
Abstract
A remarkable number of scientific papers are available in the literature about the bulk amorphous alloys and metallic glasses. Today, DSC is an essential tool for amorphous alloys research and development, and of course for quality assurance. In many cases, users seek to examine the determination of only one or two properties, although much more information can be obtained from the measurements. The research involved structural relaxation, Curie temperature, glass temperature, crystallization, phase separation, nanocrystalline volume fraction, melting point and liquidus temperature determination subjects and kinetics of microstructural transformations induced by thermal treatment. We collected and present the information that can be obtained with this technique and draws the reader’s attention to some potential problems related to data interpretation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the winter of 1782-83, Antoine Lavoisier and Pierre-Simon Laplace made the first known calorimeter, an ice-calorimeter [1]. Ever since, thermal analysis techniques, and DSC in particular, have been playing a significant role in the research and development in both non-metallic and metallic materials. Calorimetry has many advantages. Many changes in properties or structures in a material are accompanied by the liberation or consumption of energy. This energy is usually of sufficient magnitude and rate to be easily detected. Thanks to its speed, simplicity, and availability, DSC has become a universally used standard tool for characterizing thermodynamic and kinetic properties, phase transitions, and property evolution. Today DSC is the most often used thermal analysis method. Furthermore, DSC is the most commonly used device for the characterization of polymers, drugs, foods, geological materials, and metallic materials as well.
There are many types of DSCs today:
-
Heat flux DSC
-
Power compensated DSC
-
Modulated DSC
-
Pressure DSC
-
Differential fast scanning calorimetry (DFSC)
The most frequently used DSCs are the heat flux and power compensated ones. The principle of heat flux DSC is that the difference in heat flow into the sample and the reference is measured, while the temperature of the sample is changed at a constant rate. The heat flow difference between the sample and the reference is:
The heat flow difference between the sample and the reference can be either positive or negative. In an exothermic process, like a crystallization, heat is released, and, therefore, heat flow to the sample is lower than that to the reference. In an endothermic process, such as melting, the opposite is true: the heat flow is negative.
In power compensated DSC, the sample and the reference are separated into two furnaces and the temperatures of the sample and the reference are kept equal to each other, while both temperatures are increased or decreased linearly. The electric power needed to maintain equal temperatures for both the sample and the reference temperature is measured throughout the entire analysis.
For both types of equipment, the reference is an inert material such as alumina, or just an empty aluminum pan.
The results of the measurement are affected by calibration, sample pan, sample, experiment and environmental conditions, and most importantly the interpretation of the results [2,3,4].
The first metallic glass was published 60 years ago [5], since then one of the most important testing techniques of amorphous alloys is DSC. Numerous papers and reviews deal with the questions of amorphous alloys and their thermal behaviours [6,7,8]. Many glass-forming ability (GFA) factors are published in the literature. DSC is capable to measure many of thes GFA factors that include such characteristic temperatures like Tg glass temperature, Tx onset of crystallization temperature, Tl liquidus temperature. Amorphous alloys have both metallic and non-metallic properties, i.e. characteristic thermal properties of both types of materials may occur during a DSC experiment. Metallic glasses have a glass temperature similar to that for the polymers. However, the metals and alloys are very good heat conductors so that the temperature difference between the sample and reference is equalized much sooner compared to non-metallic materials. Baseline correction should also be performed differently than for polymers or drugs [3, 9]. After temperature equilibration, the samples must be heated continuously to the desired temperature, then cooled down, and followed by a second heating run as a baseline.
There are standards for drugs, polymers and organic materials, but, unfortunately, there is no standard for metals testing.
Till now, the uniform definition and measurement methods of the thermal properties of amorphous alloys is missing in the literature. There are no rules on how to measure and evaluate the measurement curves for each property of amorphous alloys.
The goal of this paper is to show, what can be deduced from the measured results on amorphous materials from a process engineering point of view.
Materials and methods
By means of the standard procedure of rapid quenching of the melt on a rotating wheel (melt-spinning method), amorphous ribbon samples were prepared. The nominal composition of the as-prepared alloy samples can be represented as follows in atomic %: Fe40Ni40B16Si4 and (Cu48Zr48Al4)96Co2Fe2. The thickness of the ribbons was approximately 50 μm. FINEMET type alloy ribbons with Fe73Si16B7Nb3Cu1 nominal composition are commercial products.
The master alloy ingots were prepared by arc-melting from a mixture of high-purity elements under argon atmosphere (min. 99.9 mass%) with a Ti-getter. The ingots were re-melted at least four times in order to ensure chemical homogeneity. Bulk amorphous samples of Cu–Zr–Ag–Al system in a wedge and rod form have been prepared by the centrifugal casting in a copper mould. The ingots were re-melted by induction melting in a quartz crucible under argon atmosphere in the centrifugal casting machine. Before melting the chamber was evacuated and purged with high-purity Ar. The dimensions of the resulting wedge are: 30 mm high, 3 mm thick, 20 mm wide. The dimensions of the rod are 25 mm length, 3 mm diameter. The thermal transformation of the samples was studied by a NETZSCH-STA449C TG/DSC, NETZSCH 204 and 404 differential scanning calorimeters (DSC) under purified Ar atmosphere. The using a continuous heating regime with heating rates β from 2 to 90 °C min-1. The temperature axis as well as the enthalpy axis was calibrated using indium, tin, bismuth, zinc, aluminum and gold standards.
Structural relaxation
The metallic glasses and bulk amorphous alloys are thermodynamically unstable materials; when heating them structural relaxation and crystallization processes take place. If one heats a quenched metallic glass below the crystallization temperature, its structure transforms to a more relaxed (close to equilibrium) state, i.e. structural relaxation takes place. This phenomenon is a thermally activated process, and it happens below the glass transition temperature. During this structural relaxation short-term atomic rearrangements occur. Many properties change irreversibly or reversibly. Almost all physical properties change continuously: free volume, density, magnetic properties, electrical properties, ductility. Several models have been developed to describe and explain the structural relaxation of metallic glasses. Egami identified three mechanisms at the atomic level during annealing: diffusion, topological and chemical rearrangements [10]. The two rearrangements are not independent of each other. Topological structural relaxation (TSRO) and chemical short-range ordering (CSRO) have been observed by X-ray and neutron scattering measurements [11, 12] and by magnetic measurements [13] during the relaxation of metallic glasses. The free volume model was proposed by Turnbull and Cohen [14] and developed by Cohen [15] and Spaepen [16], and this model is one of the most commonly used. Heating of the material during the DSC experiment leads to an increase in the atomic mobility, and the excess free volume is annihilated. The decrease in the free volume vf is accompanied by a decrease in the enthalpy of the material. As a result, at a low temperature, a broad, flat exothermic peak appears before the Tg temperature can be observed in the DSC signal at a constant heating rate as an endothermic hump prior to the exothermic event. The amorphous structure has an equilibrium amount of free volume at \({T}_{\text{g}}^{onset}\) temperature.
The exothermic signal corresponds to the enthalpy release (ΔHfv) due to the structural relaxation of the amorphous structure [17]:
where β′ is a constant and Δvf is the change in free volume per atomic volume. Preheating of the sample at a different temperature or annealing for different times below the crystallization temperature, the low-temperature exothermic effect becomes weaker and shifts toward higher temperatures with increasing preheating temperature or time [17, 18] A diagram of this phenomenon can be seen in Fig. 1. The change in material density is due to the change in free volume. Thanks to this, it can be determined that the change in free volume per atomic volume Δvf for sample was calculated as
where ρ0 is the density of the as-cast sample and ρi is the density of the relaxed one. β’ constant can be determined by knowing the linear relationship between ΔH and \(\Delta {v}_{\rm f}\).

