
Abstract
The use of experimental parameters to quantify solvent properties, for example in linear free energy relationships, is well established and several scales of solvent acidity, basicity and polarity/polarizability have been developed. The success of this approach raises questions of which molecular properties contribute to particular solvent parameters and whether these contributions are found in all parameters representing a particular solvent property. In the present study, Catalan’s hydrogen bond basicity and acidity parameters, SB and SA, and Gutmann’s acceptor number, AN, a measure of a solvent’s Lewis acidity, are correlated with molecular properties derived from computational chemistry. The results are compared with the results of similar correlations with Kamlet and Taft’s β and α Solvent Scales, Gutmann’s donor number DN) and Abraham’s B and A solute scales. The results show that measures of solvent basicity, SB, β and DN all correlate strongly with the partial charge on the most negative atom in the solvent molecule and the energy of the donor orbital and, in all cases, the parameter values for hydrogen-bonded solvents are anomalous. Abraham’s B, a measure of solute hydrogen basicity, depends only on the partial charge on the most negative atom and there is no anomaly in the values for solutes that, in the pure state, form hydrogen-bonded liquids. Similarly, all measures of solvent acidity, SA, α and AN, and Abraham’s A, a measure of solute hydrogen bond acidity, depend on the partial charge on the most positive hydrogen on the molecule.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The use of linear free energy relationships to correlate or predict chemical data in different solvents is well established and a variety of experimental parameters, intended to capture solvent properties that contribute to different solvent–solute interactions, have been developed [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16].
Essentially, for some property, X, one writes:
where the Pis represents different solvent properties, the ci is the response of X to Pi , and X0 is the value of X when all of the ciPi terms are equal to zero. Essentially, each ciPi term represents a different contribution to X. The solute–solvent interactions represented by the ciPi terms are generally taken to include acid–base interactions between specific moieties on the solute and solvent molecules and non-specific interactions, commonly described as solvent and solute polarity and polarizability. Thus, for each solvent, experimental parameters have been developed representing hydrogen bond or Lewis basicity, hydrogen bond or Lewis acidity and solvent polarity/polarizability.
The success of this approach raises two simple questions: firstly, what molecular properties contribute to a particular solvent property, as measured by these experimental parameters, and secondly, do all parameters for a particular solvent property reflect the same molecular properties.
These have been explored in a series of papers where experimental solvent and solute parameters have been correlated against a set of molecular properties derived from computational chemistry [17,18,19,20,21,22,23]. In the present paper, Catalan’s SB [14] and SA [15] measures of solvent hydrogen bond basicity and acidity, and Gutmann’s acceptor number, [5] a measure of solvent Lewis acidity, are considered and the results compared with those for Kamlet and Taft’s β [17, 18] and α [19], Gutmann’s donor number [17] and Abraham’s B [21] and A [22], measures of solute hydrogen bond basicity and acidity.
2 Computational Details
The computational methods have been discussed in some detail previously [17, 21]. Since the calculated molecular properties can depend on the method used, calculations were carried out using both the Hartree–Fock and density functional (B3LYP functional) methods both using the 6–311 + g(3df,2p) basis set; partial atomic charges were calculated using Hirshfeld’s model [24].
Seven molecular properties are considered: the partial charges on the most negative atom and on the most positive hydrogen atom of the molecule, the molecular dipole moments, quadrupolar amplitudes and polarizabilities and the energies of electron donor and acceptor orbitals.
In general, the orbital energies are taken to be those of the highest occupied and lowest unoccupied molecular orbitals; the exception to this is the case of solvents with aromatic groups. This was discussed previously [17] but, briefly, solvents with a common functional group have, for example, similar basicity parameters, whether they are aliphatic or aromatic, although, for aromatic solvents, the high energy π-bonds of the aromatic ring are clearly the HOMOs, while for aliphatic solvents, the HOMO is associated with the basic functional group. The fact that the basicity parameters are similar argues for the use of the energies of the orbitals at the interacting functional group rather than those of the aromatic ring.
All calculations were carried out using the Gaussian 09 suite of programs [25].
3 Analyses of Parameters
The approach used [17] is a simple multivariable regression of the experimental parameter, P, against molecular descriptors, Qi, representing different molecular properties. Thus:
where P0 is the value of P when all of the aiQi terms are zero and the ai is the coefficient recovered from the regression. In essence, each aiQi term represents the contribution of a particular interaction to P.
The molecular descriptors are normalized and calculated from the calculated molecular properties, qi, as:
where \(q_{i}^{{{\text{min}}}}\) and \(q_{i}^{{{\text{max}}}}\) are the minimum and maximum values of \(q_{i}^{{}}\). With the exception of the orbital energies, the values of \(q_{i}^{{{\text{min}}}}\) are taken to be zero.
In applying Eq. 1, the experimental parameter, P, was initially correlated with all seven molecular descriptors; subsequently, descriptors making negligible contributions (typically ai ≤ 0.01) were excluded and the correlation repeated with the remaining descriptors. Since both the calculated properties and experimental parameters for solvents with a common functional group are very similar, the distributions of these can’t be represented by statistics based on the normal distribution. Thus, the decision as to whether a descriptor was making a significant contribution to P was decided on the basis of the increase in the standard deviation between the calculated and experimental values of P, when the descriptor was excluded from the correlation.
4 Results and Discussion
4.1 Hydrogen Bond Basicity SB
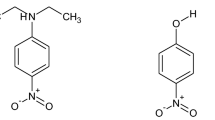
Catalan’s SB scale [14] is based on the differences in the solvatochromism of 5-nitroindoline and its homologue N-methyl-5-nitroindoline (Fig. 1) in the same way that Kamlet and Taft’s β scales of hydrogen bond basicities [3] are largely based on the differences in the solvatochromism of 4-nitroaniline and its homologue N,N-diethyl-4-nitroaniline.Footnote 1 Essentially, it is assumed that the solvatochromism of 5-nitroindoline is in two parts, one coming from hydrogen bonding at the N − H hydrogen and the second from non-specific effects on the rest of the molecule, and that the second of these can be estimated from the solvatochromism of the N-methyl-5-nitroindoline, which doesn’t hydrogen bond.

Structures of 5-nitroindoline and N-methyl-5-nitroindoline
As in the cases of Kamlet and Taft’s β and Gutmann’s donor number, the SB values of the alcohols and N–H containing solvents are anomalous and were omitted from the analyses.
The analyses of SB in the remaining solvents showed that only two of the seven molecular properties, the charge on the most negative atom and the energy of the electron donor orbital, correlated with the SB values. The coefficients recovered are listed in Table 1. Also shown are the results of analyses of Kamlet and Taft’s β, Abraham’s B and Gutmann’s donor number (as DNN which is normalized to allow comparison of the regression coefficients). Figure 2 shows a plot of the SB values calculated using the coefficients reported in Table 1 against the experimental values. In Fig. 2, the dashed lines lie one standard deviation above and below the line representing perfect agreement.

Comparison of experimental SB values and values calculated using the coefficients reported in Table 1 for properties calculated using density functional theory. Symbols represent the following: blue triangles, alkanes; red circles, halogenated alkanes and aromatics; light brown triangles, aromatics; dark brown circles, nitriles; light blue squares, esters; yellow circles, ethers; purple circles, amides; green circles, carboxylic acids; dark green diamonds, S=O, P=O compounds. The solid line represents perfect agreement and the dashed lines are 1 standard deviation above and below
The agreement among the coefficients recovered for SB, β and the normalized donor number DNN is striking, all three showing strong dependences on the partial charge on the most negative atom and the energy of the electron donor orbital. The impact of the different computational methods is also clear, with the coefficients for atomic charge and orbital energy being roughly equal when the molecular properties result from density functional calculations but the coefficients for atomic charge being substantially larger when the properties come from Hartree–Fock calculations.
Catalan et al. [26] correlated SB against a number of molecular properties, calculated at the HF/6-31G** level of theory, and found reasonable correlations with the energy of the donor orbital and the electrostatic potential minimum (calculated using the STO-5G basis set) but no correlation with the molecular dipole moment and charge on the negative centre. The last result is surprising but may reflect the use of charges based on the Mulliken population analysis, which varies considerably with changes in the basis set and seems not to converge as the basis set is improved [17].
The results indicate that, for non-hydrogen-bonded solvents, the basicity of the bulk solvent is a simple function of the properties of the isolated solvent molecules.
For hydrogen-bonded solvents such as the alcohols and solvents with N–H moieties, the situation is more complex. Figure 3 shows the same data as in Fig. 2 but also includes the SB values for alcohols and N–H containing solvents.

Comparison of experimental SB values and values calculated using the coefficients reported in Table 1 for properties calculated using density functional theory. Symbols: Data on Fig. 2, blue circles; alcohols, grey circles; solvents with N–H moieties, brown circles. The solid line represents perfect agreement and the dashed lines are 1 standard deviation above and below
It is clear that, while the values calculated from the properties of the isolated molecules show relatively little variation, the experimental values vary widely (from 0.1, trifluoroethanol to 0.966, 2-hexanol). The same is seen in plots of calculated against experimental β and DN values [17, 20].
This anomaly was recognized by Kamlet and Taft [3] who discuss the possibility of the alcohols acting as hydrogen bond donors to the oxygen atoms of the NO2 moiety but also note work by Gordon [27] and Dubuq [28] who propose that the acidity and basicity of hydrogen-bonded solvents are affected by the hydrogen bonding.
It is relevant that, for Abraham’s B coefficient [21], the values for alcohols and N–H containing molecules show no anomaly and lie on the same line as non-hydrogen bonding solvents. The B parameter is a solute, as opposed to solvent, property and was determined from the formation constants of hydrogen-bonded complexes formed in dilute solutions in dichloroethane, and so refers to the isolated molecule rather than the bulk liquid.
Thus, it is reasonable to assume that the basicity measured by SB (or β or DNN) is a property of the hydrogen-bonded network and not simply of the individual solvent molecules, as is the case for non-hydrogen-bonded solvents.
This raises the question of why Gutmann’s donor number, which is defined in terms of the enthalpy of interaction of the base with SbCl5, both at low concentrations in dichloroethane [5], shows an anomaly for values for alcohols and solvents with N–H moieties. It was observed early on that a number of spectroscopic properties correlated with the donor number (see [29] for example) and this led to a number of proxy experiments, using probe molecules, to estimate donor numbers. Such experiments are particularly attractive for solvents with acidic X–H hydrogens, because of the probable reaction with antimony pentachloride, to form HCl, which complicates the calorimetric measurements. Thus, it is probable that at least some of donor numbers reported are from experiments involving probes dissolved in the bulk solvent rather than from calorimetric experiments involving dilute solutions.
It is striking that all four of the basicity scales correlate strongly with the partial charge on the most negative atom and three also correlate with the energy of the donor orbital, while Abraham’s solute basicity, B, doesn’t correlate with the orbital energies. The reason for this latter difference isn’t obvious.
4.2 Acidity Scales
4.2.1 Gutmann’s Acceptor Number AN
Gutmann’s AN scale is based on the 31P NMR chemical shift of triethylphosphine oxide [5] and was introduced as a measure of Lewis, rather than hydrogen bond, acidity. The zero point of the scale is set at the chemical shift in hexane. In the analysis reported here, the scale, ANN, was normalized by setting the value for formic acid equal to one. The values of AN considered were taken from Marcus 1996 review [30].
The analysis of ANN showed that it correlated with only one molecular property, the charge on the most positive hydrogen atom. The coefficients recovered are listed in Table 2, along with those recovered for Catalan’s SA (see 4.2.2), Abraham’s A [22] and Kamlet and Taft’s α [19, 20].
The ANN values calculated using the coefficients reported in Table 2 are compared with the experimental values in Fig. 4.

Comparison of experimental AN values and values calculated using the coefficients reported in Table 1 for properties from Hartree–Fock calculations. Symbols represent the following: blue triangles, alkanes and benzene; red circles, halo-alkanes; purple circles, amides; green circle, acetic acid; grey circles, alcohols; brown circles, amines and pyridine. The solid line represents perfect agreement and the dashed lines are 1 standard deviation above and below
As for both Abraham’s A and Kamlet and Taft’s α, there is a marked steric effect with experimental values being lower than calculated for sterically hindered substances. In Fig. 3, the points lying to the left of the error channel are n-butanol, t-butanol and diethylamine. Those to the right are trifluoro ethanol and perfluoro n-propanol.
4.2.2 Hydrogen Bond Acidity SA
The SA scale of hydrogen bond acidity [15] is based on the differences in the solvatochromism of o-tert-butylstilbazolium and o,o'-di-tert-butylstilbazolium betaine dye (Fig. 5). As with the SB scale, it is assumed that the solvatochromism of the monosubstituted dye can be separated into contributions from hydrogen bonding at the terminal oxygen atom and the second from non-specific interactions with the rest of the molecule. It is then assumed that the second contribution to the solvatochromism can be estimated from that of the disubstituted dye, where hydrogen bonding is blocked by the second tert-butyl moiety.

o-tert-butylstilbazolium- and o,o'-di-tert-butylstilbazolium-betaine dye
The approach is similar to that adopted by Kamlet and Taft in constructing their α scale of hydrogen bond acidities [4] where the non-specific solvatochromism of Reichardt and Dimroth’s Betaine dye is approximated using the solvatochromism of the much smaller N,N-diethyl-4-nitroanaline.
The analysis of SA showed that it correlated with two molecular properties, the partial charge on the most positive hydrogen atom and the polarizability of the solvent molecule. The coefficients recovered are reported in Table 2 and the SA values calculated using the coefficients listed in Table 2 are compared with the experimental values in Fig. 6.

Comparison of experimental SA values and values calculated using the coefficients reported in Table 1 for the properties from density functional calculations. Symbols represent the following: red circles, halo-alkanes; purple circles, amides; green circles, carboxylic acids; grey circles, alcohols; brown circles, amines. The solid line represents perfect agreement and the dashed lines are 1 standard deviation above and below
The positive dependence on the partial charge of the most positive hydrogen atom on the solvent molecule is common to all of the acidity parameters but the negative dependence on the solvent polarizability is unexpected.
All experimental parameters are captives of the experiment from which they are derived. In the case of SA, it is calculated from the solvatochromism of o-tert-butylstilbazolium- and o,o'-di-tert-butylstilbazolium-betaine dye (TBSB and DTBSB) with the value for ethanol set to 0.4; this leads to:
where
where \(\tilde{v}_{{{\text{TBSB}}}}\) and \(\tilde{v}_{{{\text{DTBSB}}}}\) are the peak positions for TBSB and DTBSB, respectively [15].
Catalan et al. provide the experimental data for both dyes [15] and these were correlated with the molecular descriptors, in the same way as SA and the other experimental parameters. These analyses showed that both \(\tilde{v}_{{{\text{TBSB}}}}\) and \(\tilde{v}_{{{\text{DTBSB}}}}\) showed negative dependences on the solvent polarity but that that of TBSB is considerably greater than that of DTBSB and is not fully compensated in Eq. 4.
The negative dependence of \(\tilde{v}_{{{\text{TBSB}}}}\) and \(\tilde{v}_{{{\text{DTBSB}}}}\), and hence SA, on solvent polarizability is not unique; both SPP [23] and π* [18], the Catalan and Kamlet, Abboud and Taft measures of solvent polarity/polarizability, show negative dependences on solvent polarizability, as does Reichardