
Abstract
Atrial fibrillation (AF) is an irregular heart rhythm, characterised by chaotic atrial activation, which is promoted by remodelling. Once initiated, AF can also propagate the progression of itself in the so-called ‘‘AF begets AF’’. Several lines of investigation have shown that signalling molecules, including reactive oxygen species, angiotensin II, and phosphoinositide 3-kinases (PI3Ks), in presence or absence of cardiovascular disease risk factors, stabilise and promote AF maintenance. In particular, reduced cardiac-specific PI3K activity that is not associated with oncology is cardiotoxic and increases susceptibility to AF. Atrial-specific PI3K(p110α) transgene can cause pathological atrial enlargement. Highlighting the crucial importance of the p110α protein in a clinical problem that currently challenges the professional health care practice, in over forty (40) transgenic mouse models of AF (Table1), currently existing, of which some of the models are models of human genetic disorders, including PI3K(p110α) transgenic mouse model, over 70% of them reporting atrial size showed enlarged, greater atrial size. Individuals with minimal to severely dilated atria develop AF more likely. Left atrial diameter and volume stratification are an assessment for follow-up surveillance to detect AF. Gene therapy to reduce atrial size will be associated with a reduction in AF burden. In this overview, PI3K(p110α), a master regulator of organ size, was investigated in atrial enlargement and in physiological determinants that promote AF.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
AF is an irregular heart rhythm marked by chaotic atrial activation (fibrillatory waves on the ECG and loss of p wave) associated with irregular ventricular activation. It is the most common cardiac arrhythmia, a major clinical health problem, and a growing epidemic that manifests as a mixed disorder. It has been associated with familial inheritance due to a genetic mutation [1], can occur as ‘‘orphan’’ or idiopathic AF, and has been related to other cardiovascular diseases, underlying structural heart diseases such as cardiomyopathy [2] and most commonly to other risk factors, such as ageing [3, 4].
The incidence and prevalence of AF are rising globally [5]. The ageing population is a critical factor. The lifetime risks for development of AF were 1 in 4 at 40 years of age and above, and in the absence of antecedent congestive heart failure or myocardial infarction, the lifetime risks were 1 in 6 [6], indicating heart failure and myocardial infarction as myocardial substrate for the development of AF. Other non-modifiable risk factors in addition to ageing include sex, genetics, and race [7]. AF risk factors can also be classified as modifiable [8], and common modifiable risk factors of AF include physical activity, diabetes, obesity [9], obstructive sleep apnoea [10], alcohol [11, 12], and smoking.
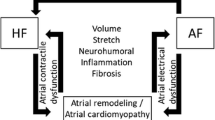
Although the precise cellular and molecular mechanisms of AF remain unclear, they are purported to involve both structural and electrical remodelling of the atria, induced by the risk factors, to maintain vulnerable atrial substrate [7]. AF formation requires a vulnerable substrate and an initiating trigger. Atrial fibrosis promotes AF perpetuation by promoting localised re-entry through slowed atrial conduction. Circus movement, leading circle, spiral wave, and multiple wavelets have all been proposed as conceptual modes of re-entrant arrhythmia. The clinical relevance of these concepts is still uncertain as their real-world application has yielded highly variable results.
Changes in the physioanatomical properties of the atria are termed atrial remodelling. Pathological stimuli and perturbation of signalling like phosphatidylinositol 3-kinase and catalytic subunit alpha (PI3K[p110α]) cause both structural and electrical remodelling of the atria. This process involves changes in protein expression, collagen deposition, abnormal Ca2+ handling and contractility, and changes in ion current densities Fig. 1; [13]. Pharmacological attenuation of PI3K(p110α) activity caused late sodium current (late INa) stimulation to induce enhanced organelle sarcoplasmic reticulum Ca2+ load and QT interval prolongation [14]. Moreover, cardiac-specific inhibition of PI3Kα robustly eliminated angiotensin II time-dependent cell shortening and changes in L-type Ca2+ currents effects [15]. This effect was specific and large enough to approximately 90% in an order of magnitude. Pathophysiological evidence supports the role of PI3K(p110α) activity in AF susceptibility, AF-associated risk factors, and the cellular and molecular mechanisms that promote AF progression and perpetuation [13]. An endogenous reduction in the activity of PI3K(p110α) on a background of mammalian sterile 20-like kinase 1 (Mst1) resulted in a more severe cardiac phenotype. The model had enlarged atrial diameter, changes in the expression of potassium channels and metabolism-related genes, left atrial thrombi, extracellular matrix deposition, and spontaneous AF [16].

Conceptualised Mechanisms of Atrial Fibrillation showing functional and structural components of remodelling that maintain AF. Re-entry requires abbreviated action potential duration (APD) and/or conduction abnormalities. Ectopic firing occurs due to early after depolarization, delay after depolarization, and action potential prolongation. Changes in autonomic nerve activation produces significant and heterogeneous changes of atrial electrophysiology. Structural remodelling can be atrial enlargement and/or fibrosis atrial enlargement determine persistent AF through re-entry. Fibrosis distorts cellular architecture, extracellular matrix composition, and physical integrity of the atria. Ca2+ handling cause DADs
Atrial enlargement is a clinical predictor of AF [17]. In about forty (40) transgenic and knockout mouse models of AF currently existing, of which some were created based upon knowledge gained from clinical mutation analysis of arrhythmias, including PI3K(p110α) transgenic mouse model, over 70% of them reporting atrial size showed enlarged, greater atrial size, or mass (Table 1). Several observational studies have identified increase in atrial size and cardiac stretch a mechanism of AF in humans [18,19,20]. Nonetheless, information is clearly required from further studies to elucidate the determinants of atrial enlargement, which are poorly understood – with a potential to increase our knowledge of pathophysiology of AF, and identify novel therapeutic targets. Starting with the types of AF and a model of remodelled atrial tissue, this review provides an overview of the potential roles of PI3K(p110α) gene, a molecular regulator of cell and organ size, in the induction of cardiac-specific pathological atrial enlargement and in physiological mechanisms of AF progression and maintenance.
