
Abstract
Acute lung injury/acute respiratory distress syndrome (ALI/ARDS) is a common disease with high morbidity and mortality, and its pathogenesis is believed to be related to oxidative stress, apoptosis, inflammation, and hypoxia. Ferroptosis is a type of nonapoptotic cell death characterized by iron-dependent lipid peroxide accumulation and is involved in many cellular physiological processes. Recent studies have confirmed that ferroptosis may be involved in the development of ALI. This review summarizes the most recent discoveries on the role of ferroptosis in ALI to provide new strategies for its prevention and treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Acute lung injury/acute respiratory distress syndrome (ALI/ARDS) is a common and critical disease caused by several factors, including infection, trauma, radiation, and ischemia-reperfusion [1,2,3,4]. Although significant advancements have been achieved, such as mechanical ventilation and corticosteroid administration, the annual mortality of ALI remains 40% [5]. Thus, efforts to develop new targets that interrupt the progression of ALI to ARDS are an attractive endeavor. The pathogenesis of ALI was previously believed to involve oxidative stress, apoptosis, inflammation, and hypoxia [6,7,8]. Recent studies have demonstrated that ferroptosis might be involved in the development of ALI [9]. In this review, the most recent discoveries regarding the role of ferroptosis in ALI are reviewed to provide new strategies for its prevention and treatment.
Ferroptosis
Ferroptosis was first found in RAS mutant tumor cells treated with erastin [10]. The morphology of cells undergoing ferroptosis is different from that of cells undergoing apoptosis, pyroptosis, and other cell death mechanisms. Ferroptosis is mainly characterized by the shrinkage of mitochondria, a decrease or disappearance of mitochondrial cristae, atrophy of the mitochondrial membrane and a lack of chromatin condensation [11]. Because the cell death mode depends on iron, accompanied by the accumulation of peroxide in cells, and can be inhibited by iron-chelating agents, it is called ferroptosis [12].
Mechanism and regulation of ferroptosis
Iron overload and ferroptosis
Intracellular free iron plays an important role in ferroptosis and reacts with H2O2 to generate free radicals through the Fenton reaction [13]. The fatty acid hydroperoxide produced by lipoxygenase also requires Fe2+ to generate free radicals. These free radicals constantly destroy the structures of cell biofilms and disrupt their function, thereby inducing ferroptosis [14]. Additionally, glutathione (GSH), an important antioxidant, can regulate the production of reactive oxygen species (ROS) by the key enzyme glutathione peroxidase 4 (GPX4), thereby inhibiting the occurrence of ferroptosis [15].
Iron overload caused by iron metabolism disorder is closely related to ferroptosis. The application of iron-chelating agents and the upregulation of ferritin levels can inhibit ferroptosis [16], suggesting that iron metabolism plays an important role in the phenomenon. The body maintains the balance of iron input and output under normal conditions; most of the iron used in the body every day derives from the rerelease of iron from aged red blood cells after phagocytosis, and a small part is obtained from the diet [17]. In the intestinal tract, Fe3+ is transformed into Fe2+ under the action of duodenal cytochrome b (Dcytb) and is then absorbed into intestinal epithelial cells through divalent metal transporter 1 (DMT1) [18, 19]. After entering the cells, the majority of Fe2+ is oxidized to Fe3+ and forms ferritin, while a small amount of nonoxidized Fe2+ can enter the plasma through the basement membranes of intestinal epithelial cells via ferroportin (Fpn) [20]. After entering the plasma, nonoxidized Fe2+ is oxidized into Fe3+ again and combines with transferrin (Tf) to form Tf-Fe3+. When Tf-Fe3+ binds to cellular transferrin receptor 1 (TFR1), Fe3+ is reduced to Fe2+ via a six-transmembrane epithelial antigen of prostate 3 (Steap3) and then enters the cytoplasm via DMT1 [21]. Some Fe2+ forms ferritin, while some forms a labile iron pool (LIP) [22], and cytoplasmic iron can also be exported to cells via the membrane iron transporter Fpn [23].
The iron metabolism balance depends on several regulators, such as Tf and hepcidin [24, 25]. Hepcidin binds to Fpn to promote the degradation of these transporters, thus inhibiting the absorption and release of iron [26]. Under inflammatory conditions, human lactoferrin (hLf) inhibits the expression of interleukin-6 (IL-6) and Fpn to alleviate intracellular iron overload [27]. In addition, lipocalin 2, a key factor involved in iron regulation, prevents bacterial siderophores from obtaining iron during infection, stabilizes the LIP, and isolates iron in the mucosa and cavity during inflammation to ensure low iron levels [28].
The stability of the intracellular iron content depends on iron regulatory protein/iron-responsive elements (IRPs/IREs), including IRP1 and IRP2. IREs are highly conserved RNA stem-loops found in the mRNAs encoding proteins involved in iron metabolism. IREs combine with the 5' iron-reactive elements of ferritin and iron transporters to prevent their synthesis, and IREs bind to the 3' iron-reactive elements of transferrin receptor 1 and DMT1 to prevent their degradation, thus increasing the intracellular iron level. When the iron level in cells is high, IRE1 is converted into cis-aconitase, and IRE2 is degraded and reduces the iron storage capacity and iron output, thus reducing the intracellular level of free iron [29]. The iron chelators deferoxamine (DFO) and ciclopirox (CPX) inhibit ferroptosis by reducing the utilization of iron and reducing the oxidative stress level in cells [30, 31].
