
Abstract
It has been demonstrated that apart from the known compounds YbVO4 and Yb8V2O17, a substitutive solid solution is also formed in the binary oxide system V2O5–Yb2O3. This solid solution is characterized by limited solubility of vanadium(V) oxide in the crystal lattice of ytterbium oxide, and its general formula is Yb2−5x□2xV3xO3, where 0.00 < x < 0.1667. The new solid solution was obtained from the mixtures of V2O5 and Yb2O3 oxides, containing vanadium(V) oxide in the amounts not exceeding 30 mol%, by high-energy ball milling in a planetary ball mill. The solid solution obtained was characterized using the methods XRD, DTA-TG, SEM, IR and UV–Vis-DR. It was found to crystallize in cubic system and to show a structure of Yb2O3. This solid solution was stable in air atmosphere at least up to ~ 800 °C, and the temperature of its decomposition decreased with increasing x, so with increasing degree of Yb3+ replacement with V5+ in the crystal lattice of Yb2O3. The energy gap estimated for this solid solution varied from ~ 2.5 eV for x = 0.0322 to ~ 2.6 eV for x = 0.1176, which means that this new solid solution is a semiconductor.
Similar content being viewed by others

Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Introduction
Intensive search for new advanced materials for application in, e.g., optics, electronics and informatics has been conducted for many years. Of great importance is to devise simple and economically and ecologically justified methods for their syntheses to meet the increasingly stringent rules for the environment protection. Such methods can be also applied for the modification of physicochemical properties of these new materials as well as other already known compounds. Different methods for syntheses of new materials have been proposed, including the solid-state reactions, co-precipitation, sol–gel method, suspension, combustion, Pechini method, hydrothermal method, sonochemical method and still too little appreciated mechanochemical method. Each of the abovementioned methods has its specific advantages and disadvantages, and the choice of the suitable one depends on the type of products and their desired properties for particular applications (morphology and size of crystallites, degree of crystallinity, absorption properties, surface area, degree of homogeneity, etc.). The mechanochemical method meets a high degree the requirements of “green chemistry” as it does not generate troublesome wastes which are difficult for utilization and it does not need the use of toxic organic solvents [1,2,3,4]. The high-energy ball milling has been successfully used for syntheses of many new functional materials such as alloys, intermetallic compounds, polymer or inorganic composites, ceramic materials [1,2,3,4,5,6]. According to the hitherto-published literature on the binary oxides of our concern, V2O5–Yb2O3, the formation of two compounds YbVO4 and Yb8V2O17 in this oxide system has been documented [7,8,9,10,11,12,13,14,15,16]. They have been obtained as a result of high-temperature treatment of the oxides mixture containing V2O5 and Yb2O3 of the contents chosen to match the stoichiometry of these compounds [11, 13,14,15,16]. To the best of our knowledge, there is no information on the solid solutions forming in the binary system of V2O5 and Yb2O3 oxides showing the structures of vanadium(V) oxide and/or ytterbium oxide. In contrast to the scarce data on ytterbium divanadate(V), the synthesis and physicochemical properties of ytterbium orthovanadate(V) have been thoroughly described [7,8,9,10,11,12]. YbVO4 has been obtained by different methods: solid-state, hydrothermal and co-precipitation [7,8,9,10,11,12] and mechanochemical method [21]. Detailed crystal structure of this compound as well as its optical, electric, magnetic, thermal and chemical properties is known [7,8,9,10,11,12]. It has been proved that as a result of YbVO4 doping with such lanthanide ions as Nd3+ and Er3+, a number of optically active laser materials can be obtained [13, 14]. The information on the other of the two compounds, i.e., Yb8V2O17, is scarce [15, 16] and ambiguous. This compound was for the first time obtained in 1973 by Brusset et al. [15] as a result of heating of a mixture of oxides Yb2O3/V2O5 at a molar ratio 4:1, up to 1550 °C. These authors claimed that it crystallized in monoclinic system and gave its elementary cell parameters [15]; however, according to our recent study, this compound obtained by the solid–solid reactions crystallizes in triclinic system and its elementary cell parameters are different from those given by Brusset et al. [16]. Recently, the authors from our group have also shown that Yb8V2O17 is the matrix (solid solvent) of the limited substitutive solid solution of the formula Yb8−xYxV2O17 and 0.0 < x < 4.0. In such a solid solution, the Y3+ ions are built into the crystalline lattice of Yb8V2O17, replacing Yb3+ ones, with no damage to the crystal structure [16].
