
Abstract
Novel H-bonded liquid crystals (HBLCs) are synthesized and examined for their mesomorphic behavior. The HBLCs are prepared with Schiff base proton acceptors containing 8 and 18 carbons in the alkyl chain and the proton donor with 4-substituted benzoic acids. All the compounds exhibit smectic A mesophases, with one of the compounds exhibiting LC properties below 298 K. It is interesting to note that the ethoxy-substituted HBLCs exhibit a wide range of thermal stability. Density functional theory calculations revealed the formation of two hydrogen bonds between the substituted acids and the pyridine ring based on the orientation of the donor and the acceptor moieties. There are no significant changes observed in the hydrogen bond length with the increase in the chain length of the proton acceptor moiety, indicating the dilution of the cores by the longer alkyl chain lengths is compensated with the (+ I) effect of alkoxy substituents. Quantum chemical modeling studies on these molecules revealed the reduction in the HOMO–LUMO energy gap by approximately 0.3 eV for oxy-containing compounds, making them more chemically reactive by donating electrons (+ I) into the aromatic cores. These materials provide a significant breakthrough in designing innovative LC materials. This work is in support of the SDG-9 of the United Nations.
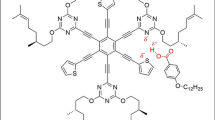
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Discovering the potential of liquid crystals (LC) is genuinely remarkable as these materials possess a unique combination of properties that make them fluid and structured [1]. New liquid crystals are designed to meet specific chemical functions suitable for applications [2]. One of the most significant soft-matter interactions is hydrogen bonding, known for its directionality and selectivity [3]. Hydrogen bonding between complementary molecules is a versatile tool for obtaining self-assembled supramolecular structures [4,5,6,7,8,9,10,11,12]. These non-covalent interactions, such as hydrogen bonds, are thermodynamically favorable for supramolecular formation [13]. Capacitors, optical filters, and light modulators utilize hydrogen-bonded liquid crystals (HBLCs) [14]. Furthermore, their stimuli-responsive [15], self-healing [16], and recyclable qualities [17] have made them valuable in other applications as ‘smart’ materials [18]. For many known categories of liquid crystals like supramolecular, polymeric, elastomer, etc., the ‘pyridinyl-carboxyl’ combination is the most prominent structural element whose self-organization has caused a significant change in mesomorphism from their starting components [19]. Acids that have alkyl substituents, such as methyl (–CH3) and ethyl (–C2H5), are known as electron-donating groups. They activate the aromatic ring by increasing the electron density through the Inductive donating effect (also known as the + I effect) [20, 21]. Several hydrogen-bonded liquid crystals have been synthesized with substituted carboxylic acids and phenols acting as proton donors, while pyridine and bipyridine moieties, stilbazole derivatives and isoquinolines act as bi-functional proton acceptors [22,23,24,25] Although there are various reports [26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42] on the synthesis of liquid crystals obtained with inherently mesogenic alkyloxy benzoic acids as proton donors (with alkyl chain length propyl and above) reports on methyl/methoxy- and ethyl/ethoxy-containing benzoic acids as proton donors are few as they are inherently non-mesogenic.
The present study demonstrates the synthesis of new series of hydrogen-bonded compounds considering non-mesogenic methyl/methoxy- and ethyl/ethoxy-containing benzoic acids as proton donors and a newly synthesized non-mesogenic proton acceptor. We have achieved synthesizing a hydrogen-bonded compound exhibiting mesomorphism below 298 K. We investigated the mesomorphic behavior of two non-mesogenic cores with varied acceptor chain lengths (8 and 18 carbons) and electron-rich donor substituents. We report the synthesis of the proton acceptor P18 viz., (4-pyridyl)-benzylidene-4'-n-octadecylaniline and 8 new HBLCs viz., P8:MBA (P8:4-methyl benzoic acid), P8:MOBA (P8:4-methoxy benzoic acid), P8:EBA (P8:4-ethyl benzoic acid), P8:EOBA (P8:4-ethoxy benzoic acid), P18:MBA (P18:4-methyl benzoic acid), P18:MOBA (P18:4-methoxy benzoic acid), P18:EBA (P18:4-ethyl benzoic acid) and P18:EOBA (P18:4-ethoxy benzoic acid). Structure–property relationships have been investigated in their LC nature with varied chain lengths and substituents. Our study focused on determining how oxy substituents affect the mesophase formations and LC temperatures. We compared the results of this research to our previous study, where we used polar substituents as donors to enhance the molecular dipole and improve molecular ordering in similar HBLCs. Density functional theory calculations provided accurate descriptions of HOMO and LUMO, molecular orbital composition, total energy, and relative energy gaps, giving us better insight into physical properties like dipole moment and hydrogen bond length.
Experimental
Materials and methods
To synthesize the proton donor, we obtained precursors such as 4-n-octadecyl aniline, and 4-pyridinecarboxaldehyde from Alfa Aesar and Merck, India. We also acquired substituted 4-alkoxy benzoic acids from Merck, India. The structure of the proton acceptor P18 has been confirmed through 1H NMR spectra obtained by the Bruker Ascend 400 MHz spectrometer. Deuterated chloroform is used as a solvent, and tetramethylsilane served as the internal standard. 13C NMR is obtained by the Bruker Ascend 75 MHz spectrometer. Deuterated chloroform is used as a solvent. Intermolecular hydrogen bonding is verified through IR spectra analysis using Shimadzu 00254 and the ATR probing method. The liquid crystal mesophases are studied using Leitz DMRXP polarizing optical microscope (POM) with METTLER HS1 hot stage controller with a cooling rate of 2°C min-1. Differential scanning calorimetry (DSC) with 10°C min-1 scans are collected using DSC 8000 PerkinElmer. Samples are crimped in weighted 30 µL aluminum pans.
Theoretical method
Density functional theory (DFT) calculations were performed for all the hydrogen-bonded compounds using the B3LYP theory and the TZVP basis set. The structures of these complexes were optimized in their ground electronic state, followed by the harmonic frequency calculation. The appearance of nonnegative frequencies in the harmonic frequency calculation ensures the optimized structure at the global minimum on the potential energy surface. All the calculations were performed with Gaussian 09 program suite [43].
Synthesis of proton acceptor and preparation of hydrogen-bonded liquid crystals
The scheme below (Fig. 1) depicts the one-step synthesis of the proton acceptor P18. Refluxing an equimolar 1:1 mixture of 4-pyridinecarboxaldehyde and 4-n-octadecyl aniline in ethanol for 6–7 h yields the proton acceptor P18. The resultant product is filtered and recrystallized with ethyl acetate. The melting point of the compound is found to be 72°C (345 K). IR, 1H NMR and 13C NMR characterization techniques are used to validate the synthesized product. The synthesis of P8 is already reported [44]. Proton acceptors with 8- and 18-carbon chain lengths and substituted 4-alkyl/4-alkoxy benzoic acids are taken in a 1:1 ratio using THF (tetrahydrofuran) as the solvent and refluxed for 1 h. As a result, hydrogen-bonded compounds are produced when the solvent evaporates, yielding supramolecules, i.e., P8:MBA, P8:MOBA, P8:EBA, P8:EOBA, P18:MBA, P18:MOBA, P18:EBA and P18:EOBA.

