
Abstract
A series of high-purity Er0.05Y1.95O3 nanopowders with different lanthanum content was prepared by modification of the Pechini sol–gel method using citric acid and ethylene glycol as the chelating agent. The microstructure of the powders was studied by means of X-ray diffraction (XRD) and Fourier-transform infrared spectroscopy. In order to evaluate the structural characteristics of the obtained gel, XRD measurements were carried out with calcination gels in selected temperatures. Simultaneous differential thermal analysis with thermal gravimeters indicates a decrease of calcination temperature with an increasing content of lanthanum ions. Morphological properties of the nano-sized powders were examined by scanning electron microscopy. Strong luminescence in near IR region was observed under 980 nm excitation at room temperature. By varying the concentration of La3+ ion, various intensities of upconversion luminescence can be easily achieved.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Recently, new types of optical devices, such as high-energy laser, optical fiber amplifiers, and optical rotators, have attracted significant attention due to their unique physical properties. Y2O3, either pure or often doped with rare-earth ions, has been widely investigated as a host material for potential applications in lasers, three dimensional volumetric displays, fluorescent labels, and luminous pipes for high-intensity discharge lamps [1,2,3,4,5,6,7,8,9,10,11,12,13]. The performance of these devices is mainly dependent on the physicochemical properties of the powders that were sintered. The powders are useful due to their good chemical and physical stability, high corrosion resistance, and broad transparency range (0.2–10 μm) [1, 2, 5]. The recent research and reports have shown the relatively low phonon energy (430–550 cm−1) [1,2,3] and high thermal conductivity [13.6 W(mK)−1] [1,2,3,4]. Pure yttrium oxide exhibits C-bixbyite, cubic, hexagonal, or monoclinic crystal structure [5,6,7,8,9,10]. Many different chemical methods, such as sol–gel EDTA, hydrothermal, co-precipitation method, spray pyrolysis, thermochemical reactions, and solid state reaction, were widely used to synthesize nano-sized powders of pure and doped Y2O3 [10,11,12,13,14,15]. Furthermore, citrate sol–gel process is one of wet chemical methods that receives attention due to its high efficiency and possibility to prepare high clean and nano-sized powders [4, 6,7,8,9, 13, 15]. Numerous studies conducted thus far indicate that the choice of preparation method influences the size, shape, and crystalline structure of powders, which determine optical, transmittance, and luminescence properties of sinters [1,2,3,4,5]. The rare-earth metal ions, e.g.: Nd, Er, Eu, and Tm, have been successfully used as an additive material in order to improve the performances of Y2O3 materials [3,4,5,6,7, 10,11,12]. Many papers have indicated that the doping of Y2O3 with erbium ions allows for luminescence emission in the infrared range [3, 14]. Additional substitution of La ions can also enhance luminescence in Er:Y2O3 by tuning the symmetry of the crystal field around the rare-earth ions [15]. Notably, doped with rare-earth ions, Y2O3 may be used as a magneto-optic ceramic potential to build a magneto-optic device [4, 16]. This device is based on Faraday phenomena, and it uses external magnetic fields. Criteria for magneto-optical material are high transparence, chemical stability, and high Verdet constant. Nano-sized yttria meets the first two criteria. The Verdet constant V may be improved by doping with diamagnetic or/and paramagnetic ions, because it is the sum of the two types of component ions: V = Vdiamagnetic (λ) + Vparamagnetic (λ; T), where λ is wavelength and T is temperature. [4, 16,17,18,19,20,21].
This paper focuses on the development of high clean Lax:Er0.5Y1.95−xO3 nanopowders with doping concentration varied from 0 to 0.1 mol. The amount of La ions was varied to improve the sintering process and luminescence properties. In addition, lanthanum (paramagnetic) and erbium (paramagnetic) ions were used to improve the Verdet constant. The presented results show that varying the amount of lanthanum ions added to the powders has a significant effect on both microstructure and luminescence properties. The structure evolution and microstructure characterization of the powders were carried out by X-ray diffraction (XRD), Fourier-transform infrared (FT-IR) spectroscopy, and scanning electron microscopy (SEM). The surface of powders was analyzed by the Brunauer–Emmett–Teller (BET) method. Strong dependence of luminescence due to the La3+ concentration was observed. High yield upconversion emissions in Er-doped Y2O3 powders were measured under the excitation of a 980 nm continuous wave diode laser.