DSC scan of Fe40Ni40B16Si4 (at.%) metallic glasse ribbon pre-heated to various temperatures: (a) as-quenched, (b) heated up to 275 °C, (c) heated up to 300 °C, (d) heated up to 325 °C (heating rate: 20 °C min-1)
According to several researchers in the literature the plasticity and free volume are closely related [19,20,21, 21]. A large amount of free volume will enhance the plasticity of metallic glasses. Several articles can be found in the literature addressing the issue of relaxation and free volume [22,23,24].
It should be noted that in condensed matter physics structural relaxation is one of the investigated metallurgical problems.
Structural relaxation and Curie transition in Fe-based metallic glasses with Cu content
The Fe-based MGs have been widely applied as soft magnetic materials and the Co-based MGs as magnetic sensors. Fe-based amorphous alloys are ferromagnetic, and their soft magnetic properties make them useful for many applications. They are the most widely studied type of metallic glass.
During the relaxation of FINEMET type metallic glasses, other processes and changes in properties take place, which does not occur in the case of non-magnetic materials. During structural relaxation, not only a ferromagnetic Curie transformation occurs, but also the formation of clusters of Cu atoms, which is extremely important in the subsequent crystallization process. During heating a FINEMET type amorphous ribbon with a constant heating rate, a very small endothermic peak is visible at about 310 °C, indicating magnetic transformation at Curie temperature (Tc) (Fig. 2).

DSC curve obtained on heating samples from an amorphous ribbon of Fe73Si16B7Nb3Cu1 alloy (heating rate: 8 °C min-1)
The Curie temperature of ferromagnetic glasses depends on the heating rate; therefore, it is recommended to use a lower rates—maximum of 80 °C min-1. Furthermore, it is highly sensitive to relaxation and stress [25]. The cooling rate influences the structure forming and in this way it affects the Curie temperature of amorphous structure. The free volume also depends on the cooling rate and thus on the subsequent annealing process which changes the free volume. Annealing causes the Tc, and the density of metallic glasses to increase [26,27,28,29], yet compression causes a decrease in Tc temperature [29]. Using a constant heating rate, a DSC measurement also provides both fast and accurate determination of TC.
Using atom-probe field ion microscopy (APFIM) and extended X-ray absorption fine structure (EXAFS) it was confirmed that Cu atoms form Cu enriched clusters prior to the onset of crystallization [30, 31]. Furthermore, the three-dimensional atom probe (3DAP) technique made it visible, that these Cu clusters serve as the heterogeneous nucleation sites for the bcc Fe(Si) primary crystals [32]. The exothermal peak of structural relaxation is broad and overlaps with the Curie temperature. Ohnuma et al. used an arbitrary baseline in order to roughly estimate the enthalpy for Cu clustering [33]. The enthalpy for clustering is less than 10% of the first crystallization enthalpy. In the case shown in Fig. 2, the enthalpy for clustering is ~ 5% of the α-Fe(Si) crystallization. The peak temperature of clustering (Tcluster) is higher in the case of lower Cu content, according to Ohnuma [33]. Our experiments showed that in the case where the difference between the Cu contents is very small (0.1 at.%), the measurement error can be compared with the degree of change. Tcluster increases with increasing heating rate. If the difference between the Tx (onset temperature of crystallization) and Tcluster is large, then there is enough time for Cu clusters to coarsen which favors obtaining the largest number of heterogeneous nucleation sites for bcc Fe(Si) crystallization.
For baseline correction, it should be taken into account that there is Curie temperature of bcc Fe(Si) crystallized phase formed during the first heating and it is visible as an endothermic signal for the 2nd measurement at above 600 °C.
Glass transition temperature
Glass temperature is defined as a single temperature which indicates the onset of the glass transition range for a glass. Chen and Turnbull already reported evidence of glass-liquid transformation in Au–Ge–Si alloy in 1968 [35]. Glass transition takes place at a temperature range rather than at a specific temperature [36, 37]. To date, the Tg temperature has been determined in several ways based on DSC measurements. The two most commonly used definitions can be observed in the inset to the Fig. 3: Tg-onset is the intersection of the baseline and the tangent with the greatest slope on the step, that is the most frequently used to indicate the glass transition temperature; the temperature lying midway between the extrapolated high- and low-temperature tangents on either side of the endothermic region indicated as Tg-mid [39]. However, conventionally Tg-onset is used as Tg temperature. Nowadays, the Tg temperature is most often and most rapidly determined by DSC measurement with continuous heating of the sample. But unfortunately, some alloys show no endothermic change in the DSC curve before crystallization, and even at higher heating rates (more than 20 °C min-1) because Tg and Tx are so close to each other. For these alloys, the Tg temperature must be determined by another method (the electrical resistivity method). The glass transition temperature, crystallization temperature, and the width of the supercooled liquid region (ΔTx = Tx-Tg) depend on the heating rate, i.e., they increase with an increase in heating rate [39, 41]. The supercooled liquid region of Fe40Ni40B16Si4 metallic glass increases from 23 to 32 °C when the heating rate increases from 10 °C min-1 to 40 °C min-1 [39, 41].

The two ways to determine the characteristic temperature of the glass transition temperature on the DSC curve of Cu36Zr48Ag8Al8 (at.%) bulk amorphous alloy (heating rate: 40 °C min-1) [34]
The characteristic thermal properties of amorphous powders produced by high energy milling generally differ from the DSC analysis of metallic glasses and bulk amorphous alloys having the same composition. The amorphous powders produced by high energy milling do not show an endothermic change in the DSC curve before crystallization as can be seen in Fig. 4. Other authors have also observed this phenomenon [40, 41]. However, the amorphous powders produced by the mechanical alloying or the process of gas-atomization behave like metallic glasses or bulk amorphous alloys.

DSC traces of the powder with 88 mass% amorphous fraction for the Ti48Cu39.5Ni10Co2.5 (at.%) alloy. The amorphous nanocrystalline powders are produced by a different ball-to-powder ratio (BPR) by mechanical milling (heating rate: 40 °C min-1) [42]
Crystallization temperature, and phase separation in the solid and liquid state
In case of non-isothermal heating, the onset of crystallization temperature (Tx) can be determined by intersecting the tangents (Fig. 3). However, there are alloy systems for which it is not so easy to determine the real temperature Tx on the DSC curve.
Phase separation can take place in the liquid and solid-state as well depending on the mixing enthalpy between the constituent element pairs. The separation occurs in a liquid state when it is large positive mixing enthalpy between the element pair. Since the discovery of amorphous alloys, phase separation has been observed in many alloy systems, and in glasses characterized by negative heat of mixing between the constituent elements or with a minor positive heat of mixing [43, 45].
The phase separation in metallic glasses characterized by negative or close to zero heat of mixing takes place mostly during heating of the as-quenched glass before the first crystallization [43, 44]. During the decomposition, nanoscale microstructure forms prior to the crystallization and this process follows the nucleation of nanocrystals on a few nm lengths [45,46,47]. The growth of nanocrystals is also an exothermic process, as is the crystallization of the amorphous structure.
Figure 5 shows the DSC curves of Cu36Zr48Ag8Al8 bulk amorphous alloy before and after relaxation. The annealing was carried out at 300 °C of about 213 °C below Tx. The heat of mixing between Cu and Ag is + 2 kJ mol-1[48]. In Fig. 5, the square area indicates the region, where the coarseness of the nanoparticles formed by phase separation.

DSC curves of Cu34.7Zr47.2Ag6.4Al11.7 (at.%) bulk amorphous alloy; as cast and annealed (heating rate: 40 °C min-1)
In the case, when a phase separation takes place before the primary crystallization in the solid state, the Tx temperature must be determined by the tangent method, but the tangent must not be drawn to the sharp exothermic peak, but to the small exothermic part (Fig. 6b). The DSC heating curve of (Cu48Zr48Al4)96Co2Fe2 alloy is shown in Fig. 6. This alloy system consists of atom pairs with a small positive heat of mixing in the liquid state: and Cu–Fe (+ 13 kJ mol-1 [48]) and Cu–Co (+ 6 kJ mol-1 [49]).

DSC curve (a) of (Cu48Zr48Al4)96Co2Fe2 (at.%) metallic glass and the enlarged part (b) of the area marked with an ellipse in the figure (a) (heating rate: 10 °C min-1)
It has long been known that in alloys with high positive mixing enthalpy, phase separation takes place in the liquid state. Below the separation temperature (Tsep) the homogeneous liquid separates into two liquids phases. These alloy systems are monotectic alloys, such as Al–Pb (+ 10 kJ mol-1 heat of mixing), and in such systems it is a stable phase separation. Nakagawa [50] first reported that liquid separation occurs in the undercooled Fe–Cu system, in which a metastable miscibility gap exists.
In metallic glasses characterized by positive heat of mixing two immiscible liquids of different composition coexist in equilibrium between separation temperature (Tsep) and monotectic temperature (TM). Bulk metallic glasses have high strength and good corrosion resistance and generally very limited ductility. When a monotectic alloy is undercooled into the miscibility gap, the liquid–liquid phase separation takes place and two liquids form. Drops of the new liquid, having a regular shape, begin generally with the nucleation of the liquid minority phase. In immiscible alloys, a composite microstructure in situ forms with the crystalline or amorphous spheres embedded in the matrix. Amorphous/amorphous or amorphous/crystalline composites can be obtained owing to the liquid separation depending on the GFA of the immiscible liquids. Liquid separation provides an opportunity to produce in situ a composite microstructure, where the separated drops are ductile crystalline phases and the matrix has an amorphous structure.
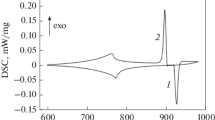
The onset temperatures of phase separation Tsep and monotectic reaction TM can be determined by DSC. The phase separation is manifested in the cooling curve (Fig. 7). The first small exothermic deviation from the baseline is attributable to liquid phase separation (Tsep) [50,51,52]. Figure 7 shows the heating and cooling curves of Cu40Zr20Ag40 master alloy. At 1000 °C (1273 K) the homogeneous liquid separates into an Ag-rich liquid and a Cu–Zr–rich liquid. The primary solidification of the m-phase (AgCu4Zr) from Ag-rich liquid starts at 986 °C (1259 K), and this is the TM temperature.

DSC curve of Cu40Zr20Ag40 (at.%) master alloy (heating and cooling rate: 5 °C min-1) [53]
Volume fraction of nanocrystals in an amorphous matrix
Nano or/and microcrystals are solidified during the casting process when the cooling rate is slower than the critical cooling rate. DSC curves of Cu36Zr48Ag8Al8 samples can be seen in Fig. 8. The three samples were cast with different cooling rates and, as a result, the samples cast with a slower cooling rate contained primary crystallized grains. The enthalpy in the first crystallization peak changes owing to nanocrystalline content, the enthalpy of the crystalline peak decreases with increasing nanocrystalline volume (Fig. 8). The peak temperature of the first crystallization slightly increases with increasing nanocrystalline volume fraction, and, because of the residual amorphous structure, it is more stable.

DSC curves of samples taken from different parts of the wedge in the case of Cu36Zr48Ag8Al8 (at.%) alloy—as the thickness increases, the cooling rate decreases
During annealing of an amorphous alloy at above the Tg temperature, nanocrystallites crystallize, and these are essential for the enhancement of the mechanical or magnetic properties of an amorphous composite. It is important to know both the quantity of crystals formed during the casting process of an amorphous alloy and the quantity of crystals formed during the partial crystallization of the amorphous matrix. Quantitative determination of the volume fraction of the primary nanocrystalline phase is generally used in the TEM, XRD, or DSC methods. However, a precise TEM study requires an expensive and difficult preparation and does not provide an overall landscape of a whole sample. DSC and XRD methods collect information about a larger volume, so these two methods are preferable to evaluate the volume fraction of primary nanocrystals.
For polymers and pharmaceuticals, the determination of the crystalline fraction by DSC measurement is accepted across the whole world [54, 55]. In the case of a sample with nanocrystallites in the amorphous matrix, the volume fraction can be estimated by using the following relationship [56, 57]:
where ΔH full amorphous is the enthalpy change of a fully amorphous sample and ΔH amorphous+nanocryst is the enthalpy change of a partially crystallized sample during crystallisation.
According to several authors [58, 59], the values of the volume fraction of crystalline determined by DSC and XRD methods generally differ. In some cases, a lower value of Vf is obtained by DSC than that by XRD [58] and otherwise, the reverse occurs [59]. In my opinion, the agreement between DSC and XRD is good for small volume percentages, but the difference is significant for larger volumes. There can be several reasons for this discrepancy. The composition of the amorphous phase changes owing to the crystals, and therefore the residual amorphous structure has different properties. Furthermore, it is also difficult to find an accurate baseline. In turn, the XRD diffraction detects very small crystallites (~ 5 nm) as an amorphous phase. Summarizing, the determination of the volume fractions of crystals in the amorphous matrix is sensitive to the nature of the amorphous structure and the change of chemical composition during the nanocrystallization process. If we have a sample with a completely amorphous structure that was found to be completely amorphous based on an additional XRD measurement, we can decide very quickly whether another sample is completely amorphous or not by comparing their DSC measurements.
Control of composition, quality assurance
DSCs are applied to determine the impurity for many types of substances, e.g. pharma materials [60], organic matter [61], metals [47, 62]. Users of amorphous products and researchers need to know whether amorphous pieces with a nominally uniform composition actually do have the same composition. DSC measurement gives the fastest answer to this question, and thus DSC is required for quality assurance.
Two DSC curves of FINEMET type ribbons with the nominally equal composition are shown in Fig. 9. It can be observed that there is a significant difference in all crystallization processes. During the first crystallization step, the bcc Fe(Si) phase crystallizes. As we mentioned, Cu clusters serve as the heterogeneous nucleation sites for the primary precipitation of bcc-Fe(Si) crystals by heat treatment from the amorphous state. The peak temperatures do not significantly differ, but the difference in enthalpy values indicates that there is a difference in the volume fraction of bcc Fe(Si) phase. Diffusion of Nb outside the crystalline phase is very slow and Nb seems to control the growth rate of bcc-Fe(Si) precipitates. In the second and third crystallization steps, the intermetallic phases containing Fe, B, Nb and Si crystallize. The difference in peak temperature values also shows that the Nb and Si content also differ in the investigated ribbons. The ICP-OES measurement confirmed this observation because there was a difference in the Nb and Si content.

DSC curves of two FINEMET type ribbons with nominally equal composition (heating rate: 8 °C min-1)
Melting temperature and liquidus temperature
Melting temperature and liquidus temperature are included in several GFA characterization study. It should be mentioned that some authors suggest that the solidus temperature should be used instead of the liquidus temperature [63]. Nonetheless, γ (γ = Tx/(Tg + Tl) parameter [64], and a reduced glass transition temperature (Trg= Tg/Tl) are included in a lot of articles.
The solidus temperature is the temperature below which there are only solid phases in equilibrium. The compositions of the best glass formers in an alloy system are quite close to the eutectic composition. In the case of non-eutectic composition, the onset temperature corresponds to melting in the eutectic reaction. If we see only one endothermic peak during the melting process, of course, it is not necessarily a pure eutectic composition. In many cases, the processes overlap and so we may see only one peak. It is highly recommended to study the first derivative of the heat flow curve and the cooling curve, because these curves help in the correct interpretation. In the Cu–Zr–Ag system, three different eutectic compositions were calculated by Kündig et al. [65], He et al. [66], and Kang et al. [67]. The DSC cooling curves of the three compositions revealed that these compositions are not eutectic (Fig. 10).

DSC curves of eutectic compositions based on calculations in Cu-Zr-Ag system (heating and cooling rate: 5 °C min-1) [68]
The liquidus (TL) is the ultimate temperature of thermodynamic equilibrium between the solid and liquid phases of any material, above which crystals are unstable. The melting process is endothermic. The melting process begins at the temperature where the DSC signal deviates from the baseline (Fig. 11). However, this is not easy to determine, so the melting temperature (which is the solidification temperature) Tonset is defined by the intersection of tangents to the curve, traced on the baseline and the low-temperature peak side. The melting process finishes at a lower temperature than the endset temperature. Conventionally, the liquidus temperature is defined as Tendset temperature (Fig. 11) and is also determined by the tangent method, but it is not a standard method and its uncertainty is difficult to estimate. A heating speed of 0.5–10 °C min-1 should normally be used for the measurement, and by no means at the same heating rate as for Tg measurement (20–40 °C min-1).

DSC curve of a Cu46Zr34Ag20 (at.%) alloy (heating rate 10 °C min-1)
Because the onset temperature corresponds to melting in a eutectic reaction, thus, it does not change with the heating rate, while the endpoint temperature varies with the heating rate and largely depends on the mass, layer thickness and thermal conductivity of the sample. Therefore, there are several methods in the literature to determine the liquidus temperature with more physical content. G.W.H. Höhne et al. [69] proposed a method that takes into account the dependence of the temperature on the heating rate. E. B. Ferreira et al. [70] found that the endset temperature is directly proportional to the heating rate and the best approximation to the liquidus temperature is the endpoint of the DSC melting peaks measured at different rates and extrapolated to 0 °C min-1. H. Nishikawa et al. [71] found that the extrapolated end temperature of the endothermic peak is proportional to the square root of the DSC heating rate.
The two proposed onset and endset temperatures for melting process determinations of Cu35Zr41.25Ag23.75 (at.%) alloy are summarized in Fig. 12. It can be observed that the Tonset temperature is independent of both the heating rate (Fig. 12a) and the square root of the heating rate (Fig. 12b). The y-axis intercepts for the peak and endset lines are very similar. The difference between the two extrapolated liquidus temperatures to 0 °C min-1 of the two models is 5.6 °C. It is worth noting that the heating rate should only be less than 20 °C min-1 because the temperature extrapolated to 0 °C min-1 measured with a heating rate of 20 °C min-1 and above differs from the value extrapolated from measurements carried out with a lower heating rate (Fig. 13). This extrapolation is also used for crystallization peaks. In Fig. 13 the crystallization temperature of Fe40Ni40B16Si4 metallic glass can be seen as a function of the heating rate. The heating rate was between 2 and 90 °C min-1. It can be observed, that the above-mentioned correlations are not particularly valid for E. B. Ferreira et al. method [70] if one uses higher heating rates as we can see in many articles.


The crystallization peak temperature of Fe40Ni40B16Si4 (at.%) metallic glass as a function of heating rate
Kinetic analysis of crystallization processes
The phase transformation processes in solids are generally thermally activated processes, during which the system sitting in the local minimum of free enthalpy passes through a potential barrier with thermal fluctuation, thus reaching a lower energy level than in the case of glass metal crystallization. The crystallization of amorphous alloys involves nucleation and growth. The knowledge of the rate of transformation is very important because heat treatment affects the properties of the product. There are many methods in the literature to determine the crystallized volume, the local activation energy and the Avrami index n, which provides information on the nucleation and growth mechanism and dimensionality. It should be noted that a method that is perfect for any process is not yet known. The reason is that the transformation rate depends exclusively on temperature, the degree of transformation and also the thermal history. Unfortunately, existing methods are also misapplied in several cases.
For both isothermal and continuous heating processes, kinetic analysis is possible. There are several kinetic models in the literature, and the most common ones will be presented here.
The Johnson, Mehl, Avrami and Kolmogorov model (JMAK) [2, 72, 73] describes the crystallization kinetics during an isothermal process. The model assumes that:
-
The crystallization is isothermal, i.e. nucleation and growth occur at a constant temperature;
-
The sample is assumed to be infinitely large and the nucleation is random throughout the bulk of the sample;
-
Growth is isotropic until crystals impinge upon one another;
-
The activation energy for crystallization and other model parameters are independent of time and temperature.
The volume fraction transformed during crystallization is written as:
where k(T) is the rate constant, t-time and n-the Avrami index. n is a parameter that depends on the nucleation and growth mechanism and dimensionality. Table 1 includes the characteristic n parameters for different reaction mechanisms as taken from Ref. [74].
The rate constant in the thermal activated process:
where the pre-exponential factor A and activation energy Ea are kinetic parameters that should not depend on the temperature T and the fractional conversion x.
A major disadvantage of this model is that the transformed fraction is given in the case of isotherms, and since measurements with DSC are often not made in isotherms, the terms must be generalized for the situation of non-isotherms. To describe non-isothermal reactions, there are several thermo-kinetic methods to evaluate the DSC curves. Kissinger [75], Takhor [76] and Ozawa [77] developed a method in which both Ea is determined at different heating rates. The basic equations of both methods are shown as Eq. 7 [75], 8 [76] and 9 [77]:
where β is the heating rate, Ea is the activation energy, R is the gas constant, Tpeak is the temperature of a peak, Tstart is the starting temperature of the peak, B0 is the rate constant. Drawing the function ln[β/T 2peak ]—1/Tpeak for the individual crystallization peaks, a straight line is obtained, the slope of which is Ea/R. In the literature, these lines are called Kissinger's straight lines. Naturally, the Kissinger method can be used only if the peak appearing on the DSC curve belongs only to one process. The kinetic analysis of the crystallization is presented in the case of Fe40Ni40B16Si4 metallic glass. Heating rate β was changed between 2 K min-1 and 99 K min-1 in 13 steps. Kissinger’s, Takhor’s and Ozawa’s lines are presented in Fig. 14.

Kissinger’s, Takhor’s and Ozawa’s straight lines of the crystallization peak temperatures determined from the DSC curves [77] in the case of Fe40Ni40B16Si4 (at.%) metallic glass
The analysis gives 296.6 ± 11.3 kJ mol-1 294.1 ± 11.3 kJ mol-1 and 302.1 ± 8.7 kJ mol-1 to the activation energy of crystallization based on the Kissinger, Takhor and Ozawa method. Only a small difference exists between the three values of the activation energy.
The crystallized volume fraction for the non-isothermal crystallization can be deduced as a function of temperature using the following equation:
where T0 and Tend are the temperatures corresponding to the onset and end of crystallization and dHc/dT is the heat capacity at constant pressure. A is the area under the DSC curve between the onset temperature and a given temperature, and A0 is the area between the onset and end of crystallization. The crystallized volume fraction is plotted as a function of temperature for Fe40Ni40B16Si4 metallic glass in Fig. 15. The variation of the crystallized volume fraction versus temperature follows a sigmoidal behavior. The crystallization process with sigmoidal curves can be divided into three parts. At the beginning (x < 0.1) and the end (x > 0.9) of the process takes place with a slow reaction rate. In the beginning, the heterogeneous nucleation of Fe atoms is required in the hole sample. In the second stage, the crystallized volume drastically increases, the surface area between the amorphous part and the nuclei increases too. In the final stage, the crystallization process slows down as a result of contact crystals inhibit further growth and the surface area between the amorphous part and the crystals decreases. At different heating rates, a small difference in the slope of the curves can be seen.

Plots of crystallized volume fraction (x) versus temperature (T) of the Fe40Ni40B16Si4 (at.%) ribbons at different heating rates
The activation energy determined by Eqs. 7, 8 and 9 is an average value of the activation energy of the whole crystallization process. Unfortunately, the activation energy depends on the volume fraction. The E(x) corresponds to the appropriate values of activation energy in a wide range of crystallized volume fraction can be evaluated from the following Kissinger–Akahira–Sunose (KAS) model [78]:
and the Ozawa–Flynn–Wall formula (OFW) [79]:
where T(x) is a temperature corresponding to the certain crystallized volume fraction β at different heating rates. Figure 16 shows the relation between the local activation energy E(x) and the crystallized volume fraction calculated by KAS and OFW. The average activation energy E(x) between x = 0.1 and 0.9 is 264.9 kJ mol-1 calculated by KAS and 263.1 kJ mol-1 calculated by OFW. These values show a very good agreement, i.e. either method can be used for this alloy. The average activation energy E(x) between x = 0.1 and 0.9 is lower than the average activation energy of the first crystallization based on Kissinger, Takhor and Ozawa method (Ea-see Fig. 14). In the initial stage, i.e., x < 0.1, E(x) increases slightly, the crystallization is slow at this stage (see Fig. 15), a barrier of crystallization must be overcome. In the second stage, i.e., 0.1 < x < 0.9, the crystallized volume fraction increases rapidly, the local activation energy decreases monotonically. In the final stage, i.e., x > 0.9, although the local activation energy decreases rapidly and reaches the lowest value, and reaches the lowest value and the degree of crystallization slows down.

