
Abstract
A procedure based on radiochemical neutron activation analysis was developed for the determination of chromium in biological materials. The procedure consists of irradiation of reference and test samples in a nuclear reactor, microwave sample digestion, selective and quantitative radiochemical separations of chromium and gamma-ray spectrometric measurement of 51Cr. Separation of chromium from the accompanying elements was done on the column packed with inorganic resin MnO2 Resin. Distribution coefficients of Cr, Zn, Co, Cs and Sc were determined in the system: MnO2 Resin—HCl, HNO3 and H2SO4. Accuracy of the procedure was checked by analysis of certified reference materials.
Similar content being viewed by others

Introduction
Chromium is an element belonging to the group of potentially toxic metals. It is a pollutant, mainly derived from human activities such as leather tanning, electroplating, wood preserving and the metallurgy industry [1–3]. Toxicity of chromium compounds strongly depends on its oxidation state [4]. Chromium can exist in several oxidation states; the most important are Cr(III) and Cr(VI). Trivalent chromium is an essential micronutrient. It is a part of the enzymes affecting the metabolism of glucose, the so-called glucose tolerance factor (GTF). Chromium deficiency can cause diabetes and cardiovascular diseases [5, 6]. Hexavalent chromium has strong oxidizing properties. It can penetrate biological membranes and has a toxic effect on humans, animals and plants. Excessive exposure to Cr(VI) can cause skin damage, respiratory problems, cancer of the kidneys or liver [7, 8]. Therefore, the content of chromium in environmental and food materials should be constantly monitored.
Accurate determination of traces of Cr in biological materials can be a serious problem and challenge. Different methods are applied, including atomic absorption spectrometry (AAS), inductively coupled plasma optical emission spectrometry (ICP-OES), inductively coupled plasma mass spectrometry (ICP-MS), spectrophotometry, chemiluminescence and neutron activation analysis (NAA). Most of the analytical techniques suffer from a number of different factors affecting the accuracy of chromium determination, such as contamination, volatility losses, incomplete dissolution, multiple oxidation states/chemical species, absorption/adsorption, interferences, incomplete separations or matrix effects [9–12]. The results of interlaboratory comparisons (ILC) frequently show the large discrepancies in chromium concentrations. This is also evidenced by the results of ILCs organized recently by the Institute of Nuclear Chemistry and Technology (INCT). The spread of results obtained for Cr (0.071–19.517 mg kg−1) supplied by the ILC participants in the case of material of plant origin, INCT-OBTL-5 made it impossible to assign a certified value [13]. Among the above-mentioned techniques applied for the determination of chromium, NAA has the highest metrological quality [14] and plays an important role in the certification of reference materials [15, 16]. NAA can be in two modes: instrumental neutron activation analysis (INAA) and radiochemical neutron activation analysis (RNAA).
RNAA is a combination of neutron activation with a post-irradiation separation of the determined element and gamma-ray spectrometry measurement [12, 13, 17, 18]. This method was used for the development of the ratio primary reference measurement procedures for the determination of cadmium, cobalt, nickel, molybdenum, uranium, iron, arsenic and selenium in biological materials [17–21]. The aim of this study was to develop a RNAA procedure for the determination of total Cr at trace levels in biological samples.
Experimental
Reagents, radioactive tracers
MnO2 Resin 100–200 mesh (Eichrom Technologies LLC) was used. A standard solution of Cr(VI) (10 mg g−1) was prepared from analytical grade K2Cr2O7 by dissolution in water. Chromium standards for irradiation were prepared by weighing aliquots of the standard solution in PE capsules (Type “V”, Vrije Universiteit, Biologisch Laboratorium, The Netherlands) and evaporating them to dryness before encapsulation. The following radioactive tracers were used: 134Cs (T1/2 = 2.06 years), 60Co (T1/2 = 5.27 years), 51Cr (T1/2 = 27.7 days), 46Sc (T1/2 = 83.8 days), 65Zn (T1/2 = 244 days). All tracers were prepared by neutron irradiation of spectrally pure oxides or salts (mostly nitrates) in a Polish nuclear reactor MARIA (neutron flux ~ 1014 cm−2 s−1). All reagents were of analytical grade. High purity water18 MΩ cm from Mili Q RG Ultra Pure Water System, Millipore Co. was used for the preparation of all solutions.
Apparatus
Micro-analytical and analytical balances, Sartorius MC5 and Sartorius BP221S, were used to prepare standards and tracer for irradiation.
A high-pressure system Anton Paar 3000 was applied to digest samples.
Gamma-ray spectroscopic measurements were performed with the aid of the 180 cm3 HPGe well-type (Canberra) with associated electronics (Ortec) (resolution 2.09 keV for 1332 keV 60Co line, efficiency approximately 35 %), coupled to the multichannel analyzer TUKAN (NCNR, Świerk, Poland).
Glass columns of I.D. 0.50 cm were used in column experiments. The resin bed of 10 cm length was rested on a glass wool plug.
Separation scheme and RNAA procedure
Biological samples (100–150 mg) and chromium standards (10 µg) in PE capsules were irradiated in nuclear reactor MARIA at thermal neutron flux of 1014 cm−2 s−1 for 50 min. After approximately 1 week of cooling, the samples were quantitatively transferred into Teflon vessels, and nonactive Cr carrier (50 µg) was added. All PE capsules were additionally washed with acid and washings were then added to the sample before mineralization process. Then the mineralization process was carried out in high pressure microwave system under controlled conditions, using a mixture of 6 mL of concentrated HNO3 and 2 mL of 30 % H2O2 and 1 mL of HF. The resulting solution was transferred into a Teflon vessel and evaporated to wet salts. The residue was dissolved in 2 mL of 0.01 M H2SO4. Finally, the obtained solution was introduced onto the top of the column filled with MnO2 Resin, 100–200 mesh (h = 10 cm, r = 0.25 cm). The column was washed with 15 mL 0.1 M HNO3 (elution of impurities, i.e. antimony, rubidium, zinc and cobalt). Next, the column was washed with 4 M HCl (elution of cesium and the rest of rubidium). The retained Cr(VI) was quantitatively eluted with 8 M H2SO4. The flow rate was maintained at 1.2–1.5 cm min−1. The chromium content was determined by gamma-ray spectrometry via the 320 keV line. The measurement time varied between 2000 and 20,000 s.
Results and discussion
NAA is one of the best analytical techniques available for determination of trace level of chromium in a variety of materials. The half-life of the radionuclide 51Cr (T = 27.7 days) gives the opportunity to apply both versions of NAA: instrumental (nondestructive) and radiochemical. However, Cr is considered to be a difficult element for instrumental neutron activation analysis due to influence of a high radioactivity of the biological matrix caused mainly by bremsstrahlung of 32P and Compton continuum from high activities of radionuclides emitting high energy gamma-rays [12]. Those effects can be eliminated by the separation of chromium from others elements. Furthermore, polyethylene capsules, commonly used as canning material, generate a signal of 51Cr (320 keV) which interferes with the signal generated by the investigated sample. PE capsules used in this work contained 0.66–2.39 μg Cr. Therefore, the Szilard–Chalmers effect was examined and the effect of nuclear recoil was found negligible. Also, the irradiated samples were removed from the capsules before processing. Another problems can be caused by possible spectral and nuclear interferences. Therefore possible influence of interfering nuclear reactions were considered. It was found that only reaction 54Fe(n, α) 51Cr can potentially have a significant impact on the result of chromium determination. Under applied irradiation conditions, 20.7 μg of Cr is formed from 1 g of Fe. Taking into account the content of iron in most biological materials, the influence of the above-mentioned reaction can be disregarded. In the case of ultratrace levels, it is most often necessary to perform radiochemical separations to isolate chromium from other elements that can interfere directly via gamma-ray overlap, or indirectly by elevating the background level of radiation under the peak of interest via Compton events and/or bremsstrahlung [22]. Radiochemical separation can solve also another problem caused by possible spectral interference from radionuclide 147Nd with gamma line 319, 4 keV (51Cr gamma line 320 keV).
An inorganic ion exchanger MnO2 Resin (manganese(IV) dioxide) was chosen for the selective separation of chromium from the biological samples. The sorbent was devised for the separation of radium from barium in natural water samples over a wide range of pH. It was also used to isolate radium from the liquid wastes from the uranium industry and sorption of radionuclides released into sea water from a nuclear reactor [23]. Recently, the resin was applied to the separation of chromium from human blood [24]. As an inorganic resin, it is resistant to high temperature and ionizing radiation, and shows high selectivity to polyvalent metals [25, 26].
In order to find the best condition for quantitative and selective separation of chromium from other elements, mass distribution coefficients of Cr, Zn, Co, Sc, Cs have been determined in the system: MnO2 Resin—HCl, HNO3 and H2SO4 in the concentration range from 0.01 to 8 M. The mass distribution coefficients (Kd) were determined by batch equilibration at room temperature using radioactive tracers of elements in question. The distribution coefficients were calculated from the equation:
where A0 is the concentration of individual tracer in the standard solution; AS is the concentration of individual tracer in an aliquot of the solution, after equilibration with the resin, loaded with a given extractant; V is the volume of the solution (mL); and mj is the mass of dry resin (g). The obtained results are shown in Table 1.