Experimental
Powder characterization
In this paper, the synthesis of polycrystalline Er0.05Y1.95O3, La0.01Er0.05Y1.94O3, La0.05Er0.05Y1.9O3, and La0.1Er0.05Y1.85O3 nano-sized powders is reported. The powders were obtained using a modified citrate sol–gel process with ethylene glycol and citric acid as fuel. Thermal analysis and evolved gas analysis of the abovementioned precursor gels were carried out in the temperature range 25–700 °C in a simultaneous thermogravimetry DTA/TG/EGA with a fast Quadrupole Mass Spectrometer (QMS) setup (Netzsch STA 449 F3 with a SDT 2960) with a heating rate 10 °C min−1 in an ambient gas atmosphere and alumina as reference.
Structural characterization and phase identification of the obtained powders were carried out using a X’Pert PANalytical X-ray diffractometer, with CuKα (1.5405 Å) radiation. XRD patterns of the gels and calcined powder were recorded over the angular range 10°–90° with step size 0.01°. Qualitative phase analysis was conducted by use of Highscore Plus software and database PCPDFWIN v.2.3.
The average crystallite size of the sample was calculated using the XRD data and the Scherrer formula:
where K = 0.9, DXRD—denotes the crystallite diameter [nm], λ—the wavelength of the incident X-rays, β—the corrected full width at half maximum (FWHM) width of the diffraction peak, and θ—the diffraction angle. A pseudo-Voigt function was used to fit the XRD curve and to calculate the FHWM value, and β was determined using Eq. (2), where Bobs is the FHWM that is related to the sample and B is the FHWM of the standard (corundum, α-Al2O3):
The dislocation density of the particulate structure can be calculated using the relation:
SEM microghaphs of the obtained powders were recorded using A FEI Nova NanoSEM 200. The powders were prepared by dispersing them in ethyl alcohol via ultra-sonication for 0.5 h. Next, the solution was poured on a commercial carbon-coated copper grid and dried at 80 °C for 3 h in a hot air oven.
Fourier-transform infrared (FT-IR) spectra of the samples were conducted using an FT-IR spectrometer (Brucker 70 V) with the KBr pellet method in the wave number range 400–4000 cm−1 with a step 3 cm−1.
The distribution of the particle (agglomerate) size of the yttria powders was determined by the laser light diffraction method (Mastersizer 2000S, Malvern Instruments) with measurement error 5%.
The specific surface area of the prepared powders was estimated by the BET (Brunauer–Emmett–Teller) method using an ASAP 2010 v4.00 G instrument with tolerance under 10%.
Powders were milled in rotary mill (600 rpm) for 1 h in an ethanol medium with ϕ = 1 mm zirconium balls and jars.
The luminescence spectroscopy of the powders was performed using a Hamamatsu NIR (0.1 nm resolution) spectrometer with continuous wave 980 nm laser diode (Spectra-Laser) as a light source.
Powder synthesis
The chemicals used were lanthanum (III) nitrate hexahydrate La(NO3)3 × 6H2O (Sigma-Aldrich; 99.9%), erbium (III) nitrate hexahydrate Er(NO3)3 × 6H2O (Sigma-Aldrich; ≥ 99.9%), and yttrium nitrate hexahydrate Y(NO3)3 × 6H2O (Sigma-Aldrich; 99.9%). Glycerin alcohol (PEG, Sigma-Aldrich) and citric acid (CA, Sigma-Aldrich) were used as starting materials. First, metal hydrates were dissolved separately in minimum quantities of distilled water. The solutions of erbium hydroxide and lanthanum hydroxide were mixed homogeneously with PEG. At the same time, a yttrium hydroxide solution was mixed with CA using a magnetic stirrer maintained at a constant rotation speed of 300 rpm for 2 h at 80 °C. After that, both solutions were mixed together by the magnetic sitter at a constant rotation speed of 300 rpm and heated at 80 °C. The stoichiometric proportion of CA to PEG was 3:7, respectively. The solution was heated at 180 °C for several hours until formation of gel took place. The precursor gel of La0.05Er0.05Y1.9O3 was calcined at different temperatures, which were selected by differential thermal analysis with thermal gravimeters (DTA/TG) results: 180, 280, 350, 500 °C, and 700 °C for 2 h in ambient air. On the basis of studies of the decomposition of the gel precursors, the optimum conditions for production of yttrium oxide doped with erbium and lanthanum powder were determined. The optimum conditions for calcination were 700 °C for 10 h in air.