Taking into account the hitherto knowledge on the compounds forming in the system V2O5–Yb2O3, and in particular, the lack of information on the synthesis of YbVO4 and Yb8V2O17 by mechanochemical method (MChS), the aim of the study reported here was to establish the type of compounds and/or phases forming in this system in the entire range of concentration of its components as a result of reactions induced by high-energy ball milling. If as a result of this process new compounds and/or solid solutions are obtained, our additional objectives will be to characterize their structures and properties, in particular thermal stability and optical properties, and to compare them with the properties of the phases obtained in the high-temperature reactions (HTS) taking place in the system studied in solid state.
Experimental
The following reagents were used in our experiments: Yb2O3, a.p. (Alfa Aesar, Germany), and V2O5, a.p. (POCh, Poland). For the experiments, 14 samples were prepared (Tables 1, 2) by using two different methods, namely:
-
(a)
Mechanochemical synthesis (MChS) using laboratory planetary ball mill Pulverisette-6 (Fritsch GmbH, Germany) with vessel and balls of zirconia, rpm = 500, BPR = 1:20, time = 3.0 and once again 3.0 h under air atmosphere.
-
(b)
High-temperature synthesis (HTS) in a horizontal tube furnace PRC 50/170/M (Czylok, Poland) equipped with a stationary optical pyrometer MARATHON MM (Raytek, Germany). Reagents weighed in suitable proportions were homogenized and calcined in air atmosphere in the temperature range 600–1500 °C. After each heating stage, the samples were gradually cooled in the furnace to room temperature and weighed (changes in their mass and their color were recorded) by the method described, e.g., in [16,17,18,19].
The powder diffraction patterns of the samples obtained were recorded on a diffractometer EMPYREAN II (PANalytical, Netherlands) using CuKα with graphite monochromator. The phases were identified on the basis of XRD (X-ray diffraction) characteristics contained in the PDF (Powder Diffraction File) cards [20].The parameters of selected unit cell solid solutions were refined using the REFINEMENT program of DHN/PDS package.
Selected samples were examined using the following methods: SEM (scanning electron microscopy) using FE-SEM Hitachi SU–70 microscope and EDX (X-ray microanalysis) using NORAN™ System 7 of Thermo Fisher Scientific (UltraDry X-ray detector). SEM analyses were performed at accelerating voltage of 5 and 15 kV, and secondary electron images were acquired. The samples were coated with palladium–gold alloy thin film using thermal evaporation PVD (physical vapor deposition) method to provide electric conductivity.
The densities of solid solutions were determined in argon (5 N purity) with the help of an Ultrapyc 1200e ultrapycnometer (Quantachrome Instruments, USA). Initial mixtures and monophasic samples were examined by IR (infrared spectroscopy). The measurements were made within the wavenumber range of 1200–250 cm−1, using a spectrophotometer SPECORD M-80 (Carl Zeiss, Jena, Germany). The technique of pressing pellets with KBr at the mass ratio of 1:300 was applied. Selected samples were also subjected to examination by the DTA–TG (differential thermal analysis and thermogravimetry) method with the use of a SDT 2960 apparatus (TA Instruments Company, USA). The measurements were taken in air atmosphere, within the temperature range 20–1400 °C, at the heating rate of 10 deg/min. The tests were conducted in corundum crucibles. The samples had the same mass of ~ 20 mg.
The UV–Vis-DR (ultraviolet–visible with diffuse reflectance) spectra were measured using a UV–Vis spectrometer V-670 (JASCO, Japan) equipped with a reflecting attachment for the solid-state investigation (integrating sphere attachment with horizontal sample platform PIV-756/(PIN-757). The spectra were recorded in the wavelength region of 200–750 nm at room temperature.