Types of AF and a model of remodelled atrial tissue
AF has been studied for over a century and the mechanism is evolving. AF can be classified clinically into different types based on duration, frequency of episodes, and manifestation. This includes i) single episode or ‘lone’ AF, ii) paroxysmal, iii) persistent, iv) long-standing persistent, and v) permanent [21].
(i) First episode- original episode of AF previously undiagnosed regardless of presence and/or severity of AF-related symptoms.
(ii) Paroxysmal- AF that terminates spontaneously, usually within 48 h. However, some episodes may persist for up to 7 days.
(iii) Persistent AF- episodes that last beyond 7 days. This type of AF is generally not self-terminating.
(iv) Long-standing persistent- episodes that last for greater than one year.
(v) Permanent AF- describes AF that is not self-terminating and does not respond to treatment or medication.
It should be noted that these definitions to some extent represent an artificial characterisation of AF syndromes for the purposes of clinical categorization particularly in the context of clinical studies. In reality, there is a spectrum of AF phenotype severity ranging from ‘lone’ AF to permanent AF.
Symptoms of AF include palpitations, fatigue, psychosocial distress, breathing difficulties, chest tightness, sleeping difficulties, and poor quality of life [22,23,24].
The underlying mechanisms of AF are still incompletely understood. An important feature of AF is very rapid and chaotic atrial activation, which can be caused by re-entry activity or spontaneous foci ectopy. AF requires re-entry and focal ectopic trigger, predominately arising from the pulmonary veins [25]; however, non-pulmonary vein triggers are also well described [26]. The initiating triggers and re-entry in addition to vulnerable atrial substrate such as atrial enlargement perpetuate AF.
Physiological mechanisms of AF and PI3K(p110α)/class IA PI3K
In the remodelled model of atrial tissue and as a physiological process, ectopic triggers (repetitive depolarization) can be due to early after depolarisations (EADs) or delay after depolarisations (DADs). EADs occur at the plateau phase (phase 2) or phase 3 of action potential duration, whereas DADs occur at phase 4 of action potential repolarisation. Triggers are regarded as abnormal secondary repolarisations and occur when DADs or EADs reach the threshold potential. Whereas, EADs are believed to be caused by slowing of repolarization, DADs are known to be caused by abnormal diastolic Ca2+ release by ryanodine receptor 2 (RyR2).
Trigger is associated with the development of arrhythmias through alterations in action potentials (APs). Enhanced sympathetic tone increase the probability of EADs. To assess the possibility of PI3Ks-induced trigger, APs were measured at different pacing frequencies in presence of increased sympathetic tone with isoproterenol (ISO), under PI3K inhibition. Control canine myocytes exposed to ISO but not class IA PI3K inhibitors had no EADs, but a decrease in APD and AP plateau height compared with untreated cells. In contrast, in the presence of 50 nM or 500 nM PI-103, ISO induced EADs in the ventricular myocytes [27]. In contrast to Lu et al., we did not observe EADs either in wild-type or Akita right atrial myocytes with reduced PIP3 signalling, in the presence of 1 µM ISO [28]. Together, the atria have both parasympathetic and sympathetic innervations, unlike the ventricle, which could be offsetting the effect of each other. It could also be that sympathetic response of the atria may differ from that of the ventricle, and in the presence of enhance sympathetic response, direct inhibition of class IA PI3K (comprising a catalytic subunit PI3K(p110α), PI3K(p110β), or PI3K(p110δ) and a p85 regulatory subunit might predispose to arrhythmias. Indeed, experimental studies in preclinical models show the essential roles of PI3Kα in the regulation of Na+ channel activity, control of the arrhythmias, and cardiac safety [29]. Although, the specific role of PI3K(p110α)-induced trigger event remains to be investigated in better details, in reduced PI3K signalling and diabetes there was slow repolarisation in both the atria and ventricle [27, 28]. The electrophysiological feature, in part, anchors rotors and wave breaks fibrillatory activities in the presence of EADs in a mouse model with spontaneous and sustained AF and enhanced persistent Na + current due to a mutation in NaV1.5 channel [30].
Besides trigger, arrhythmia at tissue level is propagated by re-entry. The concepts of re-entrant mechanisms of AF have been proposed by the elegant of works of Garrey [31, 32], Moe [33], and amongst others [34, 35], to include circus movement, leading circle, spiral wave, and multiple wavelet. Detailed discussion of these concepts is beyond the scope of this work, but these examples support their role in arrhythmogenesis – particularly, wavelength shortening and reduced conduction velocity or refractory period are present in the enlarged and remodelled atria leading to sustained re-entrant-based tachycardias [36]. Atrial enlargement as a clinical correlate of AF helps to promote AF by favouring more wavelet formation [37]. Essentially, constitutive activation of PI3K(p110α) protein-induced cardiac hypertrophy [38] and cardiac hypertrophy induces atrial and ventricular arrhythmias [39], through alteration in cardiac ion channels. In particular, dominant negative PI3K(p110α) expression has been associated with greater atrial size [16].
PI3K(p110α) mediates atrial size and AF
Drosophila having PI3K(p110α) deficiency have small cells and organs [40]. Likewise, mice deficient for cardiac-specific PI3K(p110α) expression displayed small hearts, whereas those with enhanced cardiac-specific PI3K(p110α) expression displayed large hearts [41]. These data demonstrate the importance of the PI3K regulatory pathway in physiological cell and organ growth response in invertebrate and vertebrate animals.
Atrial hypertrophy is an important feature of adverse atrial remodelling as atria respond to pathological stimuli, such as myocardial stretch. Atrial dilation, as well as enlargement, is associated with AF ([42]; Table 1) and left atrial size is a known risk factor for the development atrial fibrillation [43]. Although, there is significant lack of literature on atrial hypertrophy and chamber-specific mechanisms of hypertrophy are largely unknown, atrial and ventricular hypertrophy may have comparable mechanisms. PI3K(p110α) is a key molecular regulator of cardiac size [41], through exercise (physiological) and aortic banding (pathological) [38]. Physiological and pathological hypertrophy due to PI3K(p110α) transgene are distinct based on molecular underpinnings. Whereas physiological hypertrophy is associated with normal function, pathological hypertrophic is associated with adverse effects. Pathological atrial hypertrophic remodelling is a multiplex process involving myofibroblast differentiation, cardiac myocyte growth, and loss of myofibril content [44].