Experimental
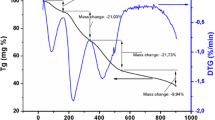
Figure 1 illustrates the DTA/TG analysis of the four dried gels, Er0.05Y1.95O3, La0.01Er0.05Y1.94O3, La0.05Er0.05Y1.9O3, and La0.1Er0.05Y1.84O3 from room temperature to 700 °C under ambient atmosphere. From the TG curves of Er0.05Y1.95O3, La0.01Er0.05Y1.94O3, and La0.1Er0.05Y1.84O3, approximately 28%, 32%, and 30% mass loss was observed in the first stage in the temperature range of 25–400 °C, which corresponds to the loss of dehydration of crystalline water molecules or the presence of the grains and organic group (PEG/CA) on the surface. The thermogravimetric curve of La0.05Er0.05Y1.9O3 presents a more steady tendency of mass loss; nevertheless, it indicates similar characteristics to rest the of the exanimate powders. In the same temperature range, mass loss was about 42% for La0.05Er0.05Y1.9O3 gel, which suggests a higher amount of water molecules. The TG curve of Er0.05Y1.95O3, La0.01Er0.05Y1.94O3, and La0.1Er0.05Y1.84O3 gels exhibits an intensive mass loss between 400 and 450 °C because of elimination of crystalline O–H groups (Fig. 2). Above 500 °C temperature, TG analysis shows sluggish mass loss up to 700 °C in the abovementioned gels, which corresponds to the elimination of rest organic groups. Total mass loss in the investigated temperature range for Er0.05Y1.95O3, La0.01Er0.05Y1.94O3, La0.05Er0.05Y1.9O3, and La0.1Er0.05Y1.84O3 gels was 60, 59, 76, and 61%, respectively. The DTA curves obtained for Er0.05Y1.95O3, La0.01Er0.05Y1.94O3, and La0.1Er0.05Y1.84O3 gels shared similar characteristics, unlike the La0.05Er0.05Y1.9O3 gel. This last gel shows splicing from three exothermic reactions at peak temperatures of 454, 502, and 537 °C. In the case of decomposition of Er0.05Y1.95O3, La0.01Er0.05Y1.94O3, and La0.1Er0.05Y1.84O3 gels, higher amounts of lanthanum decrease the temperature at the end of the crystallization process from 433 to 428 °C. The different DTA curve characteristics of La0.05Er0.05Y1.9O3 gel result from higher amounts of crystalline water molecules and organic groups, as confirmed by TG analysis (higher total mass loss than in the case of the other investigated gels).

DTA/TG analysis of Er0.05Y1.95O3, La0.01Er0.05Y1.94O3, La0.05Er0.05Y1.9O3, and La0.1Er0.05Y1.84O3 gels from room temperature to 700 °C under nitrogen atmosphere obtained by citrate process

EGA-MS analysis of evolved gases during decomposition of La0.05Er0.05Y1.9O3 precursor gel
The type of the gaseous decomposition products confirms the oxidation reactions of a gel, and consequently, the total decomposition of the materials studied. The QMS analysis allows a more precise description of the type of the volatile decomposition products emitted under oxidation. The QMS spectra of the volatile decomposition products emitted under heating of selected gel are presented Fig. 2. According to the QMS results, one can clearly see the presence of difference components: OH (m/z = 17), H2O (m/z = 18), CO (m/z = 28), NO (m/z = 30), O2 (m/z = 32), CO2 (m/z = 44), and NO2 (m/z = 46) when the gel was calcined from room temperature to 700 °C in ambient air. At the beginning, there was a release of fragments of the water molecules (H2O, OH) and NO2; next, below 200 °C, there is an indication that desorption of chemically and physically absorbed water also existed at temperatures lower than 470 °C. However as the temperature grows above 250 °C, all signals drastically increased, reaching a maximum at 290 °C. Further heating of the gel above 290 °C decreased emission of CO and NO gases and other signals were not observed. In the second temperature range above 300 °C, there was a release of all gases with a maximum at 374 °C, which can also be confirmed by the first endothermic peaks at 374 °C. At this temperature, the TG measurement indicates a strong of mass loss, as shown Fig. 1b. The temperature at the beginning of crystallization varies according to the La ion content. Finally at temperatures higher than 400 °C, the gel residue undergoes oxidation processes and thus the emission of OH, O2, H2O, CO2, and rest of the CO. Above temperatures higher than 500 °C, no gaseous decomposition products were detected.
Figure 3 shows the XRD diagrams of La0.1Er0.05Y1.84O3 gel during thermal decomposition in different temperatures (180, 280, 350, 500, and 700 °C) selected by DTA/TG analyses. The XRD pattern for the gel precursor calcinated at 180 and 350 °C indicated a completely amorphous structure. By increasing annealing temperature, the characteristic Bragg peaks related to the cubic phase appeared. The analysis of the DTA/TG curve shows that the crystallization process starts after heating the precursor gel to approx. 430 °C. Therefore, the formation of cubic phase occurs via reaction of the nitride solution present in the gel with gasses present in the air. In abovementioned temperature (ca. 500 °C), the strongest Bragg peaks related to yttrium phase appear. The XRD studies confirmed the single-phase composition of the powders after calcinations at 700 °C in ambient air.

XRD patterns of La0.1Er0.05Y1.84O3 gel during thermal decomposition in different temperature (180, 280, 350, 500, and 700 °C)
Moreover, precursor gels of Er0.05Y1.95O3, La0.01Er0.05Y1.94O3, La0.05Er0.05Y1.9O3, and La0.1Er0.05Y1.84O3 that were calcined at 500 °C contain distinct Bragg peaks associated with the cubic Y2O3 phase. All detected Bragg peaks become more intense and sharpen with decrease of lanthanum ions content in yttrium crystal lattice as present in Fig. 4. The investigated samples contained different amorphous structure, which could be explained by a higher amount of lanthanum that favors the formation of a liquid phase during heating [4, 19].

XRD patterns of Er0.05Y1.95O3, La0.01Er0.05Y1.94O3, La0.05Er0.05Y1.9O3, and La0.1Er0.05Y1.85O3 gels calcinated in 500 °C in ambient air
Figure 5 presents XRD patterns of a series of powders: Er0.05Y1.95O3, La0.01Er0.05Y1.94O3, La0.05Er0.05Y1.9O3, and La0.1Er0.05Y1.85O3 calcinated at 700 °C in ambient air. The identification of the crystalline phase of polycrystalline ceramic powders was performed using software X’pert HighScore (JCPDS card no. 98-008-1861). The theoretical model (Ical) was fitted to the experimental XRD data (Iexp). According to the Rietveld refinement results, the obtained ceramics are single phase with Ia-3 space group. No secondary phases were detected in the ceramics. The lattice parameters, such as the lattice spacing dhkl, the lattice parameter a, unit cell volume V, crystallite size Dxrd, and dislocation density ρ of the investigated sinters, are shown in Table 1. The microstructural parameters depend on the amount of La ions, suggesting that both Er and La cations have been successfully diffused into the crystal lattice of yttria. In this type of lattice, there are two distinct sites available for the substitution of trivalent lanthanide ions. Two kinds of cations exist: 8 Y3+ ions are on b-sites with symmetry C3i and 24 Y3+ ions on d-sites with point symmetry C2, while 48 oxygens are on e-sites with point symmetry C1 [20,21,22,23,24]. Hence, the effect of Er, La on lattice parameters could be correlated to a bigger ionic radii than the ionic radius of Y3+ (Y3+—0.90 Å, La3+—1.3 Å and Er3+—0.95 Å [4, 19]). Thus, the slight adjustment of Y2O3 lattice by the doped La3+ ions was observed. The position of the Bragg peaks [222] moves toward lower angles with an increase in La3+ ions (inset in Fig. 3), as shown in Table 2, indicating a change of the host lattice. With increase of lanthanum ions, the lattice parameter and unit cell volume of investigated powders increase. This agrees with FT-IR results (Fig. 8). As a consequence, the local symmetry of the crystal field around Er3+ reduces. The obtained results agree with other investigations [1,2,3,4,5,6, 8,9,10,11,12, 14,15,16].