Results and discussion
Mechanochemical synthesis
Investigation of the binary oxide system V2O5–Yb2O3 was started from preparation of mixtures of the substrates Yb2O3 and V2O5, of the compositions specified in Table 1. The compositions were chosen to represent the range of substrate concentrations in which the literature-known compounds Yb8V2O17 and YbVO4 are formed [7,8,9,10,11,12,13,14,15,16]. Mixtures of the ytterbium and vanadium oxides were homogenized and subjected to mechanochemical synthesis in a planetary ball mill in air atmosphere. XRD analysis of the phase composition revealed that already after the first stage of the 3-h milling, all the samples studied changed their phase composition. Diffractograms of samples 1–3 no longer showed the XRD reflections characteristic of one of the substrates—V2O5, but included a set of reflections evidencing the presence of Yb2O3. Analysis of these diffractograms also indicated that with an increasing content of V2O5 in the initial mixtures of the reagents, the reflections were slightly shifted toward lower 2Θ angles, which corresponds to greater interplanar distances dhkl with respect to those in the diffractograms of the pure compound Yb2O3. This result implied that in the range of concentration of up to 20.00 mol% V2O5 in the system Yb2O3–V2O5, a new solid solution, showing the structure of Yb2O3, was formed. The lack of foreign lines in XRD patterns of these samples proved that the new solid solution was substitutive, with V5+ ions taking place of Yb3+ ions in the crystal lattice of Yb2O3. Analysis of the phase composition of samples 4 and 5 revealed that after 3-h milling, these samples were biphasic and, besides the solid solution of the structure of Yb2O3, contained also the known orthovanadate YbVO4 (Table 1). The 3-h milling of the equimolar mixture of the reagents (sample 6) made the sample monophasic, with only YbVO4 in its content (Table 1) [21]. This result indicated that ytterbium orthovanadate(V) could also be obtained by the way of mechanochemical synthesis [21], besides other known methods [7,8,9,10,11,12]. Sample 7 represented the initial system composition with more than 50.00 mol% V2O5. After 3-h ball milling, the sample was biphasic and, besides the compound YbVO4, contained also small amounts of V2O5. The subsequent 3-h milling of all samples studied did not change their phase composition. The phase compositions of all samples after the last stage of their ball milling, as presented in Table 1, prove that the new substitutive solid solution forming in the system studied is characterized by limited solubility of components. The fact that V5+ ions are incorporated into the crystal lattice of ytterbium oxide replacing Yb3+ ions leads to the generation of excessive positive charge which can be compensated by the extension of the anionic lattice or by the formation of vacancies in the cationic Yb3+ sublattice. In the former case, the general formula of the solution can be written as Yb2−xVxO3+x, while in the latter case as Yb2−5x□2xV3xO3.
Charge compensation by the generation of vacancies in crystal lattice (the second formula) is supported by the values of experimental density of obtained phase in comparison with its X-ray density, as described later in this work. The data presented in Table 1 also imply that the maximum incorporation of V5+ into the lattice of Yb2O3, when expressed in the contents of components of the system studied, reaches at least 20.00 mol% (x = 0.1176) and does not exceed 30.00 mol%.
Analysis of the results obtained leads to a conclusion that in the reaction mixtures containing up to 50.00 mol% V2O5, the following reaction took place:
Figure 1 presents a fragment of XRD diffractogram of the commercial Yb2O3 (Fig. 1a) and the diffractograms of the newly obtained solid solution Yb2−5x□2xV3xO3 for x = 0.0322 (Fig. 1b), x = 0.1176 (Fig. 1c) and sample 4 containing the solid solution Yb2−5x□2xV3xO3 and YbVO4.

Fragments of diffractograms of a Yb2O3 (
 ), b Yb1.839□0.064V0.097O3 (
), b Yb1.839□0.064V0.097O3 (
 ), c Yb1.412□0.235V0.353O3 (
), c Yb1.412□0.235V0.353O3 (
 ), d Yb2−5x□2xV3xO3 (
), d Yb2−5x□2xV3xO3 (
 ) and YbVO4 (
) and YbVO4 (
 )
)
The aim of the next stage of our study was to confirm that the newly obtained solid solution Yb2−5x□2xV3xO3 shows the matrix structure, i.e., that it crystallizes in the cubic system. In order to check this, the powder diffractograms of the solution Yb2−5x□2xV3xO3 for x = 0.0322, 0.0625 or 0.1176 were refined using the program REFINEMENT. The results confirmed that the new solid solution crystallizes in the cubic system and permitted calculation of its unit cell parameters as a function of the content of incorporated V5+ replacing Yb3+ in the crystal lattice of Yb2O3. Table 2 presents the unit cell parameters, volumes and densities of Yb2−5x□2xV3xO3 with x = 0.0322, 0.0625 and 0.1176.