Lipid peroxidation and ferroptosis
Fatty acids, including saturated fatty acids, monounsaturated fatty acids (MUFAs) and polyunsaturated fatty acids (PUFAs), play several roles in cells, including blocking cell membranes, providing energy and even acting as signaling molecules [32]. Interestingly, PUFAs (especially arachidonic acid and adrenal acid) promote ferroptosis through lipid peroxidation [33]. Lipid peroxidation can be achieved via enzymatic and nonenzymatic methods, which require iron. Iron not only promotes lipid peroxidation through a nonenzymatic mechanism but also increases arachidonic acid lipoxygenase (ALOX) activity through endocrine signaling [34]. Additionally, lipid peroxide scavengers such as vitamin E, ferrostatin-1 and liproxstatin-1 can capture lipid peroxide through lipophilic free radicals and inhibit ferroptosis [35, 36]. Undeniably, additional work is needed to understand the roles of ALOX and lipid peroxide scavengers in mediating different types of ferroptosis.
GSH depletion and ferroptosis
GSH, a tripeptide that contains cysteine, is often considered the main antioxidant in the human body [37]. System XC, composed of two protein subunits, solute carrier family 7 member 11 (SLC7A11) and solute carrier 3A2 (SLC3A2), is an amino acid transporter that imports cystine and exports glutamic acid, and it exchanges intracellular glutamate with extracellular cystine to produce GSH [38]. Erastin, a small molecule that induces ferroptosis, can inhibit the transmembrane protein SLC7A11, thus inhibiting the amino acid antiporter system XC-, reducing the amount of cystine taken in by cells and decreasing the level of GSH to promote ferroptosis [39]. Buthionine sulfoximine (BSO) inhibits glutamate-cysteine ligase (GCL) and directly inhibits GSH synthesis, causing ferroptosis [40]. The utilization of GSH depends on the key enzyme GPX4, and GPX4 knockout can promote ferroptosis [41]. RSL3 can covalently bind to the active site Sec of GPX4 and directly inhibit the activity of GPX4, thus inducing ferroptosis [42]. Statins inhibit the formation of selenocysteine-tRNA (tRNA(Sec)) and inhibit the synthesis of GPX4 to induce ferroptosis [43].
Antioxidant and ferroptosis
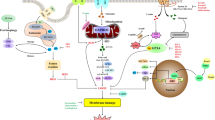
Coenzyme Q 10 (CoQ10) is an endogenous fat-soluble antioxidant, and exhaustion of CoQ10 produced by GPX4 and the mevalonate pathway by a ferroptosis inducer derived from CIL56 (FIN56) induces ferroptosis [44]. In addition, inhibiting the lipid metabolism pathway, decreasing the sources of lipid peroxides, such as modulating/inhibiting acyl-CoA synthase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3), and inhibiting lipid peroxidation involving Lox can also inhibit ferroptosis [45]. In addition, ferroptosis suppressor protein 1 (FSP1) can use nicotinamide adenine dinucleotide phosphate (NADPH) to catalyze the regeneration of CoQ10 to thereby inhibit ferroptosis [46]. Nuclear factor E2-related factor 2 (NRF2) and heat shock protein B1 were recently shown to affect the sensitivity of cells to ferroptosis inducers by regulating cellular iron metabolism [47, 48] (Fig. 1).

Mechanism and regulation of ferroptosis in alveolar epithelial cells in ALI. In alveolar epithelial cells, pathogenic factors can increase the levels of Fe3+ and H2O2. Increased Fe3+ can be reduced to Fe2+ by Steap3 and then enter the cytoplasm via DMT1 to generate free radicals through the Fenton reaction. These free radicals can oxidize membrane lipids, such as PUFAs, into lipid peroxides, constantly disrupting the structure and function of cell biofilms, thus inducing ferroptosis and leading to ALI. This process can be inhibited by regulating key proteins of the ferroptosis pathway, such as GPX4 and System XC, as well as the labile iron pool. System XC− cystine/glutamate antiporter system; PUFA polyunsaturated fatty acid; GSH glutathione; GSSG glutathione disulfide; GPX4 glutathione peroxidase 4; Fpn ferroportin; TFR1 transferrin receptor 1; STEAP3 six-transmembrane epithelial antigen of the prostate 3; LIP labile iron pool; ROS reactive oxygen species; DFO deferoxamine; CPX ciclopirox; HMOX1 heme oxygenase 1; BSO buthionine sulfoximine; DAMPS damage-associated molecular patterns; ALI acute lung injury
Ferroptosis and ALI
ALI/ARDS is a serious illness with high morbidity and mortality. Increased ROS, epithelial cell apoptosis, and inflammation are important pathogenic mechanisms of ALI [49]. Recent studies have shown that ferroptosis plays a critical role in the development of ALI (Fig. 1).
Infection-related ALI
Infection and sepsis are the main pathogenic factors of ALI/ARDS [50]. Recent studies have demonstrated that ferroptosis plays an important role in the occurrence and development of ALI caused by infection. The expression of GPX4 in the lung tissues of mice infected with Mycobacterium tuberculosis (MTB) was shown to be significantly decreased, and the production of lipid peroxide in lung parenchyma cells was increased [51]. In an ALI mouse model induced by lipopolysaccharide (LPS), the concentration of free iron in the bronchial epithelial cells of ALI mice was significantly increased, while the expression of SLC7A11 and GPX4, ferroptosis markers, was dramatically decreased [52]. However, the degree of lung injury in mice pretreated with the ferroptosis inhibitor ferrostatin-1 was significantly improved, suggesting that ferroptosis plays an important role in the occurrence and development of ALI caused by MTB and LPS [51, 52]. In addition, panaxydol can inhibit ferroptosis in ALI induced by LPS, and its effect is due to the upregulation of the Kelch-like ECH-associated protein 1 (Keap1)-Nrf2/heme oxygenase-1 (HO-1) pathway [53, 54].
Pseudomonas aeruginosa is an important pathogen that causes nosocomial infection. The ΔwspF mutant of P. aeruginosa can enhance the activity of lipoxygenase (pLOXA) to oxidize arachidonic acid-phosphatidylethanolamines (AA-PE) into 15-hydroperoxy-AA-PE (15-HOOAA-PE) to thereby induce ferroptosis, suggesting that ferroptosis mediates the occurrence of pneumonia caused by P. aeruginosa [55, 56].