XRD patterns of powders: Er0.05Y1.95O3, La0.01Er0.05Y1.94O3, La0.05Er0.05Y1.9O3, and La0.1Er0.05Y1.85O3 calcinated in 700 °C in ambient air. Right inset shows the enlarged patterns of Bragg diffraction peaks (222), and left shows the variation of lattice parameter with the different La3+ concentration
A scanning electron microscope analysis was used to investigate the particle size and morphology of powders with chemical composition: Er0.05Y1.95O3, La0.01Er0.05Y1.94O3, La0.05Er0.05Y1.9O3, and La0.1Er0.05Y1.85O3, which were calcinated at 700 °C in ambient air (Fig. 6). The typical morphological images, presented in Fig. 6, show that all of these powders have irregularly shaped individual particles with clear-cut edges, which indicates excellent crystallinity. Nano-sized particles produced micron-sized agglomerates as can be seen in Fig. 6. The surface of the agglomerates, especially of Er0.05Y1.95O3, has many cracks and pores. The SEMs of studied ceramic powders show the presence of large agglomerates in the irregular block form of approximately 2 μm and separately agglomerate over 5 μm.

SEM photographs of Er0.05Y1.95O3a, La0.01Er0.05Y1.94O3b, La0.05Er0.05Y1.9O3c, and La0.1Er0.05Y1.85O3d powders
Figure 7 shows the particle-size distribution of La0.01Er0.05Y1.94O3 and La0.1Er0.05Y1.85O3 ceramic powders (as an example) after milling for 1 h in ethanol. Dispex (BASF) medium was used as a dispersion material. The ceramic powders exhibited a mono-modal agglomerate distribution with the wide size between 0.1 and 10 μm before milling procedure and decreased 10 times from 0.01 to 1 μm after milling, which is consistence with SEM morphology (Fig. 6). The highest yield of milling process was observed for La0.1Er0.05Y1.84O3 powder. Increase amount of lanthanum ions led to increased size of agglomerates but they are easily broken during ball milling. BET parameter of investigated powders is presented in Table 3. Before milling procedure, all powders have similar BET parameter ca. 0.7, and then after milling, BET parameter is change randomly. In order to obtain a large number of a nano-sized powders, a milling procedure has to be continuous.

Powder size distributions of La0.01Er0.05Y1.94O3 and La0.1Er0.05Y1.84O3
Figure 8 shows the FT-IR spectra of the ceramic powders. The identification of functional bands referred to previous and other studies [14, 17, 19]. The FT-IR spectrum of the ceramic powder presents the characteristic bands of a Y2O3 cubic network. Three bands at 560 cm−1, 460 cm−1, and 414 cm−1 are attributed to the symmetric stretching Y–O vibrations. Bands over 750 cm−1 are assigned to the different organic stretching from KBr and are therefore not shown in Fig. 8. Detected shift of specific bands is ca. 3 cm−1, so comparable to the resolution of FT-IR device; however, this shift was confirmed by XRD measurement. It is consequence of different lattice constant. Increased quantities of lanthanum added to the lattice of Y2O3 are associated with dependences of the specific band shifts. The larger ionic radii than matrix induce stress in the powders, which increase in the Y–O bond length manifested by a corresponding change in the frequency of vibrations. The irreducible representations for the optical and acoustical modes are as follows:
where Ag, Eg, and Fg are Raman active, Fu is IR active, and A2u and Eu are inactive. In theory, 22 Raman peaks are predicted.

FT-IR spectra of the Er0.05Y1.95O3, La0.01Er0.05Y1.94O3, La0.05Er0.05Y1.9O3, and La0.1Er0.05Y1.84O3 powders
The Er:Y2O3 ceramics with various La contents (Er0.05Y1.95O3, La0.01Er0.05Y1.94O3, La0.05Er0.05Y1.9O3, and La0.1Er0.05Y1.84O3) were prepared by modification sol–gel method. Figure 9 presents the luminescence spectra of the samples in the NIR region excited by the 980 nm laser diode. Luminescence spectra are typical for Er ions doped into Y2O3 ceramic or YAG crystal [14, 15, 17]. According to Lei transition of 4I13/2 to 4I15/2, Er3+ ions can emit large band of lights from 1450 nm to 1650 nm because of energy level Stark splitting [15]. The luminescence of 1531 nm is obviously dominant in the IR spectrum and is considered as first harmonic in laser. It can be seen on Fig. 9 that the intensities of the infrared emissions increase along with the more addition of La ions (inset Fig. 9). Because of small difference of the crystallite size between the samples, its influence on the luminescent property can be neglected. To experimental points, a straight line was successful fitting with the matching factor R2 over 0.996. Based on the quantum mechanical selection rules, Er3+ ion can be excited to the 4I11/2 state by the GSA (ground state absorption) process, non-radiatively relax to 4I13/2 state, and next radiate to 4I15/2 state for the 1.5 μm emission [15]. As it is commonly known, although the f-f transitions of electric dipole are parity forbidden, they become partially allowed when the rare-earth ion is situated at a low symmetry site due to the intermixing of the intra-4f states with higher electronic configuration [25]. Hence, the probability of f-f transitions increases, leading to the raise of oscillator intensity [26]. Since the contribution of oscillator intensity f to the GSA cross section σ can be determined by:
where e electron charge, m mass, ∆v, f frequency, it may seem obvious that the raise off causes the increase of GSA cross section σ, which contributes to the 1.5 μm enhancement. As a consequence, the main mechanism for the luminescence enhancement begins to occur due to the fact that the co-doping of La3+ causes tailoring the local environment around the erbium ions. It results in the increase of the GSA cross section and increase of IR luminescence.