Scheme depicting the synthesis of P18, along with their molecular structures and sample names
Results and discussion
Proton nuclear magnetic resonance spectroscopy (1H NMR) of the proton acceptor (P18)
1H-NMR (400 MHz, CDCl3) δ (ppm) 0.77–0.81 (3H, t, –CH3), 1.18–1.23 (30H, m, –(CH2)15), 1.51–1.56 (2H, t, –CH2), 2.52–2.57 (2H, t, Ar–CH2), 7.09–7.17 (4H, m, Ar–CH), 7.64–7.66 (2H, d, Ar–CH), 8.40 (1H, s, HC = N), 8.62–8.64 (2H, d, Ar–CH).
The 1H-NMR spectra (Fig. 2a) confirm the formation of the –HC = N Schiff base P18 from 4-pyridinecarboxaldehyde and 4-n-octadecyl aniline as starting materials. The total number of hydrogens in the P18 molecule corresponds to 1H-NMR peaks. With a chemical shift of δ 0.77–0.81 ppm, the terminal –CH3 splits into triplets. The carbon skeleton –CH2 with 30H separates into multiplets with δ values ranging from 1.18 to 1.23 ppm. The near aromatic –CH2 group splits into triplets at δ 1.51–1.56 ppm, while the Ar–CH2 group splits into triplets δ 2.52–2.57 ppm values. Aldehyde Ar–CH2, 4H multiplet has a greater δ of 7.09–7.77 ppm. At δ 8.40 ppm, the Schiff base –HC = N 1H produces a singlet largely dependent on the electronegative character of the substituent pyridine ring. The pyridine ring is composed of 4H, with the 2H, Ar–CH doublet splitting at 8.62 ppm and the 2H, Ar–CH doublet splitting at 7.64–7.66 ppm. The peak at 5.22 ppm corresponds to the residual DCM.

a 1H NMR spectrum of P18; b 13C NMR spectrum of P18
Carbon-13 nuclear magnetic resonance spectroscopy (13C NMR) of the proton acceptor (P18)
13C-NMR (75 MHz, CDCl3) δ 14.40, 22.67, 29.27, 29.34, 29.67, 31.91, 35.54, 120.94, 129.25, 142.34, 143.21, 148.34, 150.18, 156.68 (Fig. 2b).
Fourier transform infra-red spectroscopy (FTIR)
FTIR of P18
The Schiff base P18 spectra (Fig. 3a) revealed no absorption band above 3100 cm−1, depicting the absence of the free –NH2 group of the starting material octadecyl aniline. Additionally, the absence of the band associated with pyridinecarboxaldehyde’s C = O stretching at the 1715–1695 cm−1 range indicates that aniline and carboxaldehyde are condensed. At 2963 cm−1, pyridine Ar–CH stretching can be observed. Bands at 2914 cm−1 and 2847 cm−1 suggest the Schiff base C–H stretches. The bands at 1601 cm−1 denotes the C = N stretching of the pyridine moiety, while the band at 1628 cm−1 denotes the C = N stretching of the Schiff base [45].

a Compiled FTIR spectrum of P18; A: 4-n-octadecyl aniline, B: 4-pyridine carboxaldehyde C: (4-pyridyl)-benzylidene-4'-n-octadecylaniline; b Compiled FTIR spectrum of hydrogen-bonded P8:MBA: A: (4-pyridyl)-benzylidene-4'-n-octylaniline (P8) B: 4-methyl benzoic acid (MBA) C: hydrogen-bonded compound of A and B; c Compiled FTIR spectrum of hydrogen-bonded P18:MBA; A: (4-pyridyl)-benzylidene-4'-octadecylaniline (P18) B: 4-methyl benzoic acid (MBA) C: hydrogen-bonded compound of A and B d FTIR spectrum of P8:MOBA; P8:EBA; P8:EOBA; e FTIR spectrum of P18:MOBA; P18:EBA; P18:EOBA. Tables 1.1 and 1.2 display the stretching frequencies of O–H, C = O, C = N, and HB (hydrogen bond)
FTIR for hydrogen-bonded P18 compounds
The precursor acid’s FTIR spectra have been compared with their hydrogen-bonded equivalents. Here is an example: Figure 3b presents the FTIR spectra of P8:MBA combined, while Fig. 3c exhibits the FTIR spectra of P18:MBA combined. The broad peak, which corresponds to hydrogen-bonded O–H stretching of MBA, is shifted to 2846 cm−1, and a decrease in the broadness is observed [46]. This suggests new hydrogen bonding interactions between MBA and P18 that are linear instead of complimentary. Figure 3d displays the FTIR spectrum of P8:MOBA, P8:EBA, and P8:EOBA, while Fig. 3e showcases the FTIR spectrum of hydrogen-bonded binary mixtures of P18:MOBA, P18:EBA, and P18:EOBA. Tables 1.1 and 1.2 in Fig. 3 list the peaks corresponding to the O–H stretching of the acid, the C = O stretching of the carboxyl group, and the C = N stretching of the pyridine.
Phase transition studies by polarizing optical microscopy (POM)
All eight synthesized novel HBLCs showed a smectic A (SmA). (Fig. 4a–d) with radiating textures from the molecules on the substrate. The only exception was the P8:MOBA combination, which displayed poorly developed SmA phases at 373.8 K and had a limited liquid crystal range. However, all the other HBLC combinations of acids with P8 exhibited evenly spaced focal conic textures [47]. Furthermore, at 297.8 K, P8:EOBA (Fig. 4b) crystallized near room temperature. On the other hand, smectic A focal conic textures were evenly distributed and well-resolved in all HBLCs containing P18 (Fig. 4c–d). During cooling at 377 K, the long-chained P18:MBA (Fig. 4c) initially took on a streak-like formation but eventually arranged into smectic batonnets, resulting in focal conic projections. It may be noticed that the orthogonal smectic A phases develop focal conic fans in the homogenous regions, while the homeotropically arranged molecules appear black corresponding to the pseudo-isotropic texture [48,49,50]. The mesomorphic temperatures of ethoxy-containing hydrogen-bonded compounds are the widest compared to other compounds of the series. This indicates that electron-rich oxygen in the molecular structure contributes to mesomorphism through the significant resonance effect and + I (inductive effect). On the contrary, the methoxy-containing hydrogen-bonded compounds have not shown a significant increase in the mesomorphism of P18 hydrogen-bonded compounds. Similarly, P8:MOBA has shown a lower mesomorphic range than P8:MBA. This indicates that methyl and methoxy substituents with one carbon atom do not contribute to the molecule’s flexibility. In contrast, ethyl and ethoxy substituents with two carbons atoms show a better contribution to the flexibility along with the + I effect.