Furthermore, ferroptosis was shown to be involved in the development of viral pneumonia. Banchini et al. found that SARS-CoV-2 could activate the hepcidin-Fpn pathway and promote ferroptosis by mobilizing iron in the vascular space, leading to acute lung inflammation [57]. Further analysis revealed that the levels of peroxide in the myocardial and renal tissues of patients with viral myocarditis of unknown cause were not significantly increased, while its levels in the myocardial cells of patients with COVID-19 were significantly increased, indicating that SARS-CoV-2-induced acute pneumonia may induce intracellular peroxide accumulation and cause ferroptosis in lung parenchyma cells to thereby induce an inflammatory reaction [58]. Regarding the important role of ferroptosis in the pathogenesis of COVID-19, iron chelators, such as lactoferrin (Lf), may have high therapeutic value [59].
Radiation-related ALI
Radiotherapy is a method used to treat malignant chest tumors, and radiation-induced lung injury (RILI) is the most common complication of chest radiotherapy [60]. Because of the heterogeneity of individuals and the high cumulative radiotherapy dose, radiation-induced lung fibrosis (RILF) often develops in the late stage of radiotherapy and seriously affects the prognosis of patients. A strategy for preventing RILI in patients undergoing chest tumor radiotherapy urgently needs to be developed [61]. Recent studies suggest that ferroptosis is a target for the prevention and treatment of RILI and pulmonary fibrosis. In an acute RILI mouse model, the expression of GPX4, a ferroptosis marker, was decreased, and mitochondria in lung tissue developed a ferroptosis phenotype, as evidenced by electron microscopy. Treatment with the ferroptosis inhibitor lipostatin-1 was shown to significantly downregulate the levels of ROS in the lung and the levels of inflammatory cytokines [tumor necrosis factor-alpha (TNF-α), IL-6, interleukin-10 (IL-10), and transforming growth factor-beta1 (TGF-β1)] in serum, indicating that ferroptosis plays a key role in RILI [62, 63].
ALI associated with ischemia–reperfusion
Ischemia–reperfusion injury is an important pathophysiological phenomenon associated with severe trauma, infection, and major surgery, and the main mediator of injury is the systemic inflammatory response [64, 65]. Ischemia–reperfusion injury in a single organ can spread to adjacent or distant tissues and organs through the transmission of inflammatory factors [66]. The lung, one of the most vulnerable organs, is often impacted by ischemia–reperfusion injury, which can lead to ALI [67]. P53, a tumor suppressor, regulates the occurrence and development of ALI by inhibiting apoptosis and ferroptosis. Recent studies have demonstrated that an inhibitor of the apoptosis-stimulating protein of p53 (iASPP) can inhibit ferroptosis and alleviate ALI caused by intestinal ischemia–reperfusion in mice. Additionally, the protective effect of the iASPP inhibitor was reduced in Nfr2-knockout mice, suggesting that the protective effect mediated by iASPP partly depends on Nfr2 signaling. In addition, ACSL4, a key enzyme involved in ferroptosis, can significantly improve ischemia–reperfusion injury [45]. A recent study showed that Nrf2 can inhibit ferroptosis by regulating SLC7A11 and HO-1 to thereby improve ALI caused by intestinal ischemia–reperfusion [68]. Thus, ferroptosis is involved in ALI induced by ischemia/reperfusion [69].
ALI caused by physical and chemical factors
In addition, physical and chemical factors commonly cause ALI. Recent experiments have proven that the levels of ROS and lipid ROS are decreased, that the mRNA expression of GPX4 is increased, and the mitochondrial membrane potential of cells is maintained in lung epithelial cells treated with seawater after treatment with the Nrf2 activator dimethyl fumarate. Additionally, the degree of lung injury observed in Nrf2-knockout mice was more serious than that observed in wild-type mice after drowning in seawater. The above results showed that Nrf2 can reduce drowning-induced ALI caused by inhibiting ferroptosis [70]. In a lung injury mouse model induced by oleic acid (OA), the content of GSH was decreased, and the level of malondialdehyde (MDA) was increased in the lung tissue. In addition, the expression of GPX4 and ferritin was decreased in the lung tissues of the OA-induced lung injury group, which indicated that ferroptosis was also involved [71]. These results suggest that ferroptosis is related to lung injury, but there is still a lack of large-scale clinical research to provide evidence for the optimal treatment plan and treatment safety.
Treatment of ALI based on ferroptosis
Increasing studies have shown that ferroptosis may play a key role in the development of ALI, indicating that the interruption of ferroptosis may be a new target for ALI treatment. In recent studies, several ferroptosis regulators, include iron steady-state regulators and lipid peroxidation inhibitors, have been shown to have therapeutic effects on ALI (Table 1).
Iron homeostasis regulator
In ALI, the levels of free iron in lung tissues are significantly increased, and the iron homeostasis of cells is out of balance, leading to ferroptosis. The iron chelators DFO and CPX can remove excessive iron from lung tissue to maintain iron homeostasis in cells and inhibit ferroptosis. In the mechanical ventilation-induced ALI model, the high-volume ventilated rats exhibited typical lung edema and histological lung injury, and the level of ROS was increased in alveolar macrophages and mitochondria. However, the level of ROS was reduced in rats preconditioned with DFO, suggesting that DFO ameliorates lung injury in the mechanically ventilated SD rat model [72]. Similarly, DFO inhibited inflammation and improved lung injury by reducing oxidative stress and mitochondrial instability in an LPS-induced ALI model [73]. Hepcidin, an iron export protein, can inhibit ferroptosis by regulating iron export mediated by iron transporters and by regulating the level of Fpn in cells. The promoter of the hepcidin gene contains the binding site of the phosphorylated signal transducer and activator of transcription 3 (STAT3) dimer. Furthermore, IL-6 can induce STAT3, and the JAK-STAT signaling pathway acts on the promoter of the hepcidin gene to regulate gene expression, thereby regulating iron homeostasis [74]. Studies have shown that early application of hepcidin downregulates the renal levels of cyclooxygenase-2 (Cox-2) and the serum levels of TNF-α in mice with LPS-induced acute kidney injury, indicating that hepcidin can alleviate acute renal failure and acute tubular necrosis in acute kidney injury induced by ischemia–reperfusion; however, no studies have confirmed its positive effect on ALI [75].