According to the data displayed in Table 2, with an increasing value of x in the formula Yb2–5x□2xV3xO3, so with an increasing number of V5+ incorporated in the crystal lattice and replacing Yb3+ ions, the crystal lattice expands, although the radius of V5+ in coordination (VI) is smaller than that of Yb3+ in the same coordination. The lattice expansion can be a result of greater electric charge of vanadium ion or generation of vacancies in the Yb3+ sublattice. The same tendency has been observed in [22]. Analysis of the same data from Table 2 also shows that the density of the new solid solution, determined by an ultrapycnometer or by X-ray method, is always smaller than that of pure Yb2O3 and decreases with increasing x, which additionally supports the assumed model of the solution structure. With an increasing x in Yb2−5x□2xV3xO3, the color of the solid solution changed from dark orange to yellow. The mixture of the substrates composed of 50 mol% Yb2O3 and 50 mol% V2O5 and the solid solution Yb2−5x□2xV3xO3 for x = 0.0625 were also studied by scanning electron microscopy (SEM/EDX). The relevant images are presented in Figs. 2 and 3.

SEM image of Yb2O3 (∆) + V2O5 (○)

SEM image of Yb2−5x□2xV3xO3 (x = 0.0625)
Figure 2 shows a clear distinction between the two types of crystallites, which can also be distinguished on the basis of their different interaction with a beam of electrons. The crystallites look like irregular deformed polyhedrons. As follows from EDX analysis, the brighter polygons correspond to Yb2O3 (∆), while the darker and slightly bigger ones to V2O5 (○). The crystallites of the solid solution Yb2−5x□2xV3xO3 presented in Fig. 3 have morphology similar to that of the matrix crystallites (Yb2O3). They look like deformed polyhedrons of irregular shapes and sizes varying from ~ 40 to ~ 270 nm (Fig. 3). X-ray microanalysis (EDX) informing about the content of metals and oxygen in the crystals of Yb2−5x□2xV3xO3 for all monophase samples is presented in Table 3.
Taking into account the average error in determination of the content of oxygen, the results provided by SEM/EDX results are in good agreement with the values calculated from the formula Yb2−5x□2xV3xO3 for x = 0.0625, which confirms its correctness.
In order to check whether the solid solution obtained shows the structure of the matrix, that is of Yb2O3, IR spectra were taken of selected monophasic samples. Figure 4, besides the IR spectrum of the commercial oxide Yb2O3 (Fig. 4a), also presents the IR spectra of the solid solution Yb2−5x□2xV3xO3 for x = 0.0322 and x = 0.1176 (Fig. 4, spectra b and c).

Fragments of IR spectra of: a Yb2O3, b Yb1.839□0.064V0.097O3 and c Yb1.412□0.235V0.353O3
The IR spectrum of Yb2O3 (Fig. 4a) shows a broad absorption band in the wavenumber range from 645 to 300 cm−1, of distinct extremes at 565, 395 and 335 cm−1 and an inflection point at ~ 500 cm−1. These bands can be assigned to the stretching vibrations of Yb–O in the deformed YbO6 octahedrons [23,24,25,26] and the stretching vibrations of the deformation bridge bonds Yb–O–Yb or O–Yb–O [23,24,25,26]. In the IR spectra of the solid solution (Fig. 4b,c), the absorption bands appear in a very narrow range of wavenumbers similar to those in the spectrum of Yb2O3, i.e., from 650 to 300 cm−1, but they are more broadened and show much less distinct maxima at 450 cm−1 (for x = 0.0322) and 465 cm−1 (for x = 0.0322). In contrast to the spectrum of Yb2O3, the spectra of the solid solution Yb2−5x□2xV3xO3 show also a broad and low-intensity band in the range 1000–600 cm−1, which on the basis of the literature can be assigned to the stretching vibrations of symmetric and asymmetric V–O bonds in VO6 octahedrons [23]. The absorption band in the range 650–300 cm−1 is interpreted as corresponding to the stretching