Smectic A textures a P8:EBA at 335.7 K (20 ×); b P8:EOBA at 297.8 K (10 ×) c P18:MBA at 337.1 K (10 ×); d P18:EOBA at 408 K (20 ×)
Thermal characterization by differential scanning calorimetry (DSC)
The DSC curves are run three times, and the results are discussed. The hydrogen-bonded compounds exhibit clear curves, confirming the mesogens’ enantiotropic nature [51]. One example, P8:MBA (Fig. 5a), melts into smectic A at 339.6 K with an enthalpy of 37.42 Jg−1, then transitions to isotropic at 376.7 K with an enthalpy change of 11.81 Jg−1. During the cooling cycle, the isotropic to smectic transition is visible at 374.6 K with an enthalpy of 11.99 Jg−1, and crystallization occurs at 311.2 K with an enthalpy change of 37.62 Jg−1. In the DSC curves of P8:MOBA (Fig. 5b), a double peak is observed in the heating cycle, while the isotropic peak is not well-resolved. The peak at 347.3 K had an enthalpy of 28.20 Jg−1, and the peak at 355.6 K had an enthalpy of 37.95 Jg−1. It can be observed that in all the hydrogen-bonded compounds of present series, the change in enthalpy during the transition to liquid crystalline phase is not more than 15.5 Jg−1. Thus, the larger change in enthalpy of 28.20 Jg−1 and 37.95 Jg−1 indicates a crystal-to-crystal transition at 347.3 K [52]. On cooling, the sample transformed to smectic A phase at 375.5 K with an enthalpy of 7.5 Jg−1 which on further heating transformed to crystal at 325.8 K with an enthalpy of 29.90 Jg−1. The P18 HBLCs are also found to display enantiotropic smectic phases (Fig. 6a–d). The hydrogen-bonded compound P18:MOBA (Fig. 6b) showed double melting behavior before transforming to liquid state. The double peaks at 348.9 K and 361.1 K with an enthalpy of 64.30 Jg−1 and 72.74 Jg−1, respectively, indicated the crystal-to-crystal transition at 348.9 K. Also, during the cooling cycle, a crystal-to-crystal transformation is indicated at 339 K with a change in enthalpy of 24.95 Jg−1. A similar crystal-to-crystal transition is observed at 348.8 K with a enthalpy change of 69.43 Jg−1 during the heating cycle of P18:EOBA (Fig. 6d). The simultaneous POM observation of the samples indicated the smectic A phase present in all the compounds with the formation of focal conic fans as shown in Fig. 4a–d.

DSC curves of hydrogen-bonded compounds of P8, including heating (h) and cooling (c) cycles recorded at 10°C min-1 (308 K min-1) a P8:MBA; b P8:MOBA; c P8:EBA; d P8:EOBA. Table 2.1 shows characteristic transition temperatures (K) and enthalpy values J g-1 and the liquid crystal temperature range (ΔT)LC of P8 hydrogen-bonded compounds

DSC curves of hydrogen-bonded compounds of P18, including heating (h) and cooling (c) cycles recorded at 10°C/min (308 K/min) a P18:MBA; b P18:MOBA; c P18:EBA; d P18:EOBA. Table 2.2 shows characteristic transition temperatures (K) and enthalpy values J g-1 and the liquid crystal temperature range (ΔT)LC of P18 hydrogen-bonded compounds
Tables 2.1 and 2.2 in Figs. 5 and 6, respectively, comprehensively summarize the DSC data for all compounds. Figure 7a depicts the comparative plots of crystallization temperatures of P8 hydrogen-bonded compounds with P18 compounds. The isotropic transition temperatures are similarly compared (Fig. 7b). The melting points of MBA, MOBA, EBA and EOBA are 450 K, 457 K, 385 K and 470 K, respectively. The melting point of the proton acceptor, P18, is 345 K, while P8 is a liquid at RT. The proton donors as well as acceptors are non-mesogenic. It is noticed that the melting points of the HB compounds lie far from the melting points of the corresponding benzoic acids. The additional interactions that prevail on the formation of HB between the two moieties are responsible for this substantial change in melting points. It may be observed that the isotropic transition temperatures of P8 and P18 compounds are similar, with P18 compounds exhibiting slightly higher isotropic temperatures than P8 compounds except for P8:EOBA. This indicates that the proton acceptor’s chain length does not influence the isotropic temperatures much. On the other hand, there is a significant variation in the crystallization temperatures of P8 and P18 compounds. P8 compounds exhibit lower crystallization temperatures than P18 compounds, indicating that the lower chain lengths favor obtaining liquid crystals toward ambient temperatures.

Phase diagrams a crystallization temperatures of P8 and P18 hydrogen-bonded compounds; b isotropic transition temperatures of P8 and P18 hydrogen-bonded compounds
The phase transition entropy changes for the transitions from crystal to smectic A and smectic A to isotropic during cooling cycle is calculated for the HBLCs as shown below. Based on the formula, ΔH = T.ΔS, the change in the entropy, ΔS (Jg−1 K), is calculated where ΔH (Jg−1) is the change in the enthalpy at the phase transition temperature, T (K) [11, 53]. The entropy changes in the HBLCs of P18 compounds are relatively higher during crystallization as compared to HBLCs of P8. It may be attributed to the long alkyl chain length of 18 carbons adding flexibility to the molecules and thus exhibiting higher entropy change as compared to P8 compounds.
ΔSiso-SmA | ΔSSmA-Cryst | ΔSiso-SmA | ΔSSmA-Cryst | ||
|---|---|---|---|---|---|
P8:MBA | 0.032 | 0.121 | P18:MBA | 0.032 | 0.283 |
P8:MOBA | 0.019 | 0.092 | P18:MOBA | 0.010 | 0.176 |
P8:EBA | 0.028 | 0.031 | P18:EBA | 0.029 | 0.255 |
P8:EOBA | 0.036 | Not resolved | P18:EOBA | 0.029 | 0.355 |
Role of electron-donating substituents on inducing mesomorphism in hydrogen-bonded compounds
To understand the influence of the oxygen atom on the mesomorphic thermal stabilities, a phase diagram of MOBA compounds of P8 and P18 is superimposed on the phase diagram of MBA compounds of P8 and P18 (Fig. 8a), and the phase diagrams of the EOBA compounds of P8 and P18 are superimposed on the EBA compounds of P8 and P18 (Fig. 8b).