Small-molecule inhibitor of ferroptosis
Ferrostatin-1 is an important small-molecule inhibitor of ferroptosis that prevents lipid membrane damage via an iron reduction mechanism [76]. Ferrostatin-1 alleviates inflammation caused by angiotensin II (Ang II) by decreasing ROS levels and activating the Nrf2/HO-1 signaling pathway [77]. In an LPS-induced ALI model, cell viability and the levels of the ferroptosis markers SLC7A11 and GPX4 were downregulated in the LPS group, while the levels of MDA, 4-hydroxynonenal (4-HNE), and total iron were increased by LPS treatment. In contrast, the cell viability in the LPS+ferrostatin-1 group was higher than that in the LPS group. In addition, the amounts of MDA, 4-HNE and total iron in the LPS+Fer-1 group were lower than those in the LPS group. These results indicated that Fer-1 exerts a therapeutic effect on LPS-induced ALI and downregulates the ferroptosis level in lung tissues [52].
Lipid peroxidation inhibitor
GPX4 is an important antioxidant enzyme, and a decrease in the level of GPX4 increases lipid peroxidation and promotes lung tissue necrosis in mice with ALI caused by MTB [51]. Nrf2 is also a key regulator of oxidative stability in cells. The target genes of Nrf2 include intracellular redox balance proteins. For example, HO-1, GPX4, and SLC7A11 are activated under high oxidative stress and play antioxidant roles [78]. In recent research, the Nrf2/HO-1 pathway was proven to be involved in ferroptosis. In this study, compared with the LPS-induced ALI group, the PX (panaxydol) + LPS group showed decreased cell viability and increased cell death and Fe2+ accumulation. Interestingly, the expression of Keap1 was significantly decreased and the expression of Nrf2 and HO-1 was increased after treatment with PX, which suggested that PX alleviates ALI by upregulating the Keap1-Nrf2/HO-1 pathway [53]. In addition, lipoastatin-1 (Lip-1), a ferroptosis-specific inhibitor, is an important lipid peroxidation inhibitor. In the ALI model after lung ischemia–reperfusion, the levels of tissue iron and lipid peroxidation were decreased after the application of lip-1, thereby alleviating lung injury induced by ischemia–reperfusion by reducing lipid peroxidation and increasing the levels of GSH and GPX4 [45].
Conclusion
Ferroptosis is a unique form of regulated cell death, and the ferroptosis process has been observed in the context of several types of acute lung injury. However, most studies on the relationship between acute lung injury and ferroptosis are qualitative, and the extent to which ferroptosis is involved in ALI remains largely unknown. Intervening with the ferroptosis process may contribute to the prevention and treatment of ALI, but the exact dose and side effects of ferroptosis inhibitors remain largely unknown, and whether respiratory-specific ferroptosis regulators exist remains unknown. Therefore, more comprehensive and in-depth studies on the relationship between acute lung injury and ferroptosis are needed to expand our understanding of acute lung injury and to regulate ferroptosis to thereby protect the lungs from damage caused by acute lung injury.
Data availability
Data sharing was not applicable to this article, as no datasets were generated or analyzed during the current study.
References
Butt Y, Kurdowska A, Allen TC (2016) Acute lung injury: a clinical and molecular review. Arch Pathol Lab Med 140(4):345–350. https://doi.org/10.5858/arpa.2015-0519-RA
Kalil AC, Patterson TF, Mehta AK, Tomashek KM, Wolfe CR, Ghazaryan V, Marconi VC, Ruiz-Palacios GM, Hsieh L, Kline S, Tapson V, Iovine NM, Jain MK, Sweeney DA, El Sahly HM, Branche AR, Regalado Pineda J, Lye DC, Sandkovsky U, Luetkemeyer AF, Cohen SH, Finberg RW, Jackson PEH, Taiwo B, Paules CI, Arguinchona H, Erdmann N, Ahuja N, Frank M, Oh MD, Kim ES, Tan SY, Mularski RA, Nielsen H, Ponce PO, Taylor BS, Larson L, Rouphael NG, Saklawi Y, Cantos VD, Ko ER, Engemann JJ, Amin AN, Watanabe M, Billings J, Elie MC, Davey RT, Burgess TH, Ferreira J, Green M, Makowski M, Cardoso A, de Bono S, Bonnett T, Proschan M, Deye GA, Dempsey W, Nayak SU, Dodd LE, Beigel JH (2021) Baricitinib plus remdesivir for hospitalized adults with Covid-19. N Engl J Med 384(9):795–807. https://doi.org/10.1056/NEJMoa2031994
Zheng L, Zhu Q, Xu C, Li M, Li H, Yi PQ, Xu FF, Cao L, Chen JY (2020) Glycyrrhizin mitigates radiation-induced acute lung injury by inhibiting the HMGB1/TLR4 signalling pathway. J Cell Mol Med 24(1):214–226. https://doi.org/10.1111/jcmm.14703
Hayase N, Doi K, Hiruma T, Matsuura R, Hamasaki Y, Noiri E, Nangaku M, Morimura N (2020) Recombinant thrombomodulin prevents acute lung injury induced by renal ischemia-reperfusion injury. Sci Rep 10(1):289. https://doi.org/10.1038/s41598-019-57205-0
Johnson ER, Matthay MA (2010) Acute lung injury: epidemiology, pathogenesis, and treatment. J Aerosol Med Pulm Drug Deliv 23(4):243–252. https://doi.org/10.1089/jamp.2009.0775
Wu G, Xu G, Chen DW, Gao WX, Xiong JQ, Shen HY, Gao YQ (2018) Hypoxia exacerbates inflammatory acute lung injury via the toll-like receptor 4 signaling pathway. Front Immunol 9:1667. https://doi.org/10.3389/fimmu.2018.01667
Xie W, Lu Q, Wang K, Lu J, Gu X, Zhu D, Liu F, Guo Z (2018) miR-34b-5p inhibition attenuates lung inflammation and apoptosis in an LPS-induced acute lung injury mouse model by targeting progranulin. J Cell Physiol 233(9):6615–6631. https://doi.org/10.1002/jcp.26274
Lei J, Wei Y, Song P, Li Y, Zhang T, Feng Q, Xu G (2018) Cordycepin inhibits LPS-induced acute lung injury by inhibiting inflammation and oxidative stress. Eur J Pharmacol 818:110–114. https://doi.org/10.1016/j.ejphar.2017.10.029
Liu X, Wang L, Xing Q, Li K, Si J, Ma X, Mao L (2021) Sevoflurane inhibits ferroptosis: a new mechanism to explain its protective role against lipopolysaccharide-induced acute lung injury. Life Sci 275:119391. https://doi.org/10.1016/j.lfs.2021.119391
Dolma S, Lessnick SL, Hahn WC, Stockwell BR (2003) Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3(3):285–296. https://doi.org/10.1016/s1535-6108(03)00050-3
Adedoyin O, Boddu R, Traylor A, Lever JM, Bolisetty S, George JF, Agarwal A (2018) Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am J Physiol Renal Physiol 314(5):F702-f714. https://doi.org/10.1152/ajprenal.00044.2017
Yu H, Guo P, Xie X, Wang Y, Chen G (2017) Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med 21(4):648–657. https://doi.org/10.1111/jcmm.13008
Qian X, Zhang J, Gu Z, Chen Y (2019) Nanocatalysts-augmented Fenton chemical reaction for nanocatalytic tumor therapy. Biomaterials 211:1–13. https://doi.org/10.1016/j.biomaterials.2019.04.023
Shen Z, Liu T, Li Y, Lau J, Yang Z, Fan W, Zhou Z, Shi C, Ke C, Bregadze VI, Mandal SK, Liu Y, Li Z, Xue T, Zhu G, Munasinghe J, Niu G, Wu A, Chen X (2018) Fenton-reaction-acceleratable magnetic nanoparticles for ferroptosis therapy of orthotopic brain tumors. ACS Nano 12(11):11355–11365. https://doi.org/10.1021/acsnano.8b06201
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149(5):1060–1072. https://doi.org/10.1016/j.cell.2012.03.042
Fang X, Cai Z, Wang H, Han D, Cheng Q, Zhang P, Gao F, Yu Y, Song Z, Wu Q, An P, Huang S, Pan J, Chen HZ, Chen J, Linkermann A, Min J, Wang F (2020) Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ Res 127(4):486–501. https://doi.org/10.1161/circresaha.120.316509
Winn NC, Volk KM, Hasty AH (2020) Regulation of tissue iron homeostasis: the macrophage “ferrostat.” JCI Insight 5(2):e132964. https://doi.org/10.1172/jci.insight.132964
Kondaiah P, Aslam MF, Mashurabad PC, Sharp PA, Pullakhandam R (2019) Zinc induces iron uptake and DMT1 expression in Caco-2 cells via a PI3K/IRP2 dependent mechanism. Biochem J 476(11):1573–1583. https://doi.org/10.1042/bcj20180939
Sandberg AS, Önning G, Engström N, Scheers N (2018) Iron supplements containing Lactobacillus plantarum 299v increase ferric iron and up-regulate the ferric reductase DCYTB in human Caco-2/HT29 MTX co-cultures. Nutrients 10(12):1949. https://doi.org/10.3390/nu10121949
Zhang DL, Wu J, Shah BN, Greutélaers KC, Ghosh MC, Ollivierre H, Su XZ, Thuma PE, Bedu-Addo G, Mockenhaupt FP, Gordeuk VR, Rouault TA (2018) Erythrocytic ferroportin reduces intracellular iron accumulation, hemolysis, and malaria risk. Science 359(6383):1520–1523. https://doi.org/10.1126/science.aal2022
Han M, Xu R, Wang S, Yang N, Ni S, Zhang Q, Xu Y, Zhang X, Zhang C, Wei Y, Ji J, Huang B, Zhang D, Chen A, Li W, Bjerkvig R, Li X, Wang J (2018) Six-transmembrane epithelial antigen of prostate 3 predicts poor prognosis and promotes glioblastoma growth and invasion. Neoplasia 20(6):543–554. https://doi.org/10.1016/j.neo.2018.04.002
Chutvanichkul B, Vattanaviboon P, Mas-Oodi S, U-pratyaWanachiwanawin YW (2018) Labile iron pool as a parameter to monitor iron overload and oxidative stress status in β-thalassemic erythrocytes. Cytom B Clin Cytom 94(4):631–636. https://doi.org/10.1002/cyto.b.21633
Billesbølle CB, Azumaya CM, Kretsch RC, Powers AS, Gonen S, Schneider S, Arvedson T, Dror RO, Cheng Y, Manglik A (2020) Structure of hepcidin-bound ferroportin reveals iron homeostatic mechanisms. Nature 586(7831):807–811. https://doi.org/10.1038/s41586-020-2668-z
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X (2015) Glutaminolysis and transferrin regulate ferroptosis. Mol Cell 59(2):298–308. https://doi.org/10.1016/j.molcel.2015.06.011
Schwartz AJ, Das NK, Ramakrishnan SK, Jain C, Jurkovic MT, Wu J, Nemeth E, Lakhal-Littleton S, Colacino JA, Shah YM (2019) Hepatic hepcidin/intestinal HIF-2α axis maintains iron absorption during iron deficiency and overload. J Clin Invest 129(1):336–348. https://doi.org/10.1172/jci122359
Santos-Silva A, Ribeiro S, Reis F, Belo L (2019) Hepcidin in chronic kidney disease anemia. Vitam Horm 110:243–264. https://doi.org/10.1016/bs.vh.2019.01.012
Rosa L, Cutone A, Lepanto MS, Paesano R, Valenti P (2017) Lactoferrin: a natural glycoprotein involved in iron and inflammatory homeostasis. Int J Mol Sci 18(9):1985. https://doi.org/10.3390/ijms18091985
Xiao X, Yeoh BS, Vijay-Kumar M (2017) Lipocalin 2: an emerging player in iron homeostasis and inflammation. Annu Rev Nutr 37:103–130. https://doi.org/10.1146/annurev-nutr-071816-064559
Holmes-Hampton GP, Ghosh MC, Rouault TA (2018) Methods for studying iron regulatory protein 1: an important protein in human iron metabolism. Methods Enzymol 599:139–155. https://doi.org/10.1016/bs.mie.2017.09.006
Yao X, Zhang Y, Hao J, Duan HQ, Zhao CX, Sun C, Li B, Fan BY, Wang X, Li WX, Fu XH, Hu Y, Liu C, Kong XH, Feng SQ (2019) Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural Regen Res 14(3):532–541. https://doi.org/10.4103/1673-5374.245480
Regdon Z, Demény MA, Kovács K, Hajnády Z, Nagy-Pénzes M, Bakondi E, Kiss A, Hegedűs C, Virág L (2021) High-content screening identifies inhibitors of oxidative stress-induced parthanatos: cytoprotective and anti-inflammatory effects of ciclopirox. Br J Pharmacol 178(5):1095–1113. https://doi.org/10.1111/bph.15344
Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, Ward CC, Cho K, Patti GJ, Nomura DK, Olzmann JA, Dixon SJ (2019) Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol 26(3):420-432.e9. https://doi.org/10.1016/j.chembiol.2018.11.016
Das UN (2019) Saturated fatty acids, MUFAs and PUFAs regulate ferroptosis. Cell Chem Biol 26(3):309–311. https://doi.org/10.1016/j.chembiol.2019.03.001
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR (2016) Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA 113(34):E4966–E4975. https://doi.org/10.1073/pnas.1603244113
Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, Pratt DA (2017) On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci 3(3):232–243. https://doi.org/10.1021/acscentsci.7b00028
Hinman A, Holst CR, Latham JC, Bruegger JJ, Ulas G, McCusker KP, Amagata A, Davis D, Hoff KG, Kahn-Kirby AH, Kim V, Kosaka Y, Lee E, Malone SA, Mei JJ, Richards SJ, Rivera V, Miller G, Trimmer JK, Shrader WD (2018) Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15-lipoxygenase. PLoS ONE 13(8):e0201369. https://doi.org/10.1371/journal.pone.0201369
Tang H, Chen D, Li C, Zheng C, Wu X, Zhang Y, Song Q, Fei W (2019) Dual GSH-exhausting sorafenib loaded manganese-silica nanodrugs for inducing the ferroptosis of hepatocellular carcinoma cells. Int J Pharm 572:118782. https://doi.org/10.1016/j.ijpharm.2019.118782
Sun L, Dong H, Zhang W, Wang N, Ni N, Bai X, Liu N (2021) Lipid peroxidation, GSH depletion, and SLC7A11 inhibition are common causes of EMT and ferroptosis in A549 cells, but different in specific mechanisms. DNA Cell Biol 40(2):172–183. https://doi.org/10.1089/dna.2020.5730
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, Xie Y, Liu J, Klionsky DJ, Kroemer G, Lotze MT, Zeh HJ, Kang R, Tang D (2018) AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system X(c)(-) activity. Curr Biol 28(15):2388-2399.e5. https://doi.org/10.1016/j.cub.2018.05.094
Sun Y, Zheng Y, Wang C, Liu Y (2018) Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis 9(7):753. https://doi.org/10.1038/s41419-018-0794-4
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, Stockwell BR (2014) Regulation of ferroptotic cancer cell death by GPX4. Cell 156(1–2):317–331. https://doi.org/10.1016/j.cell.2013.12.010
Sui X, Zhang R, Liu S, Duan T, Zhai L, Zhang M, Han X, Xiang Y, Huang X, Lin H, Xie T (2018) RSL3 drives ferroptosis through GPX4 inactivation and ROS production in colorectal cancer. Front Pharmacol 9:1371. https://doi.org/10.3389/fphar.2018.01371
Yang WS, Stockwell BR (2016) Ferroptosis: death by lipid peroxidation. Trends Cell Biol 26(3):165–176. https://doi.org/10.1016/j.tcb.2015.10.014
Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ, Stockwell BR (2016) Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol 12(7):497–503. https://doi.org/10.1038/nchembio.2079
Xu Y, Li X, Cheng Y, Yang M, Wang R (2020) Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary ischemia-reperfusion. FASEB J 34(12):16262–16275. https://doi.org/10.1096/fj.202001758R
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius E, Scheel CH, Mourão A, Buday K, Sato M, Wanninger J, Vignane T, Mohana V, Rehberg M, Flatley A, Schepers A, Kurz A, White D, Sauer M, Sattler M, Tate EW, Schmitz W, Schulze A, O’Donnell V, Proneth B, Popowicz GM, Pratt DA, Angeli JPF, Conrad M (2019) FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575(7784):693–698. https://doi.org/10.1038/s41586-019-1707-0
Qiang Z, Dong H, Xia Y, Chai D, Hu R, Jiang H (2020) Nrf2 and STAT3 alleviates ferroptosis-mediated IIR-ALI by regulating SLC7A11. Oxid Med Cell Longev 2020:5146982. https://doi.org/10.1155/2020/5146982
Shao A, Zhou Y, Yao Y, Zhang W, Zhang J, Deng Y (2019) The role and therapeutic potential of heat shock proteins in haemorrhagic stroke. J Cell Mol Med 23(9):5846–5858. https://doi.org/10.1111/jcmm.14479
Kellner M, Noonepalle S, Lu Q, Srivastava A, Zemskov E, Black SM (2017) ROS Signaling in the pathogenesis of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS). Adv Exp Med Biol 967:105–137. https://doi.org/10.1007/978-3-319-63245-2_8
Fowler AA 3rd, Truwit JD, Hite RD, Morris PE, DeWilde C, Priday A, Fisher B, Thacker LR 2nd, Natarajan R, Brophy DF, Sculthorpe R, Nanchal R, Syed A, Sturgill J, Martin GS, Sevransky J, Kashiouris M, Hamman S, Egan KF, Hastings A, Spencer W, Tench S, Mehkri O, Bindas J, Duggal A, Graf J, Zellner S, Yanny L, McPolin C, Hollrith T, Kramer D, Ojielo C, Damm T, Cassity E, Wieliczko A, Halquist M (2019) Effect of vitamin c infusion on organ failure and biomarkers of inflammation and vascular injury in patients with sepsis and severe acute respiratory failure: the CITRIS-ALI randomized clinical trial. JAMA 322(13):1261–1270. https://doi.org/10.1001/jama.2019.11825
Amaral EP, Costa DL, Namasivayam S, Riteau N, Kamenyeva O, Mittereder L, Mayer-Barber KD, Andrade BB, Sher A (2019) A major role for ferroptosis in Mycobacterium tuberculosis-induced cell death and tissue necrosis. J Exp Med 216(3):556–570. https://doi.org/10.1084/jem.20181776
Liu P, Feng Y, Li H, Chen X, Wang G, Xu S, Li Y, Zhao L (2020) Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell Mol Biol Lett 25:10. https://doi.org/10.1186/s11658-020-00205-0
Li J, Lu K, Sun F, Tan S, Zhang X, Sheng W, Hao W, Liu M, Lv W, Han W (2021) Panaxydol attenuates ferroptosis against LPS-induced acute lung injury in mice by Keap1-Nrf2/HO-1 pathway. J Transl Med 19(1):96. https://doi.org/10.1186/s12967-021-02745-1
Guo Y, Hu M, Ma J, Chinnathambi A, Alharbi SA, Shair OHM, Ge P (2021) Protective effect of panaxydol against repeated administration of aristolochic acid on renal function and lipid peroxidation products via activating Keap1-Nrf2/ARE pathway in rat kidney. J Biochem Mol Toxicol 35(1):e22619. https://doi.org/10.1002/jbt.22619
Dar HH, Tyurina YY, Mikulska-Ruminska K, Shrivastava I, Ting HC, Tyurin VA, Krieger J, St Croix CM, Watkins S, Bayir E, Mao G, Armbruster CR, Kapralov A, Wang H, Parsek MR, Anthonymuthu TS, Ogunsola AF, Flitter BA, Freedman CJ, Gaston JR, Holman TR, Pilewski JM, Greenberger JS, Mallampalli RK, Doi Y, Lee JS, Bahar I, Bomberger JM, Bayır H, Kagan VE (2018) Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J Clin Invest 128(10):4639–4653
Deshpande R, Zou C (2020) Pseudomonas aeruginosa induced cell death in acute lung injury and acute respiratory distress syndrome. Int J Mol Sci 21(15):5356. https://doi.org/10.3390/ijms21155356
Banchini F, Vallisa D, Maniscalco P, Capelli P (2020) Iron overload and Hepcidin overexpression could play a key role in COVID infection, and may explain vulnerability in elderly, diabetics, and obese patients. Acta Biomed 91(3):e2020013. https://doi.org/10.23750/abm.v91i3.9826
Jacobs W, Lammens M, Kerckhofs A, Voets E, Van San E, Van Coillie S, Peleman C, Mergeay M, Sirimsi S, Matheeussen V, Jansens H, Baar I, Vanden Berghe T, Jorens PG (2020) Fatal lymphocytic cardiac damage in coronavirus disease 2019 (COVID-19): autopsy reveals a ferroptosis signature. ESC Heart Fail 7(6):3772–3781. https://doi.org/10.1002/ehf2.12958
Habib HM, Ibrahim S, Zaim A, Ibrahim WH (2021) The role of iron in the pathogenesis of COVID-19 and possible treatment with lactoferrin and other iron chelators. Biomed Pharmacother 136:111228. https://doi.org/10.1016/j.biopha.2021.111228
Huang Y, Zhang W, Yu F, Gao F (2017) The cellular and molecular mechanism of radiation-induced lung injury. Med Sci Monit 23:3446–3450. https://doi.org/10.12659/msm.902353
Zhu X, Li X, Gu H, Yu W, Fu X (2019) Radiation-induced lung injury patterns and the misdiagnosis after SBRT of lung cancer. Eur J Radiol 121:108708. https://doi.org/10.1016/j.ejrad.2019.108708
Li X, Duan L, Yuan S, Zhuang X, Qiao T, He J (2019) Ferroptosis inhibitor alleviates Radiation-induced lung fibrosis (RILF) via down-regulation of TGF-β1. J Inflamm 16:11. https://doi.org/10.1186/s12950-019-0216-0
Li X, Zhuang X, Qiao T (2019) Role of ferroptosis in the process of acute radiation-induced lung injury in mice. Biochem Biophys Res Commun 519(2):240–245. https://doi.org/10.1016/j.bbrc.2019.08.165
Bei Y, Pan LL, Zhou Q, Zhao C, Xie Y, Wu C, Meng X, Gu H, Xu J, Zhou L, Sluijter JPG, Das S, Agerberth B, Sun J, Xiao J (2019) Cathelicidin-related antimicrobial peptide protects against myocardial ischemia/reperfusion injury. BMC Med 17(1):42. https://doi.org/10.1186/s12916-019-1268-y
Tang C, Hu Y, Gao J, Jiang J, Shi S, Wang J, Geng Q, Liang X, Chai X (2020) Dexmedetomidine pretreatment attenuates myocardial ischemia reperfusion induced acute kidney injury and endoplasmic reticulum stress in human and rat. Life Sci 257:118004. https://doi.org/10.1016/j.lfs.2020.118004
Liang S, Wang Y, Liu Y (2019) Dexmedetomidine alleviates lung ischemia-reperfusion injury in rats by activating PI3K/Akt pathway. Eur Rev Med Pharmacol Sci 23(1):370–377. https://doi.org/10.26355/eurrev_201901_16785
Riou O, Bourgier C, Brengues M, Bonnefoi N, Michaud HA, Castan F, Gourgou S, Draghici R, Farcy-Jacquet MP, Bons F, Fenoglietto P, Ozsahin M, Azria D (2019) Predictive assays for responses of tumors and normal tissues in radiation oncology. Cancer Radiother 23(6–7):666–673. https://doi.org/10.1016/j.canrad.2019.07.152
Dong H, Qiang Z, Chai D, Peng J, Xia Y, Hu R, Jiang H (2020) Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging 12(13):12943–12959. https://doi.org/10.18632/aging.103378
Li Y, Cao Y, Xiao J, Shang J, Tan Q, Ping F, Huang W, Wu F, Zhang H, Zhang X (2020) Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ 27(9):2635–2650. https://doi.org/10.1038/s41418-020-0528-x
Qiu Y, Wan B, Liu G, Wu Y, Chen D, Lu M, Chen J, Yu R, Chen D, Pang Q (2020) Nrf2 protects against seawater drowning-induced acute lung injury via inhibiting ferroptosis. Respir Res 21(1):232. https://doi.org/10.1186/s12931-020-01500-2
Zhou H, Li F, Niu JY, Zhong WY, Tang MY, Lin D, Cui HH, Huang XH, Chen YY, Wang HY, Tu YS (2019) Ferroptosis was involved in the oleic acid-induced acute lung injury in mice. Sheng Li Xue Bao 71(5):689–697. https://doi.org/10.13294/j.aps.2019.0070
Zhu W, Huang Y, Ye Y, Wang Y (2018) Deferoxamine preconditioning ameliorates mechanical ventilation-induced lung injury in rat model via ROS in alveolar macrophages: a randomized controlled study. BMC Anesthesiol 18(1):116. https://doi.org/10.1186/s12871-018-0576-7
Ritter C, da Cunha AA, Echer IC, Andrades M, Reinke A, Lucchiari N, Rocha J, Streck EL, Menna-Barreto S, Moreira JC, Dal-Pizzol F (2006) Effects of N-acetylcysteine plus deferoxamine in lipopolysaccharide-induced acute lung injury in the rat. Crit Care Med 34(2):471–477. https://doi.org/10.1097/01.ccm.0000199069.19193.89
Yang L, Wang H, Yang X, Wu Q, An P, Jin X, Liu W, Huang X, Li Y, Yan S, Shen S, Liang T, Min J, Wang F (2020) Auranofin mitigates systemic iron overload and induces ferroptosis via distinct mechanisms. Signal Transduct Target Ther 5(1):138. https://doi.org/10.1038/s41392-020-00253-0
Scindia Y, Wlazlo E, Leeds J, Loi V, Ledesma J, Cechova S, Ghias E, Swaminathan S (2019) Protective role of hepcidin in polymicrobial sepsis and acute kidney injury. Front Pharmacol 10:615. https://doi.org/10.3389/fphar.2019.00615
Miotto G, Rossetto M, Di Paolo ML, Orian L, Venerando R, Roveri A, Vučković AM, Bosello Travain V, Zaccarin M, Zennaro L, Maiorino M, Toppo S, Ursini F, Cozza G (2020) Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol 28:101328. https://doi.org/10.1016/j.redox.2019.101328
Li S, Zhou C, Zhu Y, Chao Z, Sheng Z, Zhang Y, Zhao Y (2021) Ferrostatin-1 alleviates angiotensin II (Ang II)- induced inflammation and ferroptosis in astrocytes. Int Immunopharmacol 90:107179. https://doi.org/10.1016/j.intimp.2020.107179
Dodson M, Castro-Portuguez R, Zhang DD (2019) NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol 23:101107. https://doi.org/10.1016/j.redox.2019.101107
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82000082), the Natural Science Foundation Of Anhui Province (2008085QH353), and the Fundamental Research Funds for the Central University (WK9110000124).
Author information
Authors and Affiliations
Contributions
WX and JZ developed the idea for the study, and XL wrote the paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All authors are familiar with the contents of the final draft and take responsibility for the authenticity of the data used in the paper. This manuscript is original and has not been previously published, nor has it been simultaneously submitted to any other journal.
Consent for publication
The manuscript has been approved by all authors for publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Liu, X., Zhang, J. & Xie, W. The role of ferroptosis in acute lung injury. Mol Cell Biochem 477, 1453–1461 (2022). https://doi.org/10.1007/s11010-021-04327-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-021-04327-7