The dependence of local activation energy E(x) on the crystalline volume fraction (x) for the first exothermic event of the Fe40Ni40B16Si4 ribbon
The Avrami exponent is usually used to define the mechanism of nucleation and growth behavior during the crystallization process. Based on Kissinger’s work, Benett and Augis [78] created a reworking of the Kissinger method. The Avrami index (n) can be estimated from the relation:
where ΔTFWHM is the full width at half maximum of the DSC peak, Ea is the activation energy, R is the gas constant. The Avrami exponent of Fe40Ni40B16Si4 metallic glass based on this analysis is 3.04. This relationship assumes that the activation energy is constant throughout the process and does not depend on the crystallized fraction. However, the activation energy depends on the volume fraction and consequently, the Avrami index (n) also varies depending on the crystalline fraction. The local Avrami exponent can be determined from the relation proposed by J.S. Blázquez et al. [81]:
According to Eq. (12), plotting ln[− ln(1 − x)] versus ln[(T − T0)/β] will provide the n(x) values from the slope of the curves (see Fig. 17). The slopes of the curves vary in the case of larger crystalline volume fraction (x > 0.8), which indicates that the nucleation and growth do not remain the same way during the whole crystallization process. The n(x) value reflects the crystallization mechanism. The local Avrami exponent n(x) as a function of the crystallized fraction (x) at a heating rate of 5 and 30 K min-1 is shown in Fig. 17b. The crystallization of Fe-based amorphous alloys can be controlled by diffusion [82,83,84] or phase-boundary [85].
where b is the nucleation index (b = 0 for zero nucleation rate, 0 < b < 1 for decreasing nucleation rate, b = 1 for constant nucleation rate, b > 1 for increasing nucleation rate), m is the dimensionality of the growth (m = 1, 2, 3 for one-dimensionality, two-dimensionality, three-dimensionality growth respectively), and p is the growth index (p = 0.5 for diffusion-controlled growth and p = 1 for interface-controlled growth). For diffused-controlled crystallization, n > 2.5 indicates that the crystallization process is the different kinds of growth of small particles with an increasing nucleation rate. 1.5 < n < 2.5 implies the growth of precipitates with decreasing nucleation rate, and n < 1.5 means that the growth of pre-existing nuclei [86].

Plots of ln[− ln(1 − x)] versus ln[(T − T0)/β] at different heating rates (a) and the local Avrami exponent (b)
In the case of the investigated Fe40Ni40B16Si4 ribbon, the value of the local Avrami exponent at the first crystallization process is higher than 3 during the whole crystallization, which means that the crystallization mechanism of α-Fe(Si) is dominated by three-dimensional growth with an increasing nucleation rate (b = 3; p = 0.5, considering this values, 1.5 < n < 2.5).
Comparing the value of the Avrami exponent determined by the two methods, we can conclude that the value of Avrami index determined by Benett and Augis [78] is lower than the local n(x) value determined by J.S. Blázquez et al. [81], but in both cases greater than 3.
Thermal analysis methods are widely used to study crystallization kinetics in amorphous solids. There are opinions in the literature that the JMAK model has limited validity both in isothermal and non-isothermal conditions [87]. But many improved versions have been published [88, 89]. The validity of the Kissinger method is also disputed [90] and therefore authors have further developed the Kissinger method [91, 92]. Controversial results related to the activation energy of FINEMET alloys have been also reported. For example, the kinetics of nanocrystallization of Fe73.5Si13.5B9Cu1Nb3 alloy has been investigated by several authors. According to H.A. Shivaee et al. [93] for volume fraction in the range of 0.1–0.75, activation energy increased approximately linearly with increasing crystalline fraction. In another study, W.Lu et al. [94] have found that the activation energy is approximately constant in the range of 0.3–0.7. Differences in results are probably due to several reasons: the nanocrystallization complexity of the amorphous alloy, the state of the starting material may not be the same even though the chemical composition is the same (the amorphous structure and the volume fraction that crystallizes during the devitrification depends on the cooling rate [95]) and there may be nanoscale crystals in the material that are not visible on X-ray measurement.
New developments-fast-scan DSC (FSDSC) and ultra-fast (Flash) DSC
From the second half of the twentieth century, there has been a great interest in the fundamental study of the amorphization and partial to full crystallization of materials. It is required using high cooling and heating rates. The introduction of chip-based calorimeters allowed the application of a wide range of heating and cooling rates: from 0.1 to ~ 5000 K s-1 in the case of fast-scan DSC and up to 40,000 K s-1 in the case of ultra-fast DSC (Flash DSC). Using these instruments enable in-depth study of metastability and reorganization of samples with a 1 to 1000 ng scale. In addition, FSDSC will become a crucial tool for the optimization of processing methods at high speeds like injection molding [96, 97]. The first instrument to be commercially available was Flash DSC 1 (Mettler Toledo) in 2010. The apparatus of MultiSTAR USF1 (XI-400) works with the mechanical refrigerator for the effective range of measurement from −100 to 450 °C. Nowadays it is widely used for the study of polymers, pharmaceutical materials and even less so for metallic materials. The maximum temperature in the current equipment is 800 °C, so it is understood that it is currently used even less in the testing of metallic materials. However, this technique can detect the glass temperature, small and fast phase transitions, and it is possible to analyse non-equilibrium transformations at high heating or cooling rates in the case of metallic materials.
The most obvious advantage of using fast-scan DSC is that it reduces the instrumental analytical time giving a speedier collection of data. The mass of the sample is generally less than 500 μg, so it provides information on a very small volume. The good thermal contact between a sample and the pans is also important for normal DSC, but this is especially important for fast DSC. Flat pans should always be used with helium as a purge gas, which has a higher thermal conductivity than nitrogen or air and allows better heat transfer and peak resolution. As the heating rate increases, of course, the peak temperatures shift towards higher temperatures. The onset temperature of melting greatly increases above a heating rate of 1000 °C s-1 [98]. S. Pogatscher et al. [98] determined the critical cooling rate required to reach the amorphous state from melt in the case of Au49Ag5.5Pd2.3Cu26.9Si16.3 alloy using the fast scan DSC. Due to the use of fast and ultra-fast DSC, it is possible to study several things that have not been done so far with the use of traditional DSC: J.Schawe et al. [99] found that monotropic polymorphism exists in Au-based amorphous alloys, M.Gao et al. [100] determined the delay time for primary crystallization of Al-based metallic glasses with poor glass formation ability.
Kinetical studies show that the plots of the ultrafast DSC data exhibit non-Arrhenius behavior more or less, and the value of activation energy decreases continuously when the heating rate increases [101].
Conclusions
The differential scanning calorimetry can provide a lot of information about amorphous alloys, not only the characteristic temperatures and thermal events. Of course, there are some disadvantages of the DSC test, but compared to other measurement methods, it certainly has the advantage that the experiment is not complicated, and, after a calibration that was precisely carried out, much information can be obtained from a few measurements in a short time using a small amount of material. DSC analysis supplies both thermodynamics and kinetic data on these materials analysed and also provides a better insight into the short-range order—long-range order transition. Unfortunately, the DSC analysis of amorphous alloys is not standardized. A lot of the properties determined by DSC measurement strongly depend on the measurement conditions, so a uniform test would be needed. The authors hope that the remarks in this article will be helpful for the readers in their work.
References
Guerlac H. Chemistry as a branch of physics: Laplace’s collaboration with Lavoisier. Hist Stud Phys Sci. 1976;7:193–276. https://doi.org/10.2307/27757357.
Höhne GWH, Cammenga HK, Eysel W, Gmelin E, Hemminger W. The temperature calibration of scanning calorimeters. Thermochim Acta. 1990. https://doi.org/10.1016/0040-6031(90)80235-Q.
Höhne GWH, Hemminger WF, Flammersheim H-J. Differential Scanning Calorimetry. Berlin, Heidelberg: Springer; 2003.
Boettinger William J, DTA and heat-flux DSC measurements of alloy melting and freezing. In: Methods Phase Diagr. Determ., Elsevier Science Ltd, (2007) pp. 151–221.
Klement W, Willens RH, Duwez P. Non-crystalline structure in solidified gold-silicon alloys. Nature. 1960;187:869–70. https://doi.org/10.1038/187869b0.
Inoue A, Takahashi Y, Suryanarayana C, Masumoto T. Thermal stability and crystallization behaviour of amorphous Zr-M-Si (M=IV, VIII group transition metals) alloys. J Mater Sci. 1982. https://doi.org/10.1007/BF01203492.
Buschow KHJ, Verbeek BH, Dirks AG. Thermal behaviour of amorphous alloys of Ni with Zr, Hf and Pr near 40 at% Ni. J Phys D Appl Phys. 1981. https://doi.org/10.1088/0022-3727/14/6/017.
Révész Á, Concustell A, Varga LK, Suriñach S, Baró MD. Influence of the wheel speed on the thermal behaviour of Cu60Zr20Ti20 alloys. Mater Sci Eng A. 2004. https://doi.org/10.1016/j.msea.2003.10.151.
Pak J, Qiu W, Pyda M, Nowak-Pyda E, Wunderlich B. Can one measure precise heat capacities with DSC or TMDSC? J Therm Anal Calorim. 2005. https://doi.org/10.1007/s10973-005-0935-8.
Egami T. Structural relaxation in amorphous alloys - compositional short range ordering. Mater Res Bull. 1978. https://doi.org/10.1016/0025-5408(78)90178-2.
Pethes I, Piarristeguy A, Pradel A, Michalik S, Nemausat R, Darpentigny J, Jóvári P. Short range order and topology of GexGaxTe100-2x glasses. J Alloys Compd. 2020. https://doi.org/10.1016/j.jallcom.2020.155097.
Ruppersberg H, Lee D, Wagner CNJ. Observation of chemical short-range order in an amorphous Ni40Ti60 alloy. J Phys F Met Phys. 1980. https://doi.org/10.1088/0305-4608/10/8/004.
Mohammadiparsa N, Habibi S, Dekan J, Janotová I, Švec P, Galdun L, Varga R, Kmječ T, Bujdoš M, Miglierini M. Mössbauer study and magnetic properties of Fe–Si–B–Cu amorphous systems with minor substitution of carbon. J Radioanal Nucl Chem. 2019. https://doi.org/10.1007/s10967-019-06811-3.