vibrations of Yb–O bonds in YbO6 octahedra and vibrations of the bridging bonds O–Yb–O and O–V–O [23,24,25,26]. With an increasing number of V5+ replacing Yb3+ in the crystal lattice of Yb2O3, the absorption bands are shifted toward higher wavenumbers relative to their positions in the spectrum of the matrix. The results confirmed that the solid solution Yb2−5x□2xV3xO3 has a structure of Yb2O3 and is composed of YbO6 and VO6 octahedrons. In order to establish the thermal stability of the solid solution Yb2−5x□2xV3xO3 obtained by mechanochemical method, three monophasic samples, 1–3 (Table 1), were heated in the range from 500 to 1500 ± 10 °C in air atmosphere, in a horizontal tube furnace equipped with an optical pyrometer, in a few 12-h stages. The samples heated at 500, 600, 700 and 800 °C did not show signs of melting, and their phase composition was not changed. Heating at 900 °C (12 h) caused changes in the phase composition of sample 3. The XRD diffractogram of this sample, besides the diffraction signals characteristic of Yb2O3, revealed also a set of XRD lines that, according to PDF card no. 00-017-0338, are assigned to YbVO4. The phase composition of the other samples (1 and 2) changed only after their heating at ~ 1500 °C (12 h). After this stage of heating, the samples were biphasic and besides Yb2O3 contained the literature-known compound Yb8V2O17 [15, 16]. Sample 3 after annealing at this temperature was monophasic and contained only Yb8V2O17. As follows from the results of this experiment, the solid solution obtained is thermally stable in air atmosphere to at least 800 °C (for x = 0.1176); the lower the number of V5+ ions replacing Yb3+ ions in the lattice of Yb2O3, the higher its thermal stability, and the solid solution is stable in the range 900–1500 °C. At higher temperatures, the solid solution undergoes decomposition to the oxides V2O5 and Yb2O3, which then react with formation of YbVO4. In the range of components of the system Yb2O3–V2O5, in which the oxide Yb2O3 is in excess to the stoichiometry of ytterbium orthovanadate(V), the reagents react with formation of Yb8V2O17:
The results obtained at this stage confirmed that V2O5 was initially built in Yb2O3, making the earlier unknown solid solution.
Physicochemical characterization of the matrix Yb2O3 and the solid solution Yb2−5x□2xV3xO3 was supplemented with UV–Vis-DR measurements that permitted estimation of the energy gap. Figure 5 presents the plots of K2 = f(E), where K is obtained from the Kubelka–Munk transformation (Spectra Analysis Program) and in the plot is marked as K/M. The energy gap values were read off as the points of intersection of the tangent to a given curve and the abscissa axis.

Kubelka–Munk transformation of the UV–Vis-DR spectra of Yb2O3 and the solid solution Yb2−5x□2xV3xO3 for x = 0.0322, 0.0625 and 0.1176
The energy gap (Eg) determined for Yb2O3 was ~ 5.00 eV, while the energy gap for the solid solution decreased with an decreasing x in Yb2−5x□2xV3xO3 from Eg = ~ 2.5 eV for Yb1.839□0.064V0.097O3 to ~ 2.6 eV for Yb1.412□0.235V0.353O3. On the basis of the obtained energy gap values, the solid solution Yb2−5x□2xV3xO3 was classified as a semiconductor.
High-temperature synthesis
In parallel with the mechanochemical synthesis of samples representing the system Yb2O3–V2O5, we carried out their syntheses by the classical method of high-temperature reactions taking place in solid state. The samples prepared for this procedure had the same compositions as those studied in the first part of this work, as shown in Tables 1 and 4. The mixtures of Yb2O3 and V2O5 (Table 4) after homogenization were heated in the following temperature stages: I—600 °C (24 h) → II—625 °C (24 h) → III—635 °C (24 h) → IV—1200 °C (24 h) → V—1400 °C (24 h) → VI—1450 °C (24 h).