Comparative thermal range (ΔT)LC plots a P8 and P18 MBA and MOBA b P8 and P18 EBA and EOBA
It has been reported that the lower homologs of 4-n-alkyl/alkyloxy benzoic acids, namely MBA, EBA, MOBA, and EOBA, do not exhibit liquid crystalline behavior despite their dimerization. This is due to the molecule’s insufficient length-to-breadth ratio [54, 55]. But when the above benzoic acids are combined with pyridyl moieties like P8 and P18 in a 1:1 molar ratio, they display a smectic A mesomorphism. When it comes to the terminal alkyl substituent, such as methyl, ethyl, methoxy, and ethoxy, they donate electrons inductively through a σ bond into the benzene ring. This positive inductive effect helps to decrease the acidity of the substituted benzoic acids. Additionally, the methylene (–CH2) units found in the flexible end chain of the proton acceptor moiety also have a positive inductive effect on the ring, making the proton acceptor moiety (pyridyl moiety) an electron-rich species. Proton donor and acceptor moieties with + I effect facilitate the self-assembly of molecules into supramolecular structures. This self-assembly of molecules increases the length-to-breadth (l/b) ratio, leading to the spontaneous arrangement of the proton donor and acceptor moieties with positional order along with the orientational order, resulting in exhibiting a SmA mesophase.
The alkyloxy groups in benzene have different electronegativities between the oxygen and carbon atoms. Additionally, the lone pair of electrons on the oxygen atom overlaps with the π-electron cloud of the benzene compound [56]. The P8:EOBA and P18:EOBA compounds exhibit increased mesomorphic thermal stabilities due to the overlapping of electrons, resulting in additional intermolecular interactions. The larger surface area and stronger intermolecular interactions of the ethoxy substituent, which is bulkier than the methoxy substituent, contribute to this difference.
Density functional theory calculations
The optimized molecular structure of P8:MBA is given below as a representative case (Fig. 9).

Optimized methyl substituent structure at the proton donor’s para-position
It is evident from the figure that the optimized structures are not planar. The para-substituted benzoic acid and pyridine rings are in the same plane, forming a hydrogen bond between OH and N with a hydrogen bond length of ~ 1.74 Å. Furthermore, they form another relatively weaker hydrogen bond between O and H, with a hydrogen bond length of ~ 2.4 Å. It is observed that the hydrogen bond length increases marginally from the methyl to ethoxy groups in both P8 and P18 compounds. Despite the co-planarity of the hydrogen bonding components, the aromatic ring attached to pyridine–C–N is not co-planar. The tilt angle remains largely unaffected by a substituent group at the para-position in the benzoic acid, and the average tilt angle is found to be ~ 35° (Fig. 10a–c).

Variations on tilt angles (θ) for the aromatic C–N bond and the hydrogen bond lengths OH–N and O–H in P8 and P18 compounds. a The C–N–C6H6 tilt angle; b OH–N hydrogen bond length and c O–H hydrogen bond length
Comparing the effects of electron-donating and withdrawing substituents on inducing mesomorphism in hydrogen-bonded compounds
According to studies, halogen elements like chloro or fluoro, which withdraw electrons, can decrease transition temperatures by strengthening polarization [57, 58]. Scientists have accomplished the production and isolation of low-melting LC materials by replacing the rigid rod moiety of the molecule with fluorine [59]. Using these findings, we have previously successfully developed the HB compounds of P8 using 4-chloro and 4-fluoro benzoic acids [44]. Further, we compared the mesomorphic thermal stabilities of these complexes with those of 4-alkyl/alkoxybenzoic acid compounds. Figure 11 displays the mesomorphic thermal stabilities of P8 and P18 hydrogen-bonded compounds.

Comparative mesomorphic thermal stabilities (ΔT)LC of P8 and P18 hydrogen-bonded compounds
It is noticed that the mesomorphic thermal stabilities of HBLCs with electron-withdrawing substituents exhibit lower mesomorphic thermal stability than those of the electron-donating substituents. Molecules containing chloro/fluoro substituents have an electron-withdrawing effect (-I). This effect stabilizes the carboxylate anion and increases the acidity of chloro/fluoro benzoic acids. Conversely, the methylene (–CH2) units of proton acceptor moieties in the same HBLCs have an electron-donating effect, resulting in a positive inductive effect + I [60]. Due to this positive inductive effect, the aromatic ring of the acceptor moiety (in our case, P8 and P18) becomes electron-rich. When the proton acceptor with a positive inductive effect and the proton donor with a negative inductive effect come together, they form supramolecular hetero-synthons with positional order. This leads to the spontaneous formation of an orthogonal smectic A mesophase when the isotropic liquid is cooled. All the compounds, including P8:MBA, P18:MBA, P8:MOBA, P18:MOBA, P8:EBA, P18:EBA, P18:EOBA, P18:EOBA, P8:ClBA, P18:ClBA, P8:4FBA, and P18:4FBA, exhibit a hydrogen bond that is inclined along the long molecular axis of the complex. This leads to a high longitudinal dipole moment (µl). The total dipole moment of some of the representative hydrogen-bonded compounds is summarized in Table 1.
Of all the compounds, those with chloro/fluoro substitutions in HBLCs exhibit a higher longitudinal dipole moment than those with alkyl/alkyloxy substitutions. However, our studies show chloro/fluoro benzoic acid complexes have lower mesomorphic thermal stabilities than other HBLC compounds. This may be due to the polar nature of fluoro/chloro groups, which creates repulsive forces between molecules and causes them to move away from each other, decreasing intermolecular interactions. In contrast, supramolecules that had proton donors with non-polar methyl/ethyl substituents showed more vital London dispersive forces, leading to increased thermal stability in their mesomorphic state [61]. Through mesomeric resonance, the O atom of the methoxy/ethoxy group elevates the π-cloud, resulting in increased π–π stacking when compared to methyl/ethyl groups that are hyperconjugated with the aromatic ring [62].
To validate the observation, we have meticulously computed the hydrogen bond lengths of P8’s optimized structures with various substituents positioned at the para side of the proton donor. These substituents include –OH, –CH3, –C2H5, –C3H7, –OCH3, –OC2H5, –OC3H7, –NO2, –Cl, and –F. Observe Fig. 9 for an exemplary structure featuring methyl substituent at the para-position of the proton donor. Research has shown that compounds with electron-withdrawing substituents in the proton donor have a shorter hydrogen bond length. Specifically, those with NO2, –Cl, and –F groups exhibited lengths of 1.682, 1.713, and 1.720 Å, suggesting a stronger hydrogen bond formation between the compound’s proton donor and acceptor moieties. In Figs. 12 and 13, it can be observed that the electron-donating substituent of the proton donor exhibited a longer hydrogen bond length when combined with different substituents like –OH, –CH3, –C2H5, –OCH3, –OC2H5 and –OC3H7. The bond length ranged between 1.737 and 1.745 Å. The increased length of hydrogen bonds allows for greater flexibility and reduced molecule rigidity, resulting in increased π–π stacking. This suggests that hydrogen bonding is a beneficial interaction that plays a role in achieving mesomorphism. Furthermore, the nature of the interacting components and the directionality, shape, and stability of these non-covalent interactions play a crucial role in the realization of mesomorphism.