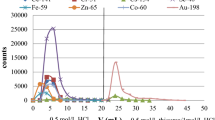
As can be seen from Table 1, the absorption of Cr(VI) takes place in the dilute solutions of hydrochloric, nitric and sulfuric acid, and decreases with increasing concentration of the acids. The distribution coefficient values of chromium in 0.01 M acids are sufficiently high to ensure its quantitative retention on the sorbent. Taking into account the distribution coefficients of the other elements in question, the optimal conditions of separation of Cr from other elements have been chosen (Fig. 1) and verified by the series of column experiments (Fig. 2). The separation of chromium on MnO2 Resin from Tea Leaves INCT-TL-1 is shown in Fig. 3, and the obtained gamma-ray spectrum of chromium fraction is shown in Fig. 4.

Scheme of the RNAA procedure for determination of Cr in biological materials

Separation of chromium from selected elements

Separation of chromium from other elements—Tea Leaves INCT-TL-1

Gamma-ray spectrum of chromium fraction isolated from Tea Leaves INCT-TL-1
The devised procedure was validated by the analysis of several CRMs: Tea Leaves INCT-TL-1, Scallop Tissue IAEA-452, Spinach Leaves NBS 1570, Oyster Tissue NIST 1566a and Orchard Leaves SRM 1571.
A comparison of the results of chromium determination obtained by the developed procedure with the certified values is shown in Table 2. All analysis were carried out repeatedly (at least twice).
As can be seen from Table 2, the results of Cr determination in biological materials, obtained by the elaborated RNAA procedure agree very well with the certified values. The obtained results confirmed also absence of spectral interferences from 147Nd, especially in the case of Tea Leaves INCT-TL-1, which contains relatively high amount of Nd (informative value 0.8 mg kg−1). This was to be excepted because Kd of scandium was very low in all used acids (Table 1) what indicated that REE should be not retained on MnO2 Resin.
The detection limit (LOD) of the procedure was calculated from Currie criterion [27] as 4.88 ng g−1 (20,000 s counting time). The combined standard uncertainty of the measurement results was evaluated according to the uncertainty propagation law, taking into account all individual uncertainty sources starting from the weighing of samples and standards, through chemical separation yield and finishing with the gamma-spectrometric measurements [14, 28]. The details of the evaluation of the standard uncertainties associated with individual sources of uncertainty have been described earlier [14–21].
As can be seen from Table 2, the values of the expanded combined uncertainty vary from 1.67 to 4.81 %.
Conclusions
The elaborated method based on RNAA can be used to the determination of total chromium content in biological materials. Chromium is selectively and quantitatively separated from other elements utilizing the inorganic sorbent MnO2 Resin. The described method gives accurate results with low levels of uncertainty. The method can be applied to verify the accuracy of the results of other methods for the determination of chromium in biological materials.
References
Edebali S, Pehlivan E (2010) Evaluation of Amberlite IRA96 and Dowex 1 × 8 ion exchange resins for the removal of Cr(VI) from aqueous solution. Chem Eng J 161:161–166
Kabata-Pendias A (2010) Trace elements in soil and plants, 4th edn. CRC Press, New York
Zhang XX, Tang SS, Land Chen M, Wang JH (2012) Iron phosphate as a novel sorbent for selective adsorption of chromium(III) and chromium speciation with detection by ETAAS. R Soc Chem 27:466–472. doi:10.1039/c2ja10292g
Sacmaci S, Kartal S, Yilmaz Y, Sacmaci M, Soykan C (2012) A new chelating resin: synthesis, characterization and application for speciation of chromium (III)/(VI) species. Chem Eng J 181–182:746–753
Bampaiti A, Noli F, Misaelides PJ (2013) Investigation of the Cr(VI) removal from aqueous solutions by stabilized iron-nanoparticles using 51Cr-tracer. Radioanal Nucl Chem 298:909–914
Buttner I, Hamm V, Knochel A, Gupta RS (1993) Development of a procedure for the determination of chromium in samples of urine and serum by neutron activation analysis. J Anal Chem 346:446–452
Manzoori JL, Mohammed H, Shemirani F (1994) Chromium speciation by a surfactant—coated alumina microcolumn using electrothermal atomic absorption spectrometry. Talanta 42:1151–1155
Zelmanov G, Semiat R (2011) Iron Fe30+ oxide/hydroxide nanoparticles—based agglomerates suspension as adsorbent for chromium (Cr60+) removal form water and recovery. Sep Purif Technol 80:330–337
Llobat-Estelles M, Mauri-Aucejo AR, Lopez-Catalan MD (2001) Spectrophotometric determination of chromium with diphenylocarbazide in the presence of vanadium, molybdenium and iron after separation by solid phase extraction. J Anal Chem 371:358–363
Langard S (1982) Biological and environmental aspects of chromium. Elsevier Biomedical Press, New York
Gomez V, Callao MP (2006) Chromium determination and speciation since 2000. Trends Anal Chem 25(10):1006–1014
Landsberger S, Peshev S (1994) Determination of chromium in biological reference materials by instrumental NAA using Compton suppression. J Radioanal Nucl Chem 181:61–70
Samczyński Z, Dybczyński RS, Polkowska-Motrenko H, Chajduk E, Pyszynska M, Danko B, Czerska E, Kulisa K, Doner K, Kalbarczyk P (2011) Preparation and certification of the new polish reference material: oriental basma tobacco leaves (INCT-OBTL-5) for inorganic trace analysis. Institute of Nuclear Chemistry and Technology, Warszawa
Greenberg RR, Bode P, De Nadai Fernandes E A (2011) Neutron activation analysis: a primary method of measurement: Spectrochim. Acta B 66:193–241
Dybczyński RS, Danko B, Kulisa K, Maleszewska E, Polkowska-Motrenko H, Samczyński Z, Szopa Z (2003) Performance and frequency of use of NAA and other techniques during the certification of two new Polish CRMs prepared by INCT. Czech J Phys 53:A171–A179
Polkowska-Motrenko H, Dybczyński RS, Chajduk E (2010) Certification of reference materials for inorganic trace analysis: the INCT approach. Accred Qual Assur 15:245–259
Dybczyński RS, Danko B, Polkowska-Motrenko H, Samczyński Z (2007) RNAA in metrology: a highly accurate (definintive) method. Talanta 71:529–536
Polkowska-Motrenko H, Danko B, Dybczyński RS (2004) Metrological assessment of the high accuracy RNAA method for determination of cobalt in biological materials. Anal Bioanal Chem 379:221–226
Dybczyński RS, Danko B, Pyszynska M, Polkowska-Motrenko H (2012) Ratio primary reference measurement procedure (RPMRP) for the determination of iron in biological materials by RNAA. Radiochim Acta 100:409–416
Chajduk E, Polkowska-Motrenko H, Dybczyński RS (2008) A definitive RNAA method for Se determination in biological samples. Uncertainty evaluation and assessment of degree of accuracy. Accred Qual Assur 13:443–451
Chajduk E, Polkowska-Motrenko H, Dybczyński RS (2008) The use of definitive methods based on RNAA for the determination of selenium and arsenic in materials used in proficiency testing. Nukleonika 53(Suppl 2):49–54
Greenberg RR, Zeisler R (1988) A radiochemical procedure for ultratrace determination of chromium in biological materials. Radianal Nucl Chem 124(1):5–20
Bigliocca C, Girardi F, Paulky J, Sabbioni E (1967) Radiochemical separations by adsorption on manganese dioxide. Anal Chem 13:1634–1639
Meloni S, Brandone A, Maxia V (1969) Chromium separation by inorganic exchanger in activation analysis of biological materials. Int J Appl Radiat Isot 20:757–760
Varga Z (2007) Preparation and characterization of manganese dioxide impregnated resin for radionuclide pre-concentration. Appl Radiat Isot 65:1095–1100
Minczewski J, Chwastowska J, Dybczyński R (1982) Separation and preconcentration methods in inorganic trace analysis. Ellis Horwood Ltd, Chichester
Currie LA (1968) Limits for qualitative detection and quantitative determination. Application to radiochemistry. Anal Chem 40:586–593
International Organization for Standarization (ISO) (1995) Guide to the expression of uncertainty in measurement. ISO, Geneva
Acknowledgment
This project is financed in the framework of Grant entitled: “Development and validation of new types of reference materials necessary for obtaining European accreditation by the Polish laboratories for industrial analytics” MODAS in the INNOTECH-K-1/I1/43/158947/NCBR/12 attributed by the National Center for Research and Development.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kużelewska, I., Polkowska-Motrenko, H. & Danko, B. Determination of chromium in biological materials by radiochemical neutron activation analysis (RNAA) using manganese dioxide. J Radioanal Nucl Chem 310, 559–564 (2016). https://doi.org/10.1007/s10967-016-4896-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-016-4896-0