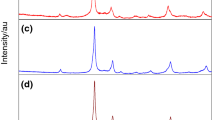
According to the XRD results, all the samples studied changed their phase composition already after the first stage of heating. The diffractograms of samples 1–7 already showed the XRD lines characteristic of YbVO4. After the next stage of heating of samples 6 and 7 at 625 °C, the oxides Yb2O3 and V2O5 were present in them in small amounts. The diffractogram of sample 6 after the third stage of its heating revealed only the set of XRD lines characteristic of YbVO4 (PDF card no. 00-017-0338) (Fig. 6d). After the same stage of heating, the diffractogram of sample 7 proved that this sample was biphasic and besides YbVO4 contained also small amounts of V2O5. Analysis of the phase composition of samples 1–5, representing the other concentration range of the system components, i.e., the concentrations over 50.00 mol% Yb2O3, proved that after stage IV of their heating (at 1200 °C) the samples contained the oxide Yb2O3 besides YbVO4 (Fig. 6c). After the next stage of heating at 1400 °C, the phase composition of samples 1–3 changed significantly. Their diffractograms no longer showed the XRD lines characteristic of YbVO4. After this stage of heating, these samples were biphasic and besides Yb2O3 contained a known compound Yb8V2O17 (Fig. 6a) [15, 16]. After the last stage of heating at 1450 °C, sample 3 was monophasic and contained only Yb8V2O17 (Fig. 6b) [16]. According to the above results of the high-temperature reaction study, besides the known compounds forming in the binary system V2O5–Yb2O3 [7,8,9,10,11,12,13,14,15,16], i.e.,YbVO4 and Yb8V2O17, no other phase was obtained. It means that the solid solution showing the structure of Yb2O3 forms only as a result of high-energy ball milling of a mixture of oxides V2O5 and Yb2O3, whereas pure YbVO4 can be obtained by the two methods. On the other hand, the compound Yb8V2O17 was obtained only as a result of the high-temperature reaction (Fig. 6b). Figure 6 presents a fragment of XRD diffractogram of the (a) Yb2O3 + Yb8V2O17, (b) Yb8V2O17, (c) Yb2O3 + YbVO4 and (d) YbVO4.

Fragments of diffractograms of a Yb2O3 (
) + Yb8V2O17 (
), b Yb8V2O17 (
), c Yb2O3 (
) + YbVO4 (
) and d YbVO4 (
)
Conclusions
-
The substitutional limited solid solution of the formula Yb2−5x□2xV3xO3 and 0.00 < x < 0.1667 is formed in the system Yb2O3–V2O5.
-
The solid solution has been obtained in air only by high-energy ball milling of the oxides: Yb2O3 and V2O5.
-
The solid solution Yb2−5x□2xV3xO3 crystallizes in the cubic system.
-
With an increasing x in Yb2−5x□2xV3xO3, the crystal lattice of solid solution expands.
-
The solid solution Yb2−5x□2xV3xO3 is stable in air atmosphere up to ~ 800 °C.
-
Yb2−5x□2xV3xO3 belongs to the group of semiconductors with the band gap energies ~ 2.5 ± 0.2 eV.
References
Baláž P, Achimovičová M, Baláž M, Billik P, Cherkezova-Zheleva Z, Manuel Criado J, Delogu F, Dutková E, Gaffet E, Jose Gotor F, Kumar R, Mitov I, Rojac T, Senna M, Streleckii A, Wieczorek-Ciurowa K (2013) Hallmarks of mechanochemistry: from nanoparticles to technology. Chem Soc Rev 42:7571–7637
James SL, Adams ChJ, Bolm C, Braga D, Collier P, Friščić T, Grepioni F, Harris KDM, Hyett G, Jones W, Krebs A, Mack J, Maini L, Orpen G, Parkin IP, Shearouse WC, Steed JW, Waddell DC (2012) Mechanochemistry: opportunities for new and cleaner synthesis. Chem Soc Rev 41:413–447
Garay AL, Pichon A, James SL (2007) Solvent-free synthesis of metal complexes. Chem Soc Rev 36(6):846–855
Pochon A, Lazuen-Garay A, James SL (2006) Solvent-free synthesis of a microporous metal-organic framework. Crys Eng Comm 8:211–214
Dulian P, Bak W, Wieczorek-Ciurowa K, Cz Kajtoch (2013) Controlled mechanochemical synthesis and properties of a selected perovskite-type electroceramics. Mater Sci 3:462–470
