardini M, Maniaci C, Hughes SJ, Testa A, Ciulli A (2019) Cereblon versus VHL: Hijacking E3 ligases against each other using PROTACs. Bioorg Med Chem 27:2466–2479. https://doi.org/10.1016/j.bmc.2019.02.048
Zheng M, Huo J, Gu X, Wang Y, Wu C, Zhang Q, Wang W, Liu Y, Liu Y, Zhou X, Chen L, Zhou Y, Li H (2021) Rational design and synthesis of novel dual PROTACs for simultaneous degradation of EGFR and PARP. J Med Chem 64:7839–7852. https://doi.org/10.1021/acs.jmedchem.1c00649
Imaide S, Riching KM, Makukhin N, Vetma V, Whitworth C, Hughes SJ, Trainor N, Mahan SD, Murphy N, Cowan AD, Chan KH, Craigon C, Testa A, Maniaci C, Urh M, Daniels DL, Ciulli A (2021) Trivalent PROTACs enhance protein degradation via combined avidity and cooperativity. Nat Chem Biol 17:1157–1167. https://doi.org/10.1038/s41589-021-00878-4
Huang Y, Yokoe H, Kaiho-Soma A, Takahashi K, Hirasawa Y, Morita H, Ohtake F, Kanoh N (2022) Design, synthesis, and evaluation of trivalent PROTACs Having a functionalization site with controlled orientation. Bioconjug Chem 33:142–151. https://doi.org/10.1021/acs.bioconjchem.1c00490
Guenette RG, Yang SW, Min J, Pei B, Potts PR (2022) Target and tissue selectivity of PROTAC degraders. Chem Soc Rev 51:5740–5756. https://doi.org/10.1039/d2cs00200k
Nguyen KM, Busino L (2020) Targeting the E3 ubiquitin ligases DCAF15 and cereblon for cancer therapy. Semin Cancer Biol 67:53–60. https://doi.org/10.1016/j.semcancer.2020.03.007
Zhang X, Crowley VM, Wucherpfennig TG, Dix MM, Cravatt BF (2019) Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat Chem Biol 15:737–746. https://doi.org/10.1038/s41589-019-0279-5
Spradlin JN, Hu X, Ward CC, Brittain SM, Jones MD, Ou L, To M, Proudfoot A, Ornelas E, Woldegiorgis M, Olzmann JA, Bussiere DE, Thomas JR, Tallarico JA, McKenna JM, Schirle M, Maimone TJ, Nomura DK (2019) Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat Chem Biol 15:747–755. https://doi.org/10.1038/s41589-019-0304-8
Ward CC, Kleinman JI, Brittain SM, Lee PS, Chung CYS, Kim K, Petri Y, Thomas JR, Tallarico JA, McKenna JM, Schirle M, Nomura DK (2019) Covalent ligand screening uncovers a RNF4 E3 ligase recruiter for targeted protein degradation applications. ACS Chem Biol 14:2430–2440. https://doi.org/10.1021/acschembio.8b01083
Wei J, Meng F, Park KS, Yim H, Velez J, Kumar P, Wang L, Xie L, Chen H, Shen Y, Teichman E, Li D, Wang GG, Chen X, Kaniskan HU, Jin J (2021) Harnessing the E3 Ligase KEAP1 for targeted protein degradation. J Am Chem Soc 143:15073–15083. https://doi.org/10.1021/jacs.1c04841
Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ (2001) Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci USA 98:8554–8559. https://doi.org/10.1073/pnas.141230798
Schneekloth JS Jr, Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, Crews CM (2004) Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc 126:3748–3754. https://doi.org/10.1021/ja039025z
Bai L, Zhou H, Xu R, Zhao Y, Chinnaswamy K, McEachern D, Chen J, Yang CY, Liu Z, Wang M, Liu L, Jiang H, Wen B, Kumar P, Meagher JL, Sun D, Stuckey JA, Wang S (2019) A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell 36:498-511 e417. https://doi.org/10.1016/j.ccell.2019.10.002
Li X, Pu W, Zheng Q, Ai M, Chen S, Peng Y (2022) Proteolysis-targeting chimeras (PROTACs) in cancer therapy. Mol Cancer 21:99. https://doi.org/10.1186/s12943-021-01434-3
Jin J, Wu Y, Chen J, Shen Y, Zhang L, Zhang H, Chen L, Yuan H, Chen H, Zhang W, Luan X (2020) The peptide PROTAC modality: a novel strategy for targeted protein ubiquitination. Theranostics 10:10141–10153. https://doi.org/10.7150/thno.46985
Ghidini A, Clery A, Halloy F, Allain FHT, Hall J (2021) RNA-PROTACs: degraders of RNA-binding proteins. Angew Chem Int Ed Engl 60:3163–3169. https://doi.org/10.1002/anie.202012330
Samarasinghe KTG, Jaime-Figueroa S, Burgess M, Nalawansha DA, Dai K, Hu Z, Bebenek A, Holley SA, Crews CM (2021) Targeted degradation of transcription factors by TRAFTACs: TRAnscription Factor TArgeting Chimeras. Cell Chem Biol 28:648-661 e645. https://doi.org/10.1016/j.chembiol.2021.03.011
Shao J, Yan Y, Ding D, Wang D, He Y, Pan Y, Yan W, Kharbanda A, Li HY, Huang H (2021) Destruction of DNA-Binding Proteins by Programmable Oligonucleotide PROTAC (O’PROTAC): Effective Targeting of LEF1 and ERG. Adv Sci (Weinh) 8:e2102555. https://doi.org/10.1002/advs.202102555
Zhang J, Che J, Luo X, Wu M, Kan W, Jin Y, Wang H, Pang A, Li C, Huang W, Zeng S, Zhuang W, Wu Y, Xu Y, Zhou Y, Li J, Dong X (2022) Structural feature analyzation strategies toward discovery of orally bioavailable PROTACs of Bruton’s tyrosine kinase for the treatment of lymphoma. J Med Chem 65:9096–9125. https://doi.org/10.1021/acs.jmedchem.2c00324
Scheepstra M, Hekking KFW, van Hijfte L, Folmer RHA (2019) Bivalent ligands for protein degradation in drug discovery. Comput Struct Biotechnol J 17:160–176. https://doi.org/10.1016/j.csbj.2019.01.006
Cao C, He M, Wang L, He Y, Rao Y (2022) Chemistries of bifunctional PROTAC degraders. Chem Soc Rev 51:7066–7114. https://doi.org/10.1039/d2cs00220e
Steinebach C, Ng YLD, Sosic I, Lee CS, Chen S, Lindner S, Vu LP, Bricelj A, Haschemi R, Monschke M, Steinwarz E, Wagner KG, Bendas G, Luo J, Gutschow M, Kronke J (2020) Systematic exploration of different E3 ubiquitin ligases: an approach towards potent and selective CDK6 degraders. Chem Sci 11:3474–3486. https://doi.org/10.1039/d0sc00167h
Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AM, Wang J, Chen X, Dong H, Siu K, Winkler JD, Crew AP, Crews CM, Coleman KG (2016) PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci USA 113:7124–7129. https://doi.org/10.1073/pnas.1521738113
Cornella-Taracido I, Garcia-Echeverria C (2020) Monovalent protein-degraders—Insights and future perspectives. Bioorg Med Chem Lett 30:127202. https://doi.org/10.1016/j.bmcl.2020.127202
Ramachandran S, Ciulli A (2021) Building ubiquitination machineries: E3 ligase multi-subunit assembly and substrate targeting by PROTACs and molecular glues. Curr Opin Struct Biol 67:110–119. https://doi.org/10.1016/j.sbi.2020.10.009
Dong G, Ding Y, He S, Sheng C (2021) Molecular glues for targeted protein degradation: from serendipity to rational discovery. J Med Chem 64:10606–10620. https://doi.org/10.1021/acs.jmedchem.1c00895
Gandhi AK, Kang J, Havens CG, Conklin T, Ning Y, Wu L, Ito T, Ando H, Waldman MF, Thakurta A, Klippel A, Handa H, Daniel TO, Schafer PH, Chopra R (2014) Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN). Br J Haematol 164:811–821. https://doi.org/10.1111/bjh.12708
Han T, Goralski M, Gaskill N, Capota E, Kim J, Ting TC, Xie Y, Williams NS, Nijhawan D (2017) Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science (New York). https://doi.org/10.1126/science.aal3755
Hanan EJ, Liang J, Wang X, Blake RA, Blaquiere N, Staben ST (2020) Monomeric targeted protein degraders. J Med Chem 63:11330–11361. https://doi.org/10.1021/acs.jmedchem.0c00093
Lu Z, Cao Y, Zhang D, Meng X, Guo B, Kong D, Yang Y (2022) Discovery of thieno[2,3-e]indazole derivatives as novel oral selective estrogen receptor degraders with highly improved antitumor effect and favorable druggability. J Med Chem 65:5724–5750. https://doi.org/10.1021/acs.jmedchem.2c00008
Loddick SA, Ross SJ, Thomason AG, Robinson DM, Walker GE, Dunkley TP, Brave SR, Broadbent N, Stratton NC, Trueman D, Mouchet E, Shaheen FS, Jacobs VN, Cumberbatch M, Wilson J, Jones RD, Bradbury RH, Rabow A, Gaughan L, Womack C, Barry ST, Robson CN, Critchlow SE, Wedge SR, Brooks AN (2013) AZD3514: a small molecule that modulates androgen receptor signaling and function in vitro and in vivo. Mol Cancer Ther 12:1715–1727. https://doi.org/10.1158/1535-7163.MCT-12-1174
He Y, Hwang DJ, Ponnusamy S, Thiyagarajan T, Mohler ML, Narayanan R, Miller DD (2021) Exploration and biological evaluation of basic heteromonocyclic propanamide derivatives as SARDs for the treatment of enzalutamide-resistant prostate cancer. J Med Chem 64:11045–11062. https://doi.org/10.1021/acs.jmedchem.1c00439
Shi S, Du Y, Zou Y, Niu J, Cai Z, Wang X, Qiu F, Ding Y, Yang G, Wu Y, Xu Y, Zhu Q (2022) Rational Design for Nitroreductase (NTR)-Responsive Proteolysis Targeting Chimeras (PROTACs) selectively targeting tumor tissues. J Med Chem 65:5057–5071. https://doi.org/10.1021/acs.jmedchem.1c02221
Moreau K, Coen M, Zhang AX, Pachl F, Castaldi MP, Dahl G, Boyd H, Scott C, Newham P (2020) Proteolysis-targeting chimeras in drug development: a safety perspective. Br J Pharmacol 177:1709–1718. https://doi.org/10.1111/bph.15014
Borad MJ, Reddy SG, Bahary N, Uronis HE, Sigal D, Cohn AL, Schelman WR, Stephenson J Jr, Chiorean EG, Rosen PJ, Ulrich B, Dragovich T, Del Prete SA, Rarick M, Eng C, Kroll S, Ryan DP (2015) Randomized phase II trial of gemcitabine plus TH-302 versus gemcitabine in patients with advanced pancreatic cancer. J Clin Oncol 33:1475–1481. https://doi.org/10.1200/JCO.2014.55.7504
Palmer BD, van Zijl P, Denny WA, Wilson WR (1995) Reductive chemistry of the novel hypoxia-selective cytotoxin 5-[N, N-bis(2-chloroethyl)amino]-2,4-dinitrobenzamide. J Med Chem 38:1229–1241. https://doi.org/10.1021/jm00007a019
Cheng W, Li S, Wen X, Han S, Wang S, Wei H, Song Z, Wang Y, Tian X, Zhang X (2021) Development of hypoxia-activated PROTAC exerting a more potent effect in tumor hypoxia than in normoxia. Chem Commun (Camb) 57:12852–12855. https://doi.org/10.1039/d1cc05715d