Optimized structures with calculated hydrogen bond lengths of P8 HBLC’s a P8:MBA; b P8:MOBA; c P8:EBA d P8:EOBA

Optimized structures with calculated hydrogen bond lengths of P18 HBLC’s a P18:MBA; b P18:MOBA; c P18:EBA d P18:EOBA
Effect of alkyl chain lengths on the mesomorphic and melting/clearing temperatures
Based on our previous research [63, 64]on HBLCs with varying alkyl chain lengths of proton acceptors as 10, 12, 14, and 16, it has been observed that the HBLC P16:4Cl has the lowest mesomorphic range of thermal stability (Table 2).
This is attributed to the dilution of the core induced by the long flexible end chain of P16, leading to decreased packing efficiency [65]. Similar is the trend observed in the hydrogen-bonded compounds of P18 in the present series.
With increasing chain length of proton acceptor from octyl to octadecyl, the melting/clearing points of the compounds have increased due to the increase in molecular mass. These results are found to agree with the reports [66] on non-HBLCs and other HBLCs [67,68,69].
Frontier molecular orbital analysis
The HOMO–LUMO calculations were performed on the compounds to locate the highly reactive position in these π–electron systems and their chemical reactivity [70]. Understanding the HOMO–LUMO energy gap is beneficial for comprehending charge transfer interactions and electrical transport. A molecule with a more significant HOMO–LUMO energy gap is more stable kinetically, with lower chemical reactivity. On the other hand, a molecule with a smaller gap is less stable and has higher chemical reactivity [71,72,73,74]. The highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbitals of the present series of compounds are shown in Figs. 14 and 15.

Optimized structures representing HOMO–LUMO energies of P8 HBLC’s a P8:MBA HOMO; b P8:MBA LUMO; c P8:MOBA HOMO; d P8:MOBA LUMO; e P8:EBA HOMO; f P8:EBA LUMO; g P8:EOBA HOMO; h P8:EOBA LUMO

Optimized structures representing HOMO- LUMO energies of P18 HBLC’s a P18:MBA HOMO; b P18:MBA LUMO; c P18:MOBA HOMO; d P18:MOBA LUMO; e P18:EBA HOMO; f P18:EBA LUMO; g P18:EOBA HOMO; h P18:EOBA LUMO
In the case of MBA- and EBA-substituted compounds, the electron density in HOMO is concentrated on the benzene ring and C = N. In contrast, for MOBA- and EOBA-substituted compounds, the electron density is on benzoic acid. The LUMO always shows the electron density on the pyridine ring and the C–N. The HOMO–LUMO energies are summarized, and the energy gaps are represented in Fig. 16.