NIR emission spectra of Er0.05Y1.95O3, La0.01Er0.05Y1.94O3, La0.05Er0.05Y1.9O3, and La0.1Er0.05Y1.85O3 powders (Inset shows the intensity of 1536 nm emission as a function of La3+ ions)
Conclusions
The novel ceramic powders: Er0.05Y1.95O3, La0.01Er0.05Y1.94O3, La0.05Er0.05Y1.9O3, and La0.1Er0.05Y1.85O3, were synthesized by the citric sol gel reaction at 700 °C for 10 h. The samples were confirmed to be single phase by XRD. FT-IR spectroscopy presents a weak detected displacement of the bands. The increasing lanthanum content leads to both a decrease of temperature crystallization of precursors and make to possible to display the Bragg peaks at lower temperature. Intensity of all emission band in NIR region could be increased by La3+ doping of the powders. Typical luminescence spectra of the powders were a strong NIR emission of Er3+ at 1.53 μm due to the (4I13/2 → 4I15/2) transition under the 980 nm excitation. The main mechanism for such enhancement was the increase of GSA cross section of Er3+ ions by the tailoring effect of La3+ ions. High content of lanthanum ions is recommended because of its ability to decrease the calcination temperature and improving the luminescence of optical ceramics. Presented doped yttria ceramic powders were full characterized and could be sintering in order to obtain a transparence bulk sample and exanimate magneto-optical properties.
References
Zhang L, Jiang Z, Yao Q, Wang Z, Zhang Y, WeiS Zhou T, BenY Sun R, Chen H. Stirring speed assisted homogenization of precipitation reaction for enhanced optical performance of Y2O3 transparent ceramics. Ceram Int. 2018;44:4967–72.
Alhaji A, Razavi RS, Ghasemi A et al. Crystallization kinetics of MgO–Y2O3 composite nanopowder synthesized via combustion sol–gel method. J Therm Anal Calorim. 2018;132:1325.
Brzyska W, Ożga W. Thermal decomposition of yttrium, lanthanum and lanthanide complexes of square acid in air atmosphere. J Therm Anal. 1987;32:2001.
Kruk A, Mrózek M, Domagała J, Brylewski T, Gawlik W. Synthesis and physicochemical properties of yttrium oxide doped with neodymium and lanthanum. J Electron Mater. 2014;43:3611–7.
Zhang L, Pan W. Structural and thermo-mechanical properties of Nd:Y2O3 transparent ceramics. J Am Ceram Soc. 2015;98:3326–31.
Biljan T, Gajovic A, Mei Z, Mestrovi´c E. Preparation, characterization and luminescence of nanocrystalline Y2O3:Ho. J Alloys Compd. 2007;431:217–20.
Li S, Liu B, Li J, Zhu X, Liu W, Pan Y, Guo J. Synthesis of yttria nano-powders by the precipitation method: the influence of ammonium hydrogen carbonate to metal ions molar ratio and ammonium sulfate addition. J Alloys Compd. 2016;678:258–66.
Srinivasan R, Yogamalar R, Chandra BA. Structural and optical studies of yttrium oxide nanoparticles synthesized by co-precipitation method. Mater Res Bull. 2010;45:1165–70.
Serivalsatit K, Kokuoz B, Yazgan-Kokuoz B, Kennedy M, Ballato J. Synthesis, processing, and properties of submicrometer—grained highly transparent yttria ceramics. J Am Ceram Soc. 2010;93:1320–5.
Biswas M, Prabhakaran K, Gokhale N, Sharma S. Synthesis of nanocrystalline yttria doped ceria powder by urea–formaldehyde polymer gel auto-combustion process. Mater Res Bull. 2007;42:609–17.
Shirakawa A, Takaichi K, Yagi H, Bisson J-F, Lu J, Musha M, Ueda K, Yanagitani T, Petrov T, Kaminskii A. Diode-pumped mode-locked Yb3+:Y2O3 ceramic laser. Opt Express. 2003;11:2911–6.
Mun JH, Novoselov A, Yoshikawa A, Boulon G, Fukuda T. Growth of Yb3+-doped Y2O3 single crystal rods by the micro-pulling-down method. Mater Res Bull. 2005;40:1235–43.
Hajizadeh-Oghaz M, Razavi RS, Barekat M, Naderi M, Malekzadeh S, Rezazadeh M. Synthesis and characterization of Y2O3 nanoparticles by sol–gel process for transparent ceramics applications. J Sol-Gel Sci Technol. 2016;78:682–91.
Qin X, Zhou G, Yang H, Yang Y, Zhang J, Wang S. Synthesis and upconversion luminescence of monodispersed, submicron-sized Er3+:Y2O3 spherical phosphors. J Alloys Compd. 2010;493:672–7.
Lei R, Wang H, Xu S, Tian Y, Yang Q. Combustion synthesis and enhanced 1.5 μm emission in Y2O3:Er3+ powders codoped with La3+ ions. J Rare Earths. 2016;34:125–9.
Kruk A. Optical and structural properties of arc melted Ce or Pr-doped Y2O3 transparent ceramics. Ceram Int. 2017;43:16909–14.
Liu B, Li J, Yavetskiy R, Ivanov M, Zeng Y, Xie T, Kou H, Zhuo S, Pan Y, Guo J. Fabrication of YAG transparent ceramics using carbonate precipitated yttria powder. J Eur Ceram Soc. 2015;35:2379–90.
Durrani S, Saeed K, Qureshi A, et al. Growth of Nd-doped YAG powder by sol spray process. J Therm Anal Calorim. 2011;104:645.
Kruk A, Bobruk M, Wajler A, Mrózek M, Gawlik W, Brylewski T. Preparation of yttria powders co-doped with Nd3+ and La3+ using EDTA gel processes for application in transparent ceramics. J Eur Ceram Soc. 2017;37:4129–40.
Atabaev T, Vu HHT, Kim H-K, Hwang Y-H. The optical properties of Eu3+ and Tm3+ codoped Y2O3 submicron particles. J Alloys Compd. 2012;525:8–13.
Kruk A, Wajler A, Mrozek M, Zych L, Gawlik W, Brylewski T. Transparent yttrium oxide ceramics as potential optical isolator materials. Opt Appl. 2015;45:585–94.
Lojpur V, Mancic L, Rabanal ME, Dramicanin MD, Tan Z, Hashishin T, Ohara S, Milosevic O. Structural, morphological and luminescence properties of nanocrystalline up-converting Y1.89Yb0.1Er0.01O3 phosphor particles synthesized through aerosol route. J Alloys Compd. 2013;580:584–91.
Zhang L, Li Z, Zhen F, Wang L, Zhang Q, Sun R, Selim FA, Wong C, Chen H. High sinterability nano-Y2O3 powders prepared via decomposition of hydroxyl-carbonate precursors for transparent ceramics. J Mater Sci. 2017;52:8556–67.
Lakshminarasappa BN, Jayaramaiah JR, Nagabhushana BM. Thermoluminescence of combustion synthesized yttrium oxide. Powder Technol. 2012;217:7–10.
Patra A, Sominska E, Ramesh S, Koltypin Y, Zhong Z, Minti H, Reisfeld R, Gedanken A. Sonochemical preparation and characterization of Eu2O3 and Tb2O3 doped in and coated on silica and alumina nanoparticles. J Phys Chem B. 1999;103:3361–5.
Patra A, Friend C, Kapoor R, Prasad P. Effect of crystal nature on upconversion luminescence in Er3+:ZrO2 nanocrystals. Appl Phys Lett. 2003;83:284.
Acknowledgements
This work was supported by the National Science Center of Poland under Grant Number 2016/23/D/ST8/00014.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kruk, A., Polnar, J. Investigation on the physicochemical properties of La-doped Er0.05Y1.95O3 nanopowders. J Therm Anal Calorim 139, 765–773 (2020). https://doi.org/10.1007/s10973-019-08499-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-019-08499-9