Turnbull D, Cohen MH. Free-volume model of the amorphous phase: Glass transition. J Chem Phys. 1961. https://doi.org/10.1063/1.1731549.
Cohen MH, Grest GS. Liquid-glass transition, a free-volume approach. Phys Rev B. 1979. https://doi.org/10.1103/PhysRevB.20.1077.
Spaepen F. A microscopic mechanism for steady state inhomogeneous flow in metallic glasses. Acta Metall. 1977. https://doi.org/10.1016/0001-6160(77)90232-2.
Slipenyuk A, Eckert J. Correlation between enthalpy change and free volume reduction during structural relaxation of Zr55Cu30Al10Ni 5 metallic glass. Scr Mater. 2004. https://doi.org/10.1016/j.scriptamat.2003.09.038.
Qiao JC, Pelletier JM. Enthalpy relaxation in Cu46Zr45Al7Y 2 and Zr55Cu30Ni5Al10 bulk metallic glasses by differential scanning calorimetry (DSC). Intermetallics. 2011. https://doi.org/10.1016/j.intermet.2010.08.042.
Murali P, Ramamurty U. Embrittlement of a bulk metallic glass due to sub-Tg annealing. Acta Mater. 2005. https://doi.org/10.1016/j.actamat.2004.11.040.
Ramamurty U, Lee ML, Basu J, Li Y. Embrittlement of a bulk metallic glass due to low-temperature annealing. Scr Mater. 2002. https://doi.org/10.1016/S1359-6462(02)00102-1.
Yokoyama Y, Akeno Y, Yamasaki T, Liaw PK, Buchanan RA, Inoue A. Evolution of mechanical properties of cast Zr50Cu40Al10 glassy alloys by structural relaxation. Mater Trans. 2005. https://doi.org/10.2320/matertrans.46.2755.
Chen LY, Fu ZD, Zhang GQ, Hao XP, Jiang QK, Wang XD, Cao QP, Franz H, Liu YG, Xie HS, Zhang SL, Wang BY, Zeng YW, Jiang JZ. New class of plastic bulk metallic glass. Phys Rev Lett. 2008. https://doi.org/10.1103/PhysRevLett.100.075501.
Xu Y, Fang J, Gleiter H, Hahn H, Li J. Quantitative determination of free volume in Pd40Ni40P20 bulk metallic glass. Scr Mater. 2010. https://doi.org/10.1016/j.scriptamat.2010.01.025.
Van Den BA, Radelaar S. On the kinetics of structural relaxation in metallic glasses. Acta Metall. 1983. https://doi.org/10.1016/0001-6160(83)90219-5.
Wang WH. Dynamic relaxations and relaxation-property relationships in metallic glasses. Prog Mater Sci. 2019. https://doi.org/10.1016/j.pmatsci.2019.03.006.
Greer AL. Effect of quench rate on the structural relaxation of a metallic glass. J Mater Sci. 1982. https://doi.org/10.1007/BF00543531.
Greer AL. The use of DSC to determine the Curie temperature of metallic glasses. Thermochim Acta. 1980. https://doi.org/10.1016/0040-6031(80)87103-6.
Blázquez JS, Lozano-Pérez S, Conde A. Enthalpy and Curie temperature relaxation effects in FeSiB-CuNb alloys prepared at different quenching rates. Mater Lett. 2000. https://doi.org/10.1016/S0167-577X(00)00113-0.
Miguel C, Kaloshkin S, Gonzalez J, Zhukov A. Curie temperature behaviour on annealing of Finemet type amorphous alloys. J Non Cryst Solids. 2003. https://doi.org/10.1016/j.jnoncrysol.2003.08.014.
Mizoguchi T. Magnetism in Amorphous Alloys. AIP Conf Proc. 1976. https://doi.org/10.1063/1.2946104.
Zhang Y, Blazquez JS, Conde A, Warren PJ, Cerezo A. Partitioning of Co during crystallisation of Fe–Co–Nb–B(–Cu) amorphous alloys. Mater Sci Eng A. 2003. https://doi.org/10.1016/s0921-5093(02)00683-4.
Borrego JM, Conde CF, Conde A, Chadwick AV, Morrison G. X-ray absorption studies of a FINEMET alloy. J Non Cryst Solids. 1998. https://doi.org/10.1016/S0022-3093(98)00420-7.
Ayers JD, Harris VG, Sprague JA, Elam WT, Jones HN. On the formation of nanocrystals in the soft magnetic alloy Fe73.5Nb3CU1Si13.5B9. Acta Mater. 1998. https://doi.org/10.1016/S1359-6454(97)00436-9.
Ohnuma M, Hono K, Linderoth S, Pedersen JS, Yoshizawa Y, Onodera H. Small-angle neutron scattering and differential scanning calorimetry studies on the copper clustering stage of Fe-Si-B-Nb-Cu nanocrystalline alloys. Acta Mater. 2000. https://doi.org/10.1016/S1359-6454(00)00277-9.
Tomolya K, Janovszky D, Sycheva A, Benke M, Erdohegyi C, Roosz A. Investigation of AgAlCuZr amorphous/crystalline structure produced by casting and milling. J Alloys Compd. 2014. https://doi.org/10.1016/j.jallcom.2012.10.155.
Chen HS, Turnbull D. Evidence of a glass-liquid transition in a gold-germanium-silicon alloy. J Chem Phys. 1968. https://doi.org/10.1063/1.1669483.
Reinker B, Dopfer M, Moske M, Samwer K. Specific heat of Zr65Al7.5Cu17.5Ni10 around the glass transition. Eur Phys J B. 1999. https://doi.org/10.1007/s100510050622.
Guo J, Zu F, Chen Z, Zheng S, Yuan Y. Exploration of a new method in determining the glass transition temperature of BMGs by electrical resistivity. Solid State Commun. 2005. https://doi.org/10.1016/j.ssc.2005.03.052.
Perera DN, Tsai AP, Perera DN. Thermal and viscoelastic properties of a strong bulk metallic glass former. J Phys D Appl Phys. 2000. https://doi.org/10.1088/0022-3727/33/16/301.
Pang J, Wang Y, Tan MJ, Liew KM. Thermal properties of metallic glasses: Heating rate dependence and their correlation. Mater Lett. 2014. https://doi.org/10.1016/j.matlet.2014.03.158.
Patel AT, Pratap A. Study of kinetics of glass transition of metallic glasses. J Therm Anal Calorim. 2012. https://doi.org/10.1007/s10973-012-2527-8.
Zhou GF, Bakker H. Solid-state amorphization of CoV crystalline intermetallic compound by mechanical milling. Intermetallics. 1994. https://doi.org/10.1016/0966-9795(94)90004-3.
Jang JSC, Koch CC. Amorphization and disordering of the Ni3Ai ordered intermetallic by mechanical milling. J Mater Res. 1990. https://doi.org/10.1557/JMR.1990.0498.
Janovszky D, Kristaly F, Miko T, Racz A, Sveda M, Sycheva A, Koziel T. Phase transformation and morphology evolution of Ti50Cu25Ni20Sn5 during mechanical milling. Materials. 2018. https://doi.org/10.3390/ma11091769.
Kim DH, Kim WT, Park ES, Mattern N, Eckert J. Phase separation in metallic glasses. Prog Mater Sci. 2013. https://doi.org/10.1016/j.pmatsci.2013.04.002.
Hays CC, Kim CP, Johnson WL. Large supercooled liquid region and phase separation in the Zr-Ti-Ni-Cu-Be bulk metallic glasses. Appl Phys Lett. 1999. https://doi.org/10.1063/1.124606.
Shen Y, Perepezko JH. Al-based amorphous alloys: Glass-forming ability, crystallization behavior and effects of minor alloying additions. J Alloys Compd. 2017. https://doi.org/10.1016/j.jallcom.2016.11.079.
Wang XL, Almer J, Liu CT, Wang YD, Zhao JK, Stoica AD, Haeffner DR, Wang WH. In situ synchrotron study of phase transformation behaviors in bulk metallic glass by simultaneous diffraction and small angle scattering. Phys Rev Lett. 2003. https://doi.org/10.1103/PhysRevLett.91.265501.
Louzguine-Luzgin DV, Jiang J, Bazlov AI, Zolotorevzky VS, Mao H, Ivanov YP, Greer AL. Phase separation process preventing thermal embrittlement of a Zr-Cu-Fe-Al bulk metallic glass. Scr Mater. 2019. https://doi.org/10.1016/j.scriptamat.2019.03.030.
Kündig AA, Ohnuma M, Ohkubo T, Hono K. Early crystallization stages in a Zr-Cu-Ni-Al-Ti metallic glass. Acta Mater. 2005. https://doi.org/10.1016/j.actamat.2005.01.022.
Takeuchi A, Inoue A. Classification of bulk metallic glasses by atomic size difference, heat of mixing and period of constituent elements and its application to characterization of the main alloying element. Mater Trans. 2005. https://doi.org/10.2320/matertrans.46.2817.
Nakagawa Y. Liquid immiscibility in copper-iron and copper-cobalt systems in the supercooled state. Acta Metall. 1958. https://doi.org/10.1016/0001-6160(58)90061-0.
Kaban I, Hoyer W. Effect of Cu and Sn on liquid-liquid interfacial energy in ternary and quaternary Al-Bi-based monotectic alloys. Mater Sci Eng A. 2008. https://doi.org/10.1016/j.msea.2007.08.097.
He J, Jiang H, Chen S, Zhao J, Zhao L. Liquid phase separation in immiscible Ag-Ni-Nb alloy and formation of crystalline/amorphous composite. J Non Cryst Solids. 2011. https://doi.org/10.1016/j.jnoncrysol.2011.07.015.
He J, Li H, Yang B, Zhao J, Zhang H, Hu Z. Liquid phase separation and microstructure characterization in a designed Al-based amorphous matrix composite with spherical crystalline particles. J Alloys Compd. 2010. https://doi.org/10.1016/j.jallcom.2009.09.102.
Janovszky D, Tomolya K, Sycheva A, Kaptay G. Stable miscibility gap in liquid Cu-Zr-Ag ternary alloy. J Alloys Compd. 2012. https://doi.org/10.1016/j.jallcom.2012.07.015.
Drzeżdżon J, Jacewicz D, Sielicka A, Chmurzyński L. Characterization of polymers based on differential scanning calorimetry based techniques. TrAC Trends Anal Chem. 2019. https://doi.org/10.1016/j.trac.2018.10.037.
Lefort R, De Gusseme A, Willart JF, Danède F, Descamps M. Solid state NMR and DSC methods for quantifying the amorphous content in solid dosage forms: an application to ball-milling of trehalose. Int J Pharm. 2004. https://doi.org/10.1016/j.ijpharm.2004.05.012.