Atrial enlargement [45] as well as fibrosis [46] are important players in AF progression. Left atrial diameter and volume stratification are an assessment for follow-up surveillance to detect AF in the clinics. Furthermore, mapping and removal of fibrotic areas and homogenisation of scares are currently emerging as rhythm control measures for AF patients. Pretorius et al. demonstrated atrial fibrosis and enlargement and increased susceptibility to AF in mice with reduced PI3K activity in the heart and Mst1 [16]. Combined assessment of left atrial fibrosis and size facilitates the identification of patients with better ablation success potential [47].
Although, atrial enlargement is an important mechanism of AF [48], the details of the molecular mechanisms of atrial size control and AF susceptibility are unknown. Atrial enlargement is part of the cellular remodelling that produces atrial substrate and AF and indicates elevated pressure and/or higher than normal blood volume in the atria. Bruton’s tyrosine kinase, a Tec family tyrosine kinase, an effector of PI3K activity, whose activation, in part, depends on the binding of PtdIns(3,4,5)P3 to the PH domain and is important for an enhanced intracellular Ca2+ signalling, caused AF in an off-target side effect, through atrial enlargement. A daily dose of selleckchem (ibrutinib), a non-specific Bruton tyrosine kinase inhibitor intraperitoneally injected for 4 weeks in mice, produced spontaneous AF, left atrial enlargement, myocardial fibrosis, and increased inflammation accompanied by prolonged atrial effective refractory periods without profound alteration in the action potential duration [49]. Although the effects were present in mice without Bruton tyrosine kinase, mice that received acalabrutinib, a specific Bruton tyrosine kinase inhibitor for 4 weeks had AF, showing an off-target side effect [49]. Chemoproteomic profiling of ibrutinib in cardiac tissue, where homogenised cardiac tissues were incubated with a biotinylated acylphosphate ATP derivative to transfer biotin to the conserved lysine residues in the ATP-binding pocket of protein kinases and other ATP-binding proteins for longer periods, identified BTK, proto-oncogene tyrosine-protein kinase (FYN), mitogen-activated protein kinase kinase 5 (MEK5), C-terminal Src kinase (CSK), and receptor-interacting serine/threonine kinase 3 as the potential targets of ibrutinib [49]. When the experiment was repeated with acalabrutinib, a second-generation ibrutinib, BTK and RIPK3 were rather identified as the targets [49]. These assessments when comparatively analysed by the authors limited the potential candidates of ibrutinib-associated AF-inducible targets to FYN, MEK5, and CSK. Consequent genetic manipulation of the three kinases in mice led to the final identification of Csk inhibition, as the mechanism of ibrutinib-associated AF, as cardiac-specific Csk knockout in mice, mimicking ibrutinib treatment predisposed to increased AF, left atrial enlargement, fibrosis, and inflammation [49]. PI3K(p110α) deficiency in mouse heterozygous for PI3K(p110α) transgene might reduce stress-induced dilation in dilated cardiomyopathy. Surprisingly, the double transgenic mouse model heterozygous for PI3K(p110α) on a background of Mst1 overexpression had AF and adverse atrial enlargement as assessed by echocardiography [16]. This is in contrast to the overexpression of PI3K(p110α) and Mst1 [16], suggesting a role for PI3K(p110α) heterozygous in atrial enlargement. To gain better insight into PI3K(p110α)-induced atrial greater size, a complete knowledge of the PI3K(p110α)-dosing effect in form of the heterozygous and homozygous transgene is required. This will improve the understanding of the likely critical roles of PI3K transgene in the control of atrial size, muscle mass, and atrial disease (Fig. 2).

A schematic of atrial hypertrophy, fibrosis, apoptosis, electrophysiological alterations, and insulin resistance as PI3K(p110α) transgene hypofunction phenotypes leading to irregular heartbeat, disease atria, and atrial fibrillation
Consistent to the upper chamber of the heart, PI3Kα-dominant negative mutant mice with heart failure pressure overload had dilated cardiomyopathy, by increased gelsolin-mediated actin severing activities in vivo. Adult cardiac stretch in PI3Kα deficiency perturbed sarcomeric actin cytoskeleton. The actin remodelling from the biomechanical stress stimuli mechanotransduction was prevented by PIP3, produced upon PI3Kα activation in feedback response. The gelsolin-driven actin cytoskeletal remodelling (depolymerisation) in heart failure was mechanistically underlined by increased expression of atrial and beta natriuretic peptides and increased cross-sectional areas of cardiomyocytes and chamber dilation [50]. The profound pathology was attenuated at the PI3Kα mutant background deletion of gelsolin [50], a Ca2+-dependent protein that regulates the dynamics of actin filament assembly and organisation and extensively expressed in many tissues, including heart, brain, and immune cells. Hence, PI3Kα/PIP3 are negative regulators of gelsolin activity. Furthermore, in experimental myocardial infarction, PI3Kα activity necessitated endothelial cell and cardiomyocytes hypertrophic response [51]. In this setting, pharmacological ablation of PI3Kα led to worsened cardiac dysfunction, profound apoptosis and inflammation, and suppressed Akt/glycogen synthase kinase 3β/endothelial nitric oxide synthetase signalling, as well as hypertrophy, post-MI [51]. In cell-specific manner, genetic PI3Kα inhibition in endothelial cells reduced coronary blood vessel density and in cardiomyocytes resulted in moderate cardiac systolic dysfunction at baseline [51]. Although these findings are novel and counterintuitive to the concept of PI3Kα hypofunction in atrial mass and enlargement and reveal potential PI3Kα inhibition cardiotoxicity, notably, cardiac hypertrophic risk of PI3Kα is dose dependent of its activity, relies on cell-specific communication effects and paracrine signalling, and has not been completely deciphered in better details.
Molecular mechanism of PI3K(P110α)-induced atrial enlargement
Several elegant studies show that cardiac cells require active PI3K/Akt signalling to maintain proliferation. Mice homozygous for 110-kDa catalytic subunit isoform (Pik3cα), demonstrating loss of expression of PI3K(p110α), had embryonic lethality at day 9.5 due to a severe defect in the proliferative capacity of the embryo. The defect was demonstrated by the observation that the mouse embryonic fibroblasts from the explants of PI3K(p110α) homozygous embryos but not those of wild-type and the PI3K(p110α) heterozygous embryos failed to replicate in Dulbecco’s modified Eagle’s medium and foetal calf serum, even with supplemental growth factors [52]. How dose-dependent (heterozygous and homozygous) effects of PI3K(p110α) may regulate atrial cells size leading to AF is unclear. We know that PI3K biological signalling network maintain cell viability and proliferation, reduce apoptosis, and respond to constantly changing external and internal conditions to maintain dynamic equilibrium state. When the signalling is adjusted by way of dosing, the network could be acutely or chronically altered. For instance, chronic stimulation of tissue-resident cells with growth factors can cause aberrant shift from resting to actively proliferating cells.
In response to growth factor receptor activation, PI3K(p110α) signalling begins leading to the synthesis of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) from phosphatidylinositol (4,5)-trisphosphate (PIP2) and translocation of Akt to cell membrane. Phosphorylation and activation of Akt leads to inactivation of tuberous sclerosis (TSC) 1 and 2 and activation of Ras homolog enriched in brain (Rheb) and the mammalian target of rapamycin (mTOR1). Through this process, PI3K and its downstream signalling effectors, such as Akt, PIP3, mammalian target of rapamycin, GSK3, and PDK1, regulate cell growth and survival [53]. It has been suggested that PI3K promotes cardiac cell proliferation through the inhibition of the GSK3 and mitogen-activated protein kinases/extracellular signal-regulated kinase (MAPK/ERK). This is consistent with the finding that PI3K signalling is accentuated during suppression of MAPK activation in stress-related growth of neonatal heart [54]. PI3K activation leads to inhibition of GSK3. Downstream GSK3 inhibition as a consequence leads to activation of D- and E-type cyclins, glycogen synthase, mTORC1, and nuclear factor of activated T-cells, a regulator of hypertrophy. GSK3 inactivation additionally occurs through p38. GSK3 regulates the canonical Wnt signalling. β-catenin activation by GSK3 results in ubiquitination and degradation of β-catenin by proteasome to stop gene expression. β-catenin is stable and translocates to the nucleus, when GSK3 is inhibited, resulting in gene expression. β-catenin modulates a host of events through fibroblast differentiation and fibrosis to cardiomyocyte hypertrophy (Fig. 3, right).

Mechanisms of PI3K(p110α)-induced atrial enlargement. Whereas, inactivation of PI3K would result in the nuclear accumulation of GSK3, and GSK3 inhibition by PI3K activation on the other hand or p38 can mediate β-catenin activity to regulate cell cycle activity, DNA content, and nucleation. Cell cycle activity, DNA content and nucleation, and mitochondria dysfunction that result from an initial molecular activity of a PI3K transgene hypofunction would be guided by post-translational modification and gene expression to myocyte growth, disarray, and fibrosis—phenotypical features and AF (right). PI3K(p110α)-induced atrial enlargement can also occur through Ang II-induced TGF-β1 release in enlargement cardiomyocytes to trigger paracrine signalling between the cardiomyocytes and fibroblast leading to proliferation of fibroblast and further enlargement, structural and morphological alteration before AF (left)
Furthermore, PI3K(p110α) transgene-induced hypertrophy is a feature of stretched cardiomyocytes and stretch as pathological stimuli results in angiotensin II (Ang II) release, which triggers the activation of transforming growth factor beta (TGF-β) [55], leading to a series of transcriptomic changes and post-translational modification and phenotype manifestation (Fig. 3, left). Cells interact with one another within their environment of occupation, through paracrine signalling that favours fibrosis. How the cells respond to the signalling may differ depending on cell type. For instance, Ang II-induced TGF-β release in cardiomyocyte may lead to cardiomyocyte death and hypertrophy, whereas in fibroblast, it may cause fibroblast proliferation. Cardiomyocyte death and hypertrophy and fibroblast proliferation lead to fibrosis and regeneration [56].
PI3K(p110α) as a target for prevention of atrial enlargement
Conventional therapy for cardiac arrhythmias is limited and it is time to think of biological therapies (gene therapy, cell therapy, or both), as an alternative to the present therapeutic regime, which rely on pharmacology or resource heavy interventional approaches [57]. Gene therapy is the use of genetic material to modify the genetic codes of the cell of the patient carrying inherited and/or acquired disease by transfer of genetic material into that cell for cure or to improve function. The genetic material can be transferred through nanoparticles, vectors, or plasmids to target specific traits of a disease. This approach may present advantage to AF management because the method can be tissue specific with minimal or no off-target effects. However, AF is a mixed disorder and single gene modification may not be insufficient even in the setting of a valid therapeutic target. Nonetheless, a single gene validated to have pleotropic effect on the numerous substrates for AF may alleviate the challenge and be a good choice for gene therapy for AF. The cardio protective role of PI3K(p110α) could be utilised to customise therapy for AF, particularly in this era of personalised medicine. Based on our molecular understanding of the atrial substrates and AF pathophysiology, gene therapy targets for AF include atrial enlargement, apoptosis, fibrosis, hyper innervation of the autonomic nervous system, ion channel, and gap junction alteration. As discussed above, constitutive PI3K(p110α) expression attenuates the targets to ensure cardio protection, highlighting a potential non-pharmacological relevance of a moderate dose of PI3K(p110α) gene in the pathological atrial remodelling. It is therefore mechanistically feasible that PI3K(p110α) gene may prevent atrial enlargement when identified as a risk factor, even before conventional treatment is required. The attractive potentials, nonetheless, drawbacks can be foreseen for a cardiac-targeted PI3K(p110α) gene therapy. PI3K-targeted gene therapy might be complicated with respect to impacts on genes of the targeted cells, delivery and activation, and immune system response. Although, drugs can be given to temporarily suppress the immune system response and lowest doses of effective viruses or viruses with reduced susceptibility to cause immune response can be used, it is still a concern—with a potential to cause debilitating illness or even death that immune systems fight to ward off foreign matters, such as bacteria and viruses, when introduced in the system. Introduced gene moulds itself to become a permanent part of an entire genome. This process can disrupt another gene or lead to an inappropriate location of the gene. Unguided delivery, activation, and integration of the PI3K gene to unspecific places of the genome can occur and would be carcinogenic. The role of PI3K in carcinogenesis is well known.
Conclusion
Pathological increase in atrial muscle size, otherwise known as ‘‘atrial enlargement’’ is a mechanism of AF. AF consequently induces atrial enlargement, suggesting a process through which AF promotes itself. Individuals with minimal to severely dilated atria may be more likely to develop AF than those with normal atrial size. A reduction in atrial size with gene therapy as a non-interventional therapy will be associated with a reduced AF burden. It will also be associated with primordial prevention of AF, suggesting huge potential in identify and treat risk factors (i.e. risk factor prevention), before the disease occurs. A better understanding of AF molecular mechanisms is required to improve treatment strategies and management of AF. Evidence for molecular mechanisms of PI3K(p110α)-induced atrial enlargement as a clinical correlate of AF is crucial, and studies elucidating cellular mechanisms of atrial enlargement are needed. Advancing our knowledge of the role of PI3K(p110α) gene in the symptoms, pathophysiology, AF-associated risk factors, and in the incidence of AF will help to provide new preventive and treatment measures and reduce the public health burden of AF. The works reviewed in this study highlight that PI3K(p110α) is very likely a master regulator of atrial size, yet its implications remain to be defined with respect to atrial size control and therapeutic strategies for AF management.
References
Hodgson-Zingman DM, Karst ML, Zingman LV, Heublein DM, Darbar D, Herron KJ et al (2008) Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N Engl J Med 359:158–165
Rowin EJ, Hausvater A, Link MS, Abt P, Gionfriddo W, Wang W et al (2017) Clinical profile and consequences of atrial fibrillation in hypertrophic cardiomyopathy. Circulation 136:2420–2436
Murphy NF, Simpson CR, Jhund PS, Stewart S, Kirkpatrick M, Chalmers J et al (2007) A national survey of the prevalence, incidence, primary care burden and treatment of atrial fibrillation in Scotland. Heart 93:606–612
Rodriguez CJ, Soliman EZ, Alonso A, Swett K, Okin PM, Goff DC Jr et al (2015) Atrial fibrillation incidence and risk factors in relation to race-ethnicity and the population attributable fraction of atrial fibrillation risk factors: the multi-ethnic study of atherosclerosis. Ann Epidemiol 25(71–6):6.e1
Lip GYH, Brechin CM, Lane DA (2012) The global burden of atrial fibrillation and stroke: a systematic review of the epidemiology of atrial fibrillation in regions outside North America and Europe. Chest 142:1489–1498
Lloyd-Jones DM, Wang TJ, Leip EP, Larson MG, Levy D, Vasan RS et al (2004) Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation 110:1042–1046
Staerk L, Sherer JA, Ko D, Benjamin EJ, Helm RH (2017) Atrial fibrillation: epidemiology, pathophysiology, and clinical outcomes. Circ Res 120:1501–1517
Lau DH, Nattel S, Kalman JM, Sanders P (2017) Modifiable risk factors and atrial fibrillation. Circulation 136:583–596
Abed HS, Samuel CS, Lau DH, Kelly DJ, Royce SG, Alasady M et al (2013) Obesity results in progressive atrial structural and electrical remodeling: implications for atrial fibrillation. Heart Rhythm 10:90–100
Dimitri H, Ng M, Brooks AG, Kuklik P, Stiles MK, Lau DH et al (2012) Atrial remodeling in obstructive sleep apnea: implications for atrial fibrillation. Heart Rhythm 9:321–327
Voskoboinik A, Costello BT, Kalman E, Prabhu S, Sugumar H, Wong G et al (2018) Regular alcohol consumption is associated with impaired atrial mechanical function in the atrial fibrillation population: a cross-sectional MRI-based study. JACC Clinical electrophysiology 4:1451–1459
Voskoboinik A, Prabhu S, Ling LH, Kalman JM, Kistler PM (2016) Alcohol and atrial fibrillation: a sobering review. J Am Coll Cardiol 68:2567–2576
Ezeani M, Prabhu S (2021) Pathophysiology and therapeutic relevance of PI3K(p110alpha) protein in atrial fibrillation: a non-interventional molecular therapy strategy. Pharmacol Res 165:105415
Zhabyeyev P, McLean B, Chen X, Vanhaesebroeck B, Oudit GY (2019) Inhibition of PI3Kinase-alpha is pro-arrhythmic and associated with enhanced late Na(+) current, contractility, and Ca(2+) release in murine hearts. J Mol Cell Cardiol 132:98–109
Liang W, Oudit GY, Patel MM, Shah AM, Woodgett JR, Tsushima RG et al (2010) Role of phosphoinositide 3-kinase {alpha}, protein kinase C, and L-type Ca2+ channels in mediating the complex actions of angiotensin II on mouse cardiac contractility. Hypertension 56:422–429
Pretorius L, Du XJ, Woodcock EA, Kiriazis H, Lin RC, Marasco S et al (2009) Reduced phosphoinositide 3-kinase (p110alpha) activation increases the susceptibility to atrial fibrillation. Am J Pathol 175:998–1009
Henry WL, Morganroth J, Pearlman AS, Clark CE, Redwood DR, Itscoitz SB et al (1976) Relation between echocardiographically determined left atrial size and atrial fibrillation. Circulation 53:273–279
Debonnaire P, Joyce E, Hiemstra Y, Mertens BJ, Atsma DE, Schalij MJ et al (2017) Left atrial size and function in hypertrophic cardiomyopathy patients and risk of new-onset atrial fibrillation. Circ Arrhythm Electrophysiol. https://doi.org/10.1161/CIRCEP.116.004052
Morillo CA, Klein GJ, Jones DL, Guiraudon CM (1995) Chronic rapid atrial pacing: structural, functional, and electrophysiological characteristics of a new model of sustained atrial fibrillation. Circulation 91:1588–1595
Rosenberg MA, Manning WJ (2012) Diastolic dysfunction and risk of atrial fibrillation: a mechanistic appraisal. Circulation 126:2353–2362
Charitos EI, Purerfellner H, Glotzer TV, Ziegler PD (2014) Clinical classifications of atrial fibrillation poorly reflect its temporal persistence: insights from 1,195 patients continuously monitored with implantable devices. J Am Coll Cardiol 63:2840–2848
Peinado R, Arribas F, Ormaetxe JM, Badia X (2010) Variation in quality of life with type of atrial fibrillation. Rev Esp Cardiol 63:1402–1409
Sears SF, Serber ER, Alvarez LG, Schwartzman DS, Hoyt RH, Ujhelyi MR (2005) Understanding atrial symptom reports: objective versus subjective predictors. Pacing Clin Electrophysiol 28:801–807
Steg PG, Alam S, Chiang CE, Gamra H, Goethals M, Inoue H et al (2012) Symptoms, functional status and quality of life in patients with controlled and uncontrolled atrial fibrillation: data from the RealiseAF cross-sectional international registry. Heart 98:195–201
Lin WS, Tai CT, Hsieh MH, Tsai CF, Lin YK, Tsao HM et al (2003) Catheter ablation of paroxysmal atrial fibrillation initiated by non-pulmonary vein ectopy. Circulation 107:3176–3183
Haissaguerre M, Jais P, Shah DC, Takahashi A, Hocini M, Quiniou G et al (1998) Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med 339:659–666
Lu Z, Wu CY, Jiang YP, Ballou LM, Clausen C, Cohen IS et al (2012) Suppression of phosphoinositide 3-kinase signaling and alteration of multiple ion currents in drug-induced long QT syndrome. Sci Transl Med. 4:131ra50
Polina I, Jansen HJ, Li T, Moghtadaei M, Bohne LJ, Liu Y et al (2020) Loss of insulin signaling may contribute to atrial fibrillation and atrial electrical remodeling in type 1 diabetes. Proc Natl Acad Sci USA 117:7990–8000
Zhabyeyev P, Chen X, Vanhaesebroeck B, Oudit GY (2019) PI3Kalpha in cardioprotection: Cytoskeleton, late Na(+) current, and mechanism of arrhythmias. Channels (Austin) 13:520–532
Avula UMR, Abrams J, Katchman A, Zakharov S, Mironov S, Bayne J et al (2019) Heterogeneity of the action potential duration is required for sustained atrial fibrillation. JCI Insight. https://doi.org/10.1172/jci.insight.128765
Garrey WE (1914) The nature of fibrillary contraction of the heart: its relation to tissue mass and form. Am J Physiol. 33:397–414
Garrey WE (1924) Auricular fibrillation. Physiol Rev 4:215–50
Moe GK, Rheinboldt WC, Abildskov JA (1964) A computer model of atrial fibrillation. Am Heart J 67:200–220
Allessie MA, Bonke FI, Schopman FJ (1977) Circus movement in rabbit atrial muscle as a mechanism of tachycardia III: the “leading circle” concept: a new model of circus movement in cardiac tissue without the involvement of an anatomical obstacle. Circ Res. 41:9–18
Mines GR (1914) On circulating excitation on heart muscles and their possible relation to tachycardia and fibrillation. Trans R Soc Can 4:43–53
Nattel S (2002) New ideas about atrial fibrillation 50 years on. Nature 415:219–226
Flaker GC, Fletcher KA, Rothbart RM, Halperin JL, Hart RG (1995) Clinical and echocardiographic features of intermittent atrial fibrillation that predict recurrent atrial fibrillation: stroke prevention in atrial fibrillation (SPAF) investigators. Am J Cardiol. 76:355–358
McMullen JR, Shioi T, Zhang L, Tarnavski O, Sherwood MC, Kang PM et al (2003) Phosphoinositide 3-kinase(p110alpha) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc Natl Acad Sci U S A 100:12355–12360
Stewart MH, Lavie CJ, Shah S, Englert J, Gilliland Y, Qamruddin S et al (2018) Prognostic implications of left ventricular hypertrophy. Prog Cardiovasc Dis 61:446–455
Leevers SJ, Weinkove D, MacDougall LK, Hafen E, Waterfield MD (1996) The Drosophila phosphoinositide 3-kinase Dp110 promotes cell growth. EMBO J 15:6584–6594
Shioi T, Kang PM, Douglas PS, Hampe J, Yballe CM, Lawitts J et al (2000) The conserved phosphoinositide 3-kinase pathway determines heart size in mice. EMBO J 19:2537–2548
Takemoto Y, Ramirez RJ, Kaur K, Salvador-Montanes O, Ponce-Balbuena D, Ramos-Mondragon R et al (2017) Eplerenone reduces atrial fibrillation burden without preventing atrial electrical remodeling. J Am Coll Cardiol 70:2893–2905
Abhayaratna WP, Seward JB, Appleton CP, Douglas PS, Oh JK, Tajik AJ et al (2006) Left atrial size: physiologic determinants and clinical applications. J Am Coll Cardiol 47:2357–2363
Hanif W, Alex L, Su Y, Shinde AV, Russo I, Li N et al (2017) Left atrial remodeling, hypertrophy, and fibrosis in mouse models of heart failure. Cardiovasc Pathol 30:27–37
Fornengo C, Antolini M, Frea S, Gallo C, Grosso Marra W, Morello M et al (2015) Prediction of atrial fibrillation recurrence after cardioversion in patients with left-atrial dilation. Eur Heart J Cardiovasc Imaging 16:335–341
Marrouche NF, Wilber D, Hindricks G, Jais P, Akoum N, Marchlinski F et al (2014) Association of atrial tissue fibrosis identified by delayed enhancement MRI and atrial fibrillation catheter ablation: the DECAAF study. JAMA 311:498–506
den Uijl DW, Delgado V, Bertini M, Tops LF, Trines SA, van de Veire NR et al (2011) Impact of left atrial fibrosis and left atrial size on the outcome of catheter ablation for atrial fibrillation. Heart 97:1847–1851
Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA (1995) Atrial fibrillation begets atrial fibrillation: a study in awake chronically instrumented goats. Circulation. 92:1954–68
Xiao L, Salem JE, Clauss S, Hanley A, Bapat A, Hulsmans M et al (2020) Ibrutinib-mediated atrial fibrillation attributable to inhibition of C-terminal SRC kinase. Circulation 142:2443–2455
Patel VB, Zhabyeyev P, Chen X, Wang F, Paul M, Fan D et al (2018) PI3Kalpha-regulated gelsolin activity is a critical determinant of cardiac cytoskeletal remodeling and heart disease. Nat Commun 9:5390
Chen X, Zhabyeyev P, Azad AK, Vanhaesebroeck B, Grueter CE, Murray AG et al (2021) Pharmacological and cell-specific genetic PI3Kalpha inhibition worsens cardiac remodeling after myocardial infarction. J Mol Cell Cardiol 157:17–30
Bi L, Okabe I, Bernard DJ, Wynshaw-Boris A, Nussbaum RL (1999) Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3-kinase. J Biol Chem 274:10963–10968
Shiojima I, Walsh K (2006) Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev 20:3347–3365
Porrello ER et al (2009) Heritable pathologic cardiac hypertrophy in adulthood is preceded by neonatal cardiac growth restriction. Am J Physiol Regul Integr Comp Physiol. 296:R672–R80
Rosenkranz S (2004) TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc Res 63:423–432
Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC (2016) Cardiac Fibrosis: The Fibroblast Awakens. Circ Res 118:1021–1040
Cho HC, Marban E (2010) Biological therapies for cardiac arrhythmias: can genes and cells replace drugs and devices? Circ Res 106:674–685
Wei L, Taffet GE, Khoury DS, Bo J, Li Y, Yatani A et al (2004) Disruption of Rho signaling results in progressive atrioventricular conduction defects while ventricular function remains preserved. FASEB J 18:857–859
Sah VP, Minamisawa S, Tam SP, Wu TH, Dorn GW 2nd, Ross J Jr et al (1999) Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J Clin Investig 103:1627–1634
Hong CS, Cho MC, Kwak YG, Song CH, Lee YH, Lim JS et al (2002) Cardiac remodeling and atrial fibrillation in transgenic mice overexpressing junctin. FASEB J 16:1310–1312
Hong CS, Kwon SJ, Cho MC, Kwak YG, Ha KC, Hong B et al (2008) Overexpression of junctate induces cardiac hypertrophy and arrhythmia via altered calcium handling. J Mol Cell Cardiol 44:672–682
Arad M, Moskowitz IP, Patel VV, Ahmad F, Perez-Atayde AR, Sawyer DB et al (2003) Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Parkinson-White syndrome in glycogen storage cardiomyopathy. Circulation 107:2850–2856
Kirchhof P, Fabritz L, Fortmuller L, Matherne GP, Lankford A, Baba HA et al (2003) Altered sinus nodal and atrioventricular nodal function in freely moving mice overexpressing the A1 adenosine receptor. Am J Physiol Heart Circ Physiol 285:H145–H153
Fabritz L, Kirchhof P, Fortmuller L, Auchampach JA, Baba HA, Breithardt G et al (2004) Gene dose-dependent atrial arrhythmias, heart block, and brady-cardiomyopathy in mice overexpressing A(3) adenosine receptors. Cardiovasc Res 62:500–508
Chen HH, Baty CJ, Maeda T, Brooks S, Baker LC, Ueyama T et al (2004) Transcription enhancer factor-1-related factor-transgenic mice develop cardiac conduction defects associated with altered connexin phosphorylation. Circulation 110:2980–2987
Xiao HD, Fuchs S, Campbell DJ, Lewis W, Dudley SC Jr, Kasi VS et al (2004) Mice with cardiac-restricted angiotensin-converting enzyme (ACE) have atrial enlargement, cardiac arrhythmia, and sudden death. Am J Pathol 165:1019–1032
Li J, McLerie M, Lopatin AN (2004) Transgenic upregulation of IK1 in the mouse heart leads to multiple abnormalities of cardiac excitability. Am J Physiol Heart Circ Physiol 287:H2790–H2802
Temple J, Frias P, Rottman J, Yang T, Wu Y, Verheijck EE et al (2005) Atrial fibrillation in KCNE1-null mice. Circ Res 97:62–69
Sampson KJ, Terrenoire C, Cervantes DO, Kaba RA, Peters NS, Kass RS (2008) Adrenergic regulation of a key cardiac potassium channel can contribute to atrial fibrillation: evidence from an I Ks transgenic mouse. J Physiol 586:627–637
Schrickel JW, Stockigt F, Krzyzak W, Paulin D, Li Z, Lubkemeier I et al (2010) Cardiac conduction disturbances and differential effects on atrial and ventricular electrophysiological properties in desmin deficient mice. J Interv Card Electrophysiol 28:71–80
Muller FU, Lewin G, Baba HA, Boknik P, Fabritz L, Kirchhefer U et al (2005) Heart-directed expression of a human cardiac isoform of cAMP-response element modulator in transgenic mice. J Biol Chem 280:6906–6914
Kirchhof P, Marijon E, Fabritz L, Li N, Wang W, Wang T et al (2013) Overexpression of cAMP-response element modulator causes abnormal growth and development of the atrial myocardium resulting in a substrate for sustained atrial fibrillation in mice. Int J Cardiol 166:366–374
Li N, Chiang DY, Wang S, Wang Q, Sun L, Voigt N et al (2014) Ryanodine receptor-mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation 129:1276–1285
Kehat I, Heinrich R, Ben-Izhak O, Miyazaki H, Gutkind JS, Aronheim A (2006) Inhibition of basic leucine zipper transcription is a major mediator of atrial dilatation. Cardiovasc Res 70:543–554
Adam O, Frost G, Custodis F, Sussman MA, Schafers HJ, Bohm M et al (2007) Role of Rac1 GTPase activation in atrial fibrillation. J Am Coll Cardiol 50:359–367
Schrickel JW, Brixius K, Herr C, Clemen CS, Sasse P, Reetz K et al (2007) Enhanced heterogeneity of myocardial conduction and severe cardiac electrical instability in annexin A7-deficient mice. Cardiovasc Res 76:257–268