Xue G, Wang K, Zhou D, Zhong H, Pan Z (2019) Light-induced protein degradation with photocaged PROTACs. J Am Chem Soc 141:18370–18374. https://doi.org/10.1021/jacs.9b06422
Jin YH, Lu MC, Wang Y, Shan WX, Wang XY, You QD, Jiang ZY (2020) Azo-PROTAC: novel light-controlled small-molecule tool for protein knockdown. J Med Chem 63:4644–4654. https://doi.org/10.1021/acs.jmedchem.9b02058
Lebraud H, Wright DJ, Johnson CN, Heightman TD (2016) Protein degradation by in-cell self-assembly of proteolysis targeting chimeras. ACS Cent Sci 2:927–934. https://doi.org/10.1021/acscentsci.6b00280
Buckley DL, Raina K, Darricarrere N, Hines J, Gustafson JL, Smith IE, Miah AH, Harling JD, Crews CM (2015) HaloPROTACS: use of small molecule PROTACs to induce degradation of halotag fusion proteins. ACS Chem Biol 10:1831–1837. https://doi.org/10.1021/acschembio.5b00442
Nabet B, Ferguson FM, Seong BKA, Kuljanin M, Leggett AL, Mohardt ML, Robichaud A, Conway AS, Buckley DL, Mancias JD, Bradner JE, Stegmaier K, Gray NS (2020) Rapid and direct control of target protein levels with VHL-recruiting dTAG molecules. Nat Commun 11:4687. https://doi.org/10.1038/s41467-020-18377-w
Prozzillo Y, Fattorini G, Santopietro MV, Suglia L, Ruggiero A, Ferreri D, Messina G (2020) Targeted protein degradation tools: overview and future perspectives. Biology (Basel). https://doi.org/10.3390/biology9120421
Clift D, McEwan WA, Labzin LI, Konieczny V, Mogessie B, James LC, Schuh M (2017) A method for the acute and rapid degradation of endogenous proteins. Cell 171:1692-1706 e1618. https://doi.org/10.1016/j.cell.2017.10.033
Clift D, So C, McEwan WA, James LC, Schuh M (2018) Acute and rapid degradation of endogenous proteins by Trim-Away. Nat Protoc 13:2149–2175. https://doi.org/10.1038/s41596-018-0028-3
Liu J, Chen H, Liu Y, Shen Y, Meng F, Kaniskan HU, Jin J, Wei W (2021) Cancer selective target degradation by folate-caged PROTACs. J Am Chem Soc 143:7380–7387. https://doi.org/10.1021/jacs.1c00451
Maneiro MA, Forte N, Shchepinova MM, Kounde CS, Chudasama V, Baker JR, Tate EW (2020) Antibody-PROTAC conjugates enable HER2-dependent targeted protein degradation of BRD4. ACS Chem Biol 15:1306–1312. https://doi.org/10.1021/acschembio.0c00285
Dragovich PS, Pillow TH, Blake RA, Sadowsky JD, Adaligil E, Adhikari P, Chen J, Corr N, Dela Cruz-Chuh J, Del Rosario G, Fullerton A, Hartman SJ, Jiang F, Kaufman S, Kleinheinz T, Kozak KR, Liu L, Lu Y, Mulvihill MM, Murray JM, O’Donohue A, Rowntree RK, Sawyer WS, Staben LR, Wai J, Wang J, Wei B, Wei W, Xu Z, Yao H, Yu SF, Zhang D, Zhang H, Zhang S, Zhao Y, Zhou H, Zhu X (2021) Antibody-mediated delivery of chimeric BRD4 degraders. Part 2: improvement of in vitro antiproliferation activity and in vivo antitumor efficacy. J Med Chem 64:2576–2607. https://doi.org/10.1021/acs.jmedchem.0c01846
Dragovich PS, Pillow TH, Blake RA, Sadowsky JD, Adaligil E, Adhikari P, Bhakta S, Blaquiere N, Chen J, Dela Cruz-Chuh J, Gascoigne KE, Hartman SJ, He M, Kaufman S, Kleinheinz T, Kozak KR, Liu L, Liu L, Liu Q, Lu Y, Meng F, Mulvihill MM, O’Donohue A, Rowntree RK, Staben LR, Staben ST, Wai J, Wang J, Wei B, Wilson C, Xin J, Xu Z, Yao H, Zhang D, Zhang H, Zhou H, Zhu X (2021) Antibody-mediated delivery of chimeric BRD4 degraders. Part 1: exploration of antibody linker, payload loading, and payload molecular properties. J Med Chem 64:2534–2575. https://doi.org/10.1021/acs.jmedchem.0c01845