Dutta H, Pradhan SK (2002) Microstructure characterization of high energy ball milled nanocrystalline V2O5 by Rietveld analysis. Mater Chem Phys 77:868–877
Zhang H, Yu Y, Cheng Y, Liu J, Li H, Ge W, Cheng X, Xu X, Wang J, Jiang M (2005) Growth of YbVO4 stoichiometric crystal. J Crys Growth 283:438–443
Xu Z, Zhao Q, Liu T, Wang L, Bian S (2013) Uniform and well-dispersed YbVO4 hierarchical nanoarchitectures: synthesis and luminescence properties. J Nanosci Nanotechnol 13:344–350
Jindal R, Sinha MM (2013) Gupta HC Lattice vibrations of AVO4 crystals (A = Lu, Yb, Dy, Tb, Ce). Spectrochim Acta Part A 113:286–290
Brusset H, Madaule-Aubry F, Blanck B, Glaziou JP, Laude JP (1971) Etude des oxydes mixtes de lanthanides et de vanadium(V). Can J Chem 49:3700–3707
Fotiev AA, Slobodin BV, Khodos MY, Pletnev RN (1988) Vanadates: composition, synthesis, structure, properties. Izd. Nauka, Moskwa
Gao Y, Fan M, Fang Q, Han W (2013) Controllable synthesis, morphology evolution and luminescence properties of YbVO4 microcrystals. New J Chem 37:670–678
Liu FQ, Sun SQ, Gao ChY, Xu JQ (2015) Optical properties of Nd:YbVO4 crystal. Opt Appl 45:63–70
Cheng Y, Zhang H, Zhang K, Xin Z, Yang X, Xu X, Gao W, Li D, Zhao Ch, Xu J (2009) Growth and spectroscopic characteristics of Er3+:YbVO4 crystal. J Crys Growth 311:3963–3968
Brusset H, Mahe R, Laude JP (1973) Studies of rare-earth vanadates with formula T8V2O17. Bull Soc Chim Fr 2:495–499
Piz M, Filipek E (2017) Synthesis and homogeneity range of Yb8-xYxV2O17 in the Yb8V2O17–Y8V2O17 system. J Therm Anal Calorim 130:277–283
Filipek E, Wieczorek-Ciurowa K (2009) Comparison between the synthesis in molybdenum and antimony oxides system by high-temperature treatment and high-energy ball milling. J Therm Anal Calorim 97:105–110
Groń T, Filipek E, Piz M, Duda H (2014) Conductance in Nb6VSb3O25–a new ceramic compound. Mater Res Bull 51:105–108
Filipek E, Walczak J, Tabero P (1998) Synthesis and some properties of the phase Cr2V4O13. J Alloys Compd 265:121–124
Powder Diffraction File (1989) International Center for Diffraction Data, Swarthmore USA (PDF-4 +), No. 00-017-0338, No. 00-043-1037
Tojo T, Zhang Q, Saito F (2007) Mechanochemical synthesis of rare earth orthovanadates from R2O3 (R = rare earth elements) and V2O5 powders. J Alloys Compd 427:219–222
Tomaszewicz E, Piątkowska M, Pawlikowska M, Groń T, Oboz M, Sawicki B, Urbanowicz P (2016) New vacancied and Dy3+-doped molybdates—Their structure, thermal stability, electrical and magnetic properties. Ceram Int 42:18357–18367
Filipek E, Dąbrowska G (2007) Synthesis and selected properties of CrSbVO6 and phase relations in the V2O5–Cr2O3–α-Sb2O4 system in the solid state. J Mater Sci 42:4905–4915. https://doi.org/10.1007/s10853-006-0539-0
Atkinson SC (2013) Crystal structures and phase transitions in the rare earth oxides School of Computing. Science and Engineering University of Salford, Salford
Vasundhara K, Achary SN, Patwe SJ, Sahu AK, Manoj N, Tyagi AK (2014) Structural and oxide ion conductivity studies on Yb1−xBixO1.5 (0.00 ≤ x≤0.50) composites. J Alloys Compd 596:151–157
Heiba ZK, Akin Y, Sigmund W, Hascicek YS (2003) X-ray structure and microstructure determination of the mixed sesquioxides (Eu1−xYbx)2O3 prepared by a sol-gel process. J Appl Crystallogr 36:1411–1416
Acknowledgements
The authors are grateful to PhD DSc. Piotr Tabero for valuable discussion. This work was partly supported by Ministry of Science and Higher Education (Poland), No. BMN 517-10-020-3604/17.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Piz, M., Dulian, P., Filipek, E. et al. Characterization of phases in the V2O5–Yb2O3 system obtained by high-energy ball milling and high-temperature treatment. J Mater Sci 53, 13491–13500 (2018). https://doi.org/10.1007/s10853-018-2449-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-018-2449-3