Saba S, Janczewski AM, Baker LC, Shusterman V, Gursoy EC, Feldman AM et al (2005) Atrial contractile dysfunction, fibrosis, and arrhythmias in a mouse model of cardiomyopathy secondary to cardiac-specific overexpression of tumor necrosis factor-{alpha}. Am J Physiol Heart Circ Physiol 289:H1456–H1467
Sawaya SE, Rajawat YS, Rami TG, Szalai G, Price RL, Sivasubramanian N et al (2007) Downregulation of connexin40 and increased prevalence of atrial arrhythmias in transgenic mice with cardiac-restricted overexpression of tumor necrosis factor. Am J Physiol Heart Circ Physiol 292:H1561–H1567
Ogata T, Ueyama T, Isodono K, Tagawa M, Takehara N, Kawashima T et al (2008) MURC, a muscle-restricted coiled-coil protein that modulates the Rho/ROCK pathway, induces cardiac dysfunction and conduction disturbance. Mol Cell Biol 28:3424–3436
Zhang X, Chen S, Yoo S, Chakrabarti S, Zhang T, Ke T et al (2008) Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell 135:1017–1027
Zhang Z, He Y, Tuteja D, Xu D, Timofeyev V, Zhang Q et al (2005) Functional roles of Cav1.3(alpha1D) calcium channels in atria: insights gained from gene-targeted null mutant mice. Circulation. 112:1936–44
Mancarella S, Yue Y, Karnabi E, Qu Y, El-Sherif N, Boutjdir M (2008) Impaired Ca2+ homeostasis is associated with atrial fibrillation in the alpha1D L-type Ca2+ channel KO mouse. Am J Physiol Heart Circ Physiol 295:H2017–H2024
Levin MD, Lu MM, Petrenko NB, Hawkins BJ, Gupta TH, Lang D et al (2009) Melanocyte-like cells in the heart and pulmonary veins contribute to atrial arrhythmia triggers. J Clin Investig 119:3420–3436
Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG et al (2009) Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Investig 119:1940–1951
Hirose M, Takeishi Y, Niizeki T, Shimojo H, Nakada T, Kubota I et al (2009) Diacylglycerol kinase zeta inhibits G(alpha)q-induced atrial remodeling in transgenic mice. Heart Rhythm 6:78–84
Chinchilla A, Daimi H, Lozano-Velasco E, Dominguez JN, Caballero R, Delpon E et al (2011) PITX2 insufficiency leads to atrial electrical and structural remodeling linked to arrhythmogenesis. Circ Cardiovasc Genet 4:269–279
Cunha SR, Hund TJ, Hashemi S, Voigt N, Li N, Wright P et al (2011) Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation 124:1212–1222
Watanabe H, Yang T, Stroud DM, Lowe JS, Harris L, Atack TC et al (2011) Striking In vivo phenotype of a disease-associated human SCN5A mutation producing minimal changes in vitro. Circulation 124:1001–1011
Xie W, Santulli G, Reiken SR, Yuan Q, Osborne BW, Chen BX et al (2015) Mitochondrial oxidative stress promotes atrial fibrillation. Sci Rep 5:11427
Li N, Wang T, Wang W, Cutler MJ, Wang Q, Voigt N et al (2012) Inhibition of CaMKII phosphorylation of RyR2 prevents induction of atrial fibrillation in FKBP12.6 knockout mice. Circ Res. 110:465–70
Choi EK, Chang PC, Lee YS, Lin SF, Zhu W, Maruyama M et al (2012) Triggered firing and atrial fibrillation in transgenic mice with selective atrial fibrosis induced by overexpression of TGF-beta1. Circ J 76:1354–1362
Rosenberg MA, Das S, Quintero Pinzon P, Knight AC, Sosnovik DE, Ellinor PT et al (2012) A novel transgenic mouse model of cardiac hypertrophy and atrial fibrillation. J Atr Fibrillation 4:415
Glukhov AV, Kalyanasundaram A, Lou Q, Hage LT, Hansen BJ, Belevych AE et al (2015) Calsequestrin 2 deletion causes sinoatrial node dysfunction and atrial arrhythmias associated with altered sarcoplasmic reticulum calcium cycling and degenerative fibrosis within the mouse atrial pacemaker complex1. Eur Heart J 36:686–697
Ozcan C, Battaglia E, Young R, Suzuki G (2015) LKB1 knockout mouse develops spontaneous atrial fibrillation and provides mechanistic insights into human disease process. J Am Heart Assoc 4:e001733
Wan E, Abrams J, Weinberg RL, Katchman AN, Bayne J, Zakharov SI et al (2016) Aberrant sodium influx causes cardiomyopathy and atrial fibrillation in mice. J Clin Investig 126:112–122
Moreira LM, Takawale A, Hulsurkar M, Menassa DA, Antanaviciute A, Lahiri SK et al (2020) Paracrine signalling by cardiac calcitonin controls atrial fibrogenesis and arrhythmia. Nature 587:460–465
Kunzel SR, Hoffmann M, Weber S, Kunzel K, Kammerer S, Gunscht M et al (2021) Diminished PLK2 induces cardiac fibrosis and promotes atrial fibrillation. Circ Res 129:804–820
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. This study was supported by the Australian Government Research Training Programme Scholarship and Monash University, Faculty of Medicine, Nursing and Health Sciences Excellence Award, awarded to ME.
Author information
Authors and Affiliations
Contributions
ME conceived the idea and developed concepts, performed research and collated literature, and then wrote and edited the paper. SP wrote and edited the paper.
Corresponding author
Ethics declarations
Conflict of interest
We declare no conflict of interest and we do not have any industrial relations to declare; Dr Sandeep Prabhu MBBS (Hons), LLB (Hons), FRACP, PhD is Clinical Associate Prof at the University Melbourne and a Cardiologist and Electrophysiologist at The Alfred, Melbourne Australia; Martin Ezeani MSc, PhD is the Managing Editor, AF Issue, Frontiers in Bioscience.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ezeani, M., Prabhu, S. PI3K(p110α) as a determinant and gene therapy for atrial enlargement in atrial fibrillation. Mol Cell Biochem 478, 471–490 (2023). https://doi.org/10.1007/s11010-022-04526-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-022-04526-w