Adlwarth-Dieball R, Ryder PL. Crystallization kinetics of amorphous CuNiP alloys. Mater Sci Eng A. 1991. https://doi.org/10.1016/0921-5093(91)90087-4.
Xing LQ, Bertrand C, Dallas JP, Cornet M. Nanocrystal evolution in bulk amorphous Zr57Cu20Al10Ni8Ti5 alloy and its mechanical properties. Mater Sci Eng A. 1998. https://doi.org/10.1016/s0921-5093(97)00489-9.
Yang HW, Wen J, Quan MX, Wang JQ. Evaluation of the volume fraction of nanocrystals devitrified in Al-based amorphous alloys. J Non Cryst Solids. 2009. https://doi.org/10.1016/j.jnoncrysol.2008.12.001.
Wesseling P, Ko BC, Lewandowski JJ. Quantitative evaluation of α-Al nano-particles in amorphous Al87Ni7Gd6 - comparison of XRD, DSC, and TEM. Scr Mater. 2003. https://doi.org/10.1016/S1359-6462(03)00127-1.
Görög S. Critical review of reports on impurity and degradation product profiling in the last decade. TrAC - Trends Anal Chem. 2018. https://doi.org/10.1016/j.trac.2017.09.012.
Khoshooei MA, Fazlollahi F, Maham Y. A review on the application of differential scanning calorimetry (DSC) to petroleum products: characterization and kinetic study. J Therm Anal Calorim. 2019. https://doi.org/10.1007/s10973-019-08244-2.
Marti EE. Purity determination by differential scanning calorimetry. Thermochim Acta. 1972. https://doi.org/10.1016/0040-6031(72)85022-6.
Zhang Y, Li Y, Tan H, Chen GL. Effect of liquidus temperature depression on glass forming ability criteria for La-Al-(Cu, Ni) alloys. Intermetallics. 2007. https://doi.org/10.1016/j.intermet.2006.10.023.
Lu ZP, Liu CT. A new glass-forming ability criterion for bulk metallic glasses. Acta Mater. 2002. https://doi.org/10.1016/S1359-6454(02)00166-0.
Kündig AA, Ohnuma M, Ohkubo T, Abe T, Hono K. Glass formation and phase separation in the Ag-Cu-Zr system. Scr Mater. 2006. https://doi.org/10.1016/j.scriptamat.2006.05.012.
He XC, Wang H, Liu HS, Jin ZP. Thermodynamic description of the Cu-Ag-Zr system. Calphad Comput Coupling Phase Diagrams Thermochem. 2006. https://doi.org/10.1016/j.calphad.2006.09.001.
Kang DH, Jung IH. Critical thermodynamic evaluation and optimization of the Ag-Zr, Cu-Zr and Ag-Cu-Zr systems and its applications to amorphous Cu-Zr-Ag alloys. Intermetallics. 2010. https://doi.org/10.1016/j.intermet.2009.12.013.
Janovszky D, Sycheva A, Tomolya K, Geiger J, Solyom J, Roosz A. Solidification processes in Cu-Zr-Ag amorphisable alloy system. J Alloys Compd. 2014. https://doi.org/10.1016/j.jallcom.2013.09.095.
Ferreira EB, Lima ML, Zanotto ED. DSC method for determining the liquidus temperature of glass-forming systems. J Am Ceram Soc. 2010. https://doi.org/10.1111/j.1551-2916.2010.03976.x.
Nishikawa H, Hamada Y, Takemoto T. Estimation method for liquidus temperature of lead-free solder using differential scanning calorimetry profiles. J Electron Mater. 2009. https://doi.org/10.1007/s11664-009-0921-1.
Johnson WA. Reaction kinetics in processes of nucleation and growth. Trans Am Inst Min Met Eng. 1939;135:416.
Avrami M. Granulation, phase change, and microstructure kinetics of phase change. III J Chem Phys. 1941. https://doi.org/10.1063/1.1750872.
Kolmogorov AN. On the Statistical Theory of Crystallization of Metals [in Russian]. Izv Akad Nauk SSSR Ser Mat. 1937;3:355–9.
Sestak J. Comprehensive Analytical Chemistry. Amsterdam: Elsevier; 1984.
Kissinger HE. Reaction kinetics in differential thermal analysis. Anal Chem. 1957. https://doi.org/10.1021/ac60131a045.
Takhor R.L. Advances in Nucleation and Crystallization of Glasses. Am Ceram Soc. 1972; 166.
Ozawa T. Kinetic analysis of derivative curves in thermal analysis. J Therm Anal. 1970. https://doi.org/10.1007/BF01911411.
Akahira T, Sunosa T, Trans, . Joint convention of four electrical institutes. T Res Rep Chiba Inst Technol. 1971;16:22–31.
Flynn JH. The isoconversional method for determination of energy of activation at constant heating rates - corrections for the Doyle approximation. J Therm Anal. 1983. https://doi.org/10.1007/BF01907325.
Augis JA, Bennett JE. Calculation of the Avrami parameters for heterogeneous solid state reactions using a modification of the Kissinger method. J Therm Anal. 1978. https://doi.org/10.1007/BF01912301.
Blázquez JS, Conde CF, Conde A. Non-isothermal approach to isokinetic crystallization processes: application to the nanocrystallization of HITPERM alloys. Acta Mater. 2005. https://doi.org/10.1016/j.actamat.2005.01.037.
Stoica M, Li R, Yavari AR, Vaughan G, Eckert J, Van Steenberge N, Romera DR. Thermal stability and magnetic properties of FeCoBSiNb bulk metallic glasses. J Alloys Compd. 2010. https://doi.org/10.1016/j.jallcom.2010.04.013.
Santos DSD, Santos DRD. Crystallization kinetics of Fe-B-Si metallic glasses. J Non Cryst Solids. 2002. https://doi.org/10.1016/S0022-3093(02)01004-9.
Tang L, Peng K, Wu Y, Zhang W, Effect of ion bombardment on the crystallization kinetics of FeSiNbBCu amorphous alloys. 2016; https://doi.org/10.1016/j.jallcom.2016.11.057.
Chen Z, Zhu Q, Zhang K, Jiang Y. The non-isothermal and isothermal crystallization behavior and mechanism of Fe–Ni alloys. Cryst Growth Des. 2020. https://doi.org/10.1021/acs.cgd.9b01059.
Ramanan VRV, Fish GE. Crystallization kinetics in Fe−B−Si metallic glasse. J Appl Phys. 1982. https://doi.org/10.1063/1.330797.
Malek J. Kinetic analysis of crystallization processes in amorphous materials. Thermochim Acta. 2000. https://doi.org/10.1016/S0040-6031(00)00449-4.
Farjas J, Roura P. Modification of the Kolmogorov-Johnson-Mehl-Avrami rate equation for non-isothermal experiments and its analytical solution. Acta Mater. 2006. https://doi.org/10.1016/j.actamat.2006.07.037.
Očenášek J, Novák P, Agbo S. Finite-thickness effect on crystallization kinetics in thin films and its adaptation in the Johnson-Mehl-Avrami-Kolmogorov model. J Appl Phys. 2014. https://doi.org/10.1063/1.4862858.
Holba P, Šesták J. Imperfections of Kissinger evaluation method and crystallization kinetics. Glas Phys Chem. 2014. https://doi.org/10.1134/S1087659614050058.
Ozawa T. Non-isothermal kinetics and generalized time. Thermochim Acta. 1986. https://doi.org/10.1016/0040-6031(86)87053-8.
Ozawa T. Estimation of activation energy by isoconversion methods. Thermochim Acta. 1992. https://doi.org/10.1016/0040-6031(92)85192-X.
Shivaee HA, Hosseini HRM. Advanced isoconversional kinetics of nanocrystallization in Fe73.5Si13.5B9Nb3Cu 1 alloy. Thermochim Acta. 2009. https://doi.org/10.1016/j.tca.2009.04.020.
Lu W, Yang L, Yan B, Huang WH. Nanocrystallization kinetics of amorphous Fe73.5Cu1Nb3Si13.5B9 alloy. J Alloys Compd. 2006. https://doi.org/10.1016/j.jallcom.2005.10.049.
Gheiratmand T, Hosseini HRM, Davami P, Ostadhossein F, Song M, Gjoka M. On the effect of cooling rate during melt spinning of FINEMET ribbons. Nanoscale. 2013. https://doi.org/10.1039/c3nr01213a.
Zhuravlev E, Schick C. Fast scanning power compensated differential scanning nano-calorimeter: 1 the device. Thermochim Acta. 2010. https://doi.org/10.1016/j.tca.2010.03.019.
Schick C, Mathot V. Fast scanning calorimetry. New York: Springer; 2016.
Pogatscher S, Leutenegger D, Hagmann A, Uggowitzer PJ, Löffler JF. Characterization of bulk metallic glasses via fast differential scanning calorimetry. Thermochim Acta. 2014. https://doi.org/10.1016/j.tca.2014.06.007.
Schawe JEK, Pogatscher S, Löffler JF. Thermodynamics of polymorphism in a bulk metallic glass: heat capacity measurements by fast differential scanning calorimetry. Thermochim Acta. 2020. https://doi.org/10.1016/j.tca.2020.178518.
Gao M, Perepezko JH. Al-based amorphous metallic plastics. Adv Eng Mater. 2019. https://doi.org/10.1002/adem.201800930.
Chen Y, Pan H, Mu S, Wang G, Wang R, Shen X, Wang J, Dai S, Xu T. Intermediate crystallization kinetics in Germanium-Tellurides. Acta Mater. 2019. https://doi.org/10.1016/j.actamat.2018.10.051.
Acknowledgements
This work was carried out as part of the GINOP-2.3.2-15-2016-00027 project implemented in the framework of the Szechenyi 2020 program. The realization of this project is supported by the European Union.
Funding
Open access funding provided by University of Miskolc.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by [Piotr Bala], [Maria Sveda], [Anna Sycheva] and [Ferenc Kristaly]. The first draft of the manuscript was written by [Dora Janovszky] and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Janovszky, D., Sveda, M., Sycheva, A. et al. Amorphous alloys and differential scanning calorimetry (DSC). J Therm Anal Calorim 147, 7141–7157 (2022). https://doi.org/10.1007/s10973-021-11054-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-021-11054-0