Ji CH, Kwon YT (2017) Crosstalk and interplay between the ubiquitin-proteasome system and autophagy. Mol Cells 40:441–449. https://doi.org/10.14348/molcells.2017.0115
Ding Y, Xing D, Fei Y, Lu B (2022) Emerging degrader technologies engaging lysosomal pathways. Chem Soc Rev 51:8832–8876. https://doi.org/10.1039/d2cs00624c
Takahashi D, Moriyama J, Nakamura T, Miki E, Takahashi E, Sato A, Akaike T, Itto-Nakama K, Arimoto H (2019) AUTACs: cargo-specific degraders using selective autophagy. Mol Cell 76:797-810 e710. https://doi.org/10.1016/j.molcel.2019.09.009
Li Z, Wang C, Wang Z, Zhu C, Li J, Sha T, Ma L, Gao C, Yang Y, Sun Y, Wang J, Sun X, Lu C, Difiglia M, Mei Y, Ding C, Luo S, Dang Y, Ding Y, Fei Y, Lu B (2019) Allele-selective lowering of mutant HTT protein by HTT–LC3 linker compounds. Nature 575:203–209. https://doi.org/10.1038/s41586-019-1722-1
Banik SM, Pedram K, Wisnovsky S, Ahn G, Riley NM, Bertozzi CR (2020) Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 584:291–297. https://doi.org/10.1038/s41586-020-2545-9
Lv M, Hu W, Zhang S, He L, Hu C, Yang S (2022) Proteolysis-targeting chimeras: a promising technique in cancer therapy for gaining insights into tumor development. Cancer Lett 539:215716. https://doi.org/10.1016/j.canlet.2022.215716
Lu M, Liu T, Jiao Q, Ji J, Tao M, Liu Y, You Q, Jiang Z (2018) Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur J Med Chem 146:251–259. https://doi.org/10.1016/j.ejmech.2018.01.063
Chatterjee P, Ponnapati M, Kramme C, Plesa AM, Church GM, Jacobson JM (2020) Targeted intracellular degradation of SARS-CoV-2 via computationally optimized peptide fusions. Commun Biol 3:715. https://doi.org/10.1038/s42003-020-01470-7
Haniff HS, Tong Y, Liu X, Chen JL, Suresh BM, Andrews RJ, Peterson JM, O’Leary CA, Benhamou RI, Moss WN, Disney MD (2020) Targeting the SARS-CoV-2 RNA genome with small molecule binders and Ribonuclease Targeting Chimera (RIBOTAC) degraders. ACS Cent Sci 6:1713–1721. https://doi.org/10.1021/acscentsci.0c00984
Słabicki M, Kozicka Z, Petzold G, Li YD, Manojkumar M, Bunker RD, Donovan KA, Sievers QL, Koeppel J, Suchyta D, Sperling AS, Fink EC, Gasser JA, Wang LR, Corsello SM, Sellar RS, Jan M, Gillingham D, Scholl C, Fröhling S, Golub TR, Fischer ES, Thomä NH, Ebert BL (2020) The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 585:293–297. https://doi.org/10.1038/s41586-020-2374-x
Author information
Authors and Affiliations
Contributions
NY wrote the main manuscript text. BK, ZZ, and FH prepared the articles collection. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yang, N., Kong, B., Zhu, Z. et al. Recent advances in targeted protein degraders as potential therapeutic agents. Mol Divers 28, 309–333 (2024). https://doi.org/10.1007/s11030-023-10606-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-023-10606-w