HOMO–LUMO energy difference in eV and tabulated HOMO–LUMO energies of hydrogen-bonded compounds
The overall energy gap of < 4 eV for the π–electron systems indicate their high chemical reactivity. The energy gap is nearly the same for methyl- and ethyl-substituted compounds. For P8:MBA, P8:EBA, P18:MBA, and P18:EBA, it is 3.94 eV, whereas for MOBA (3.73 eV)- and EOBA (3.71 eV)-substituted compounds, the energy gap decreased marginally. The lowest energy gap exhibited by ethoxy-substituted compounds indicates its higher reactivity.
Conclusions
A new series of HBLCs has been successfully synthesized with non-mesogenic proton acceptor and donor combinations. These compounds display smectic A mesomorphism at a consistent working temperature and a supercooled phase. Oxygen containing substituents with two carbon chain on the proton donor contributed to a higher range of mesomorphism compared to other substituents. Additionally, substituents that release electrons on the proton donors are more effective in inducing mesomorphism than those that withdraw electrons from the proton donors. It is possible to decrease the crystallization temperatures by reducing the chain length of the proton acceptors.
The compounds in this series exhibit lower dipole moments and longer hydrogen bond lengths, indicating higher π–π stacking ability and lower rigidity. Methoxy- and ethoxy-substituted HBLCs exhibit higher chemical reactivity than methyl- and ethyl-substituted compounds, according to HOMO–LUMO calculations.
The oxy substituents in the molecular structure increased the mesomorphic thermal range. Ethoxy substituents with a larger surface area indicated the more vital intermolecular force of attraction contributing to mesomorphism. In the P8 and 18 compounds, hydrogen bonds were formed between OH and N, with a bond length of 1.74 Å, and O&H, with a bond length of 2.4 Å. The bond length increased slightly from the methyl to the ethoxy group.
Comparative studies showed that liquid crystals with electron-withdrawing groups stabilize carboxylate anions and increase acidity. In contrast, electron-donating groups decrease acidity and create an orthogonal smectic phase with positional order. Furthermore, electron-withdrawing groups with a polar nature generate repulsive forces that result in the molecules moving away from each other, causing fewer intermolecular interactions compared to electron-rich substituents. Adding oxygen boosts the effectiveness of π–π stacking compared to methyl and ethyl groups. HOMO–LUMO calculations show that electron density of these groups is concentrated on the benzene ring, with oxygen reducing the energy gap and resulting in more reactive substituents with broader mesophase ranges.
References
Beeckman J, Neyts K, Vanbrabant PJM. Liquid-crystal photonic applications. Opt Eng. 2011. https://doi.org/10.1117/1.3565046.
Hu YX, Hao X, Xu L, Xie X, Xiong B, Hu Z, Sun H, Yin GQ, Li X, Peng H, Yang HB. Construction of supramolecular liquid crystalline metallacycles for holographic storage of colored images. J Am Chem Soc. 2020. https://doi.org/10.1021/jacs.0c00698.
Johnson ER, Keinan S, Mori-Sánchez P, Contreras-García J, Cohen AJ, Yang W. Revealing noncovalent interactions. J Am Chem Soc. 2010. https://doi.org/10.1021/ja100936w.
Kato T, Kihara H, Yu T, Ujiie S, Iimura K, Fréchet JMJ, Kumar U. Hydrogen-bonded ferroelectric liquid-crystalline complexes based on a chiral benzoic acid and stilbazoles. Induction of chiral smectic C phases by molecular self-assembly. Ferroelectrics. 1993. https://doi.org/10.1080/00150199308019942.
He W, Pan G, Yang Z, Zhao D, Niu C, Huang W, Yuan X, Cuo J, Cao H, Yang H. Wide blue phase range in a hydrogen-bonded self-assembled complex of chiral fluoro-substituted benzoic acid and pyridine derivative. Adv Mater. 2009. https://doi.org/10.1002/adma.200802927.
Naoum MM, Fahmi AA, Alaasar MA. Supramolecular liquid crystals induced by hydrogen-bonding interactions between non-mesomorphic compounds. I. 4-(4′-Pyridylazophenyl)-4″-substituted benzoates and 4-substituted benzoic acids. Mol Cryst Liq Cryst. 2009. https://doi.org/10.1080/15421400902841403.
Naoum MM, Fahmi AA, Alaasar MA, Salem RA. Supramolecular liquid crystals in binary and ternary systems. Thermochim Acta. 2011. https://doi.org/10.1016/j.tca.2011.01.033.
Han J, Geng Q, Chen W, Zhu L, Wu Q, Wang Q. Self-assembled liquid crystals formed by hydrogen bonding between non-mesogenic 1,3,4-oxadiazole-based pyridines and substituted benzoic acids. Supramol Chem. 2012. https://doi.org/10.1080/10610278.2011.638378.
Rajanandkumar R, Pongali SPN, Madhu Mohan MLN. Investigations on hydrogen-bonded liquid crystals formed by p-n-alkyl benzoic acids and dodecane dicarboxylic acids. Mol Cryst Liq Cryst. 2016. https://doi.org/10.1080/15421406.2015.1106879.
Missaoui T, Amor IB, Soltani T, Ouada HB, Jeanneau E, Chevalier Y. Dielectric and electro-optic properties of cybotactic nematic phase in hydrogen-bonded liquid crystals. J Mol Liq. 2020. https://doi.org/10.1016/j.molliq.2020.112726.
Alhaddad OA, Ahmed HA, Hagar M. Experimental and theoretical approaches of new nematogenic chair architectures of supramolecular H-bonded liquid crystals. Molecules. 2020. https://doi.org/10.3390/molecules25020365.
Walker R, Pociecha D, Crawford CA, Storey JMD, Gorecka E, Imrie CT. Hydrogen bonding and the design of twist-bend nematogens. J Mol Liq. 2020. https://doi.org/10.1016/j.molliq.2020.112630.
Kato T, Fukumasa M, Fréchet JMJ. Supramolecular liquid-crystalline complexes exhibiting room-temperature mesophases and electrooptic effects. Hydrogen-bonded mesogens derived from alkylpyridines and benzoic acids. Chem Mater. 1995. https://doi.org/10.1021/cm00050a021.
Madhu Mohan MLN. Diversified applications of hydrogen bond liquid crystals. IOP Conf Ser Mater Sci Eng. 2021. https://doi.org/10.1088/1757-899X/1084/1/012089.
Shogo Yamane TK, Kana T, Yoshimitsu S. Stimuli responsive photoluminescent liquid crystals. Top Curr Chem. 2011. https://doi.org/10.1007/128_2011_275.
Yang X, Zhong X, Zhang J, Gu J. Intrinsic high thermal conductive liquid crystal epoxy film simultaneously combining with excellent intrinsic self-healing performance. J Mater Sci Technol. 2021. https://doi.org/10.1016/j.jmst.2020.08.027.
Chang CA, Cheng C, Yeh JA. Analysis and modeling of liquid-crystal tunable capacitors. IEEE Trans Electron Devices. 2006. https://doi.org/10.1109/TED.2006.875818.
Lugger SJD, Houben SJA, Foelen Y, Debije MG, Schenning APHJ, Mulder DJ. Hydrgen-bonded supramolecular liquid crystal polymers: smart materials with stimuli-responsive, self-healing, and recyclable properties. Chem Rev. 2022. https://doi.org/10.1021/acs.chemrev.1c00330.
Fredrickson DD, Hilberg BA, Lasure KK, Tessner JD, Waner AE, Zenner MD, Wiegel KN. Supramolecular main-chain liquid crystalline polymers and networks with competitive hydrogen bonding: a study of rigid networking agents in supramolecular systems. Liq Cryst. 2012. https://doi.org/10.1080/02678292.2012.714484.
Katritzky AR, Topsom RD. The σ-and π-inductive effects. J Chem Educ. 1971. https://doi.org/10.1021/ed048p427.
March J. Advanced organic chemistry. 4th ed. Hoboken: Wiley, Adelphi University; 2005.
Kato T, Kubota Y, Nakano M, Uruyu T. Doubly hydrogen-bonded liquid crystalline complexes obtained by supramolecular self-assembly of 2,6-diacyclaminopyridine and 4-alkoxybenzoic acids. Chem Lett. 1995. https://doi.org/10.1246/cl.1995.1127.
Kato T, Ihata O, Ujiie S, Tokita M, Watanabe J. Self-assembly of liquid-crystalline polyamide complexes through the formation of double hydrogen bonds between a 2,6-bis(amino)pyridine moiety and benzoic acids. Macromolecules. 1998. https://doi.org/10.1021/ma9719014.
Lin H, Ko C, Guo K, Cheng T. Supramolecular liquid crystals containing isoquinoline hydrogen-bonded acceptors. Liq Cryst. 1999. https://doi.org/10.1080/026782999205083.
Kihara H, Kato T, Uryu T, Ujiie S, Kumar U, Fréchet JMJ, Bruce DW, Price DJ. Supramolecular ferroelectric liquid crystals. Hydrogen-bonded complexes between benzoic acids and chiral stilbazoles. Liq Cryst. 1996. https://doi.org/10.1080/02678299608033792.
Gray GW, Hartley JB, Jones B. Mesomorphism and chemical constitution. Part V. The mesomorphic properties of the 4′-n-alkoxydiphenyl-4-carboxylic acids and their simple alkyl esters. J Chem Soc (Resumed). 1955. https://doi.org/10.1039/JR9550001412.
Fukumasa M, Kato T, Uryu T, Fréchet JMJ. The Simplest structure of the hydrogen-bonded mesogen built from 4-alkoxybenzoic acid and 4-alkylpyridine. Chem Lett. 1993. https://doi.org/10.1246/cl.1993.65.
Sideratou Z, Tsiourvas D, Paleos CM, Skoulios A. Liquid crystalline behaviour of hydrogen bonded complexes of a non-mesogenic anil with p-n-alkoxybenzoic acids. Liq Cryst. 1997. https://doi.org/10.1080/026782997209676.
Bernhardt H, Weissflog W, Kresse H. Preliminary communication hydrogen-bonded ionic liquid crystals. Liq Cryst. 1998. https://doi.org/10.1080/026782998206731.
Fukumasa M, Takeuchi K, Kato T. Preliminary communication-miscibility of a hydrogen-bonded mesogenic complex with normal liquid crystals. Liq Cryst. 1998. https://doi.org/10.1080/026782998207505.
Kraft A, Reichert A, Kleppinger R. Supramolecular liquid crystals with columnar mesophases through self-assembly of carboxylic acids around a tribasic core. Chem Comm. 2000. https://doi.org/10.1039/b003091k.
Lai LL, Yang YG, Chen JJ, Chen WY, Wang E. Mesogenic study of the H-bonded complexes of N, N-disubstituted aminophenylazo-(4)-1,3,4-thiadiazole with p-alkoxybenzoic acids. Liq Cryst. 2002. https://doi.org/10.1080/02678290210145193.
Parra M, Alderete J, Zuñiga C, Jimenez V, Hidalgo P. Hydrogen-bonded complexes between mesogenic heterocyclic Schiff’s bases and mesogenic 4-n-nonyloxybenzoic acid: Mesomorphic behaviour, FTIR study and PM3 semi-empirical calculations. Liq Cryst. 2003. https://doi.org/10.1080/0267829031000078827.
Song X, Li J, Zhang S. Supramolecular liquid crystals induced by intermolecular hydrogen bonding between benzoic acid and 4-(alkoxyphenylazo) pyridines. Liq Cryst. 2003. https://doi.org/10.1080/0267829031000071284.
Sridevi B, Chalapathi PV, Srinivasulu M, Pisipati VGKM, Potukuchi DM. Influence of hydrogen bonding on phase abundance in ferroelectric liquid crystals. Liq Cryst. 2004. https://doi.org/10.1080/02678290410001648705.
Prabu NPS, Madhu Mohan MLN. Thermal and dielectric investigations on supramolecular hydrogen bonded liquid crystals. Mol Cryst Liq Cryst. 2012. https://doi.org/10.1080/15421406.2012.703035.
Mahalingam T, Venkatachalam T, Jayaprakasam R, Vijayakumar VN. Design and synthesis of hydrogen bonded binary mixture liquid crystals. Ferroelectrics. 2016. https://doi.org/10.1080/00150193.2016.1234913.
Vasanthi T, Subhasri P, Jayaprakasam R, Vijayakumar VN. Experimental and computational studies on induced thermochromic effect and re-entrant smectic phase in linear double hydrogen-bonded binary liquid crystal mixtures. Phase Transit. 2019. https://doi.org/10.1080/01411594.2019.1566546.
Fouzai M, Guesmi A, Hamadi NB, Soltani T. Fluoro-substitution in hydrogen bonding liquid crystal benzoic acid dielectric, electro-optic and optical proprieties and inducing polar nematic phase. Liq Cryst. 2020. https://doi.org/10.1080/026782922019.1679900.
Subhapriya P, Sadasivam K, Madhu Mohan MLN, Vijayanand PS. Experimental and theoretical investigation of p–n alkoxy benzoic acid based liquid crystals–A DFT approach. Spectrochimica Acta Part A: Mol Biomol Spectrosc. 2014. https://doi.org/10.1016/j.saa.2014.01.074.
Fouzai M, Hamdi R, Ghrab S, Soltani T, Ionescu A, Othman T. Properties of binary mixtures derived from hydrogen bonded liquid crystals. J Mol Liq. 2018. https://doi.org/10.1016/j.molliq.2017.11.128.
Das P, Lakshmi PP. Structure and phase behavior of alkoxy benzoic acids in DMSO for thermodynamic applications: theoretical investigation. J Mol Struc. 2021. https://doi.org/10.1016/j.molstruc.2021.130137.
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg LJ, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta Jr JE, Ogliaro F, Bearpark M, Heyd J J, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ , Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, and Fox DJ, Gaussian 09, Revision D.01,Gaussian, Inc., Wallingford CT, 2013.
Sonali MK, Bhagavath P, Srinivasulu M, Sinha RK, Swamynathan K. Induced mesomorphism in supramolecular structures of H-bonded binary mixtures containing fluoro and chloro substituted benzoic acids. J Fluor Chem. 2022. https://doi.org/10.1016/j.jfluchem.2022.110002.
Silverstein RM, Webster FX. Spectrometric identification of organic compounds. 7th ed. New Jercy: Wiley; 2005.
Paterson DA, Felipe AM, Jansze SM, Marcelis ATM, Storey JMD, Imrie CT. New insights into the liquid crystal behaviour of hydrogen-bonded mixtures provided by temperature dependent FTIR spectroscopy. Liq Cryst. 2015. https://doi.org/10.1080/02678292.2015.1037122.
Dierking I. Textures of liquid crystals. Weinheim: Wiley; 2003.
Gray GW, Goodby JW. Smectic liquid crystals-textures and structures. London: Leonard Hill; 1984.
Muniprasad M, Srinivasulu M, Chalapathi PV, Potukuchi DM. Induction of liquid crystalline phases and influence of chain length of fatty acids in linear hydrogen bonded liquid crystal complexes. Mol Cryst Liq Cryst. 2012. https://doi.org/10.1080/15421406.2011.636662.
Sangeetha GB, Girish SR, Poornima B, Mahabaleshwara S, Potukuchi DM, Srinivasulu M. Self-assembled liquid crystalline materials with fatty acids. J Therm Anal Calorim. 2018. https://doi.org/10.1007/s10973-017-6879-y.
Doran MC, Sharma D. Plotting DSC results using logger pro for a binary liquid crystals system (BLCS). Int J Res Eng Sci. 2022;10:449–61.
Padmaja S, Ajitha N, Srinivasulu M, Girish SR, Pisipati VGKM, Potukuchi DM. Crystallization kinetics in liquid crystals with hexagonal precursor phases by calorimetry. Z Naturforsch. 2010;65a:733–44.
Mustafa O, Şükrü O. Thermal and mesomorphic properties of ternary mixtures of some hydrogen-bonded liquid crystals. Liq Cryst. 2014. https://doi.org/10.1080/02678292.2014.919669.
Kang SK, Samulski ET. Liquid crystals comprising hydrogen-bonded organic acids I Mixtures of non-mesogenic acids. Liq Cryst. 2000. https://doi.org/10.1080/026782900202822.
Kang SK, Samulski ET. Liquid crystals comprising hydrogen-bonded organic acids II. Heterodimers in mixed mesogenic acids. Liq Cryst. 2000. https://doi.org/10.1080/026782900202831.
Petrov VF. Alkoxylation in achiral calamitic liquid crystals. Liq Cryst. 2002. https://doi.org/10.1080/02678290210133114.
Goodby JW. The nanoscale engineering of nematic liquid crystals for displays. Liq Cryst. 2011. https://doi.org/10.1080/02678292.2011.614700.
Kašpar M, Bílková P, Bubnov A, Hamplová V, Novotná V, Glogarová M, Knižek K, Pociecha D. New chlorine-substituted liquid crystals possessing frustrated TGBA and SmQ phases. Liq Cryst. 2008. https://doi.org/10.1080/02678290802056212.
Gray GW, Hogg C, Lacey D. The synthesis and liquid crystal properties of some laterally fluorinated trans-cyclohexane-1-carboxylate and benzoate esters. Mol Cryst Liq Cryst. 1981. https://doi.org/10.1080/00268948108070871.
Morrison RT, Boyd RN. Organic Chemistry. 6th ed. New Delhi: Pearson Education Inc; 2004.
Parra M, Hidalgo P, Barbera J, Alderete J. Properties of thermotropic liquid crystals induced by hydrogen bonding between pyridyl-1,2,4-oxadiazole derivatives and benzoic acid, 4-chlorobenzoic acid or 4-methylbenzoic acids. Liq Cryst. 2005. https://doi.org/10.1080/02678290500115617.
Mohammady SZ, Aldhayan DM, Hagar M. Preparation and DFT study for new three-ring supramolecular H-bonded induced liquid crystal complexes. Front Chem. 2021. https://doi.org/10.3389/fchem.2021.679528.
Bhagavath P, Bhat SG, Mahabaleshwara S, Girish SR, Potukuchi DM, Srinivasulu M. Induced Smectic-A phase at low temperatures through self-assembly. J Mol Struct. 2013. https://doi.org/10.1016/j.molstruc.2013.01.053.
Bhagavath P, Bhat SG, Mahabaleshwara S, Girish SR, Potukuchi DM, Chalapathi PV, Srinivasulu M. Mesomorphic thermal stabilities in supramolecular liquid crystals: influence of the size and position of a substituent. J Mol Liq. 2013. https://doi.org/10.1016/j.molliq.2013.05.013.
Berdague P, Bayle JP, Ho MS, Fung BM. New laterally aromatic branched liquid crystal materials with large nematic ranges. Liq Cryst. 1993. https://doi.org/10.1080/02678299308027746.
Coates D. The effect of lateral substitution on smectic C formation. Liq Cryst. 1987. https://doi.org/10.1080/02678298708086299.
Mustafa O. Synthesis and characterization of hydrogen bonded liquid crystal complexes by 4-octyloxy benzoic acid and some dicarboxylic acids. J Mol Liq. 2018. https://doi.org/10.1016/j.molliq.2018.06.111.
Mustafa O. Thermal characterisation of binary mixture of some supramolecular liquid crystals. J Therm Anal Calorim. 2015. https://doi.org/10.1007/s10973-015-4488-1.
Mustafa O, Hasan E, Murat S, Şükrü Ö. Mesogenic properties of PAA/6BA binary liquid crystal complexes. J Mol Struc. 2018. https://doi.org/10.1016/j.molstruc.2018.10.049.
Fukui K, Yonezawa T, Shingu H. A molecular orbital theory of reactivity in aromatic hydrocarbons. J Chem Phys. 1952. https://doi.org/10.1063/1.1700523.
Aihara J. Reduced HOMO-LUMO gap as an index of kinetic stability for polycyclic aromatic hydrocarbons. J Phys Chem A. 1999. https://doi.org/10.1021/jp990092i.
Manolopoulos DE, May JC, Down SE. Theoretical studies of the fullerenes: C34 to C70. Chem Phys Lett. 1991. https://doi.org/10.1016/0009-2614(91)90340-F.
Morales YR. HOMO-LUMO gap as an index of molecular size and structure for polycyclic aromatic hydrocarbons (PAHs) and asphaltenes: a theoretical study. I J Phys Chem A. 2002. https://doi.org/10.1021/jp021152e.
Miar M, Shiroudi A, Pourshamsian K, Oliaey AR, Hatamjafari F. Theoretical investigations on the HOMO–LUMO gap and global reactivity descriptor studies, natural bond orbital, and nucleus-independent chemical shifts analyses of 3-phenylbenzo [d] thiazole-2(3H)-imine and its para-substituted derivatives: solvent and substituent effects. J Chem Res. 2021. https://doi.org/10.1177/1747519820932091.
Acknowledgements
We thank Dr Srinivasulu Maddasani, Associate Professor (Senior Scale) at Department of Chemistry, MIT, Manipal, for his guidance and support. We thank the Centre for Nano and Soft Matter Sciences (CeNS) Bengaluru for providing advanced liquid crystal characterization resources. Intramural grants from the Manipal Academy of Higher Education have enabled us to pursue research with renewed vigor.
Funding
Open access funding provided by Manipal Academy of Higher Education, Manipal.
Author information
Authors and Affiliations
Contributions
Conceptualization was done by Poornima Bhagavath; methodology was done by Poornima Bhagavath and Sonali M K; formal analysis was done by Poornima Bhagavath, Sonali M K and Rajeev K Sinha; investigation was done by Sonali M K and Poornima Bhagavath; writing—original draft were done by Poornima Bhagavath, Sonali M K and Rajeev K Sinha; writing—review and editing were done by Poornima Bhagavath, Sonali M K and Rajeev K Sinha; funding acquisition was done by Poornima Bhagavath; resources were done by Poornima Bhagavath; supervision was done by Poornima Bhagavath.
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sonali, M.K., Sinha, R.K. & Bhagavath, P. New hydrogen-bonded liquid crystal supramolecular systems: role of (+ I)-alkoxy substituents in promoting molecular ordering. J Therm Anal Calorim (2024). https://doi.org/10.1007/s10973-024-13254-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10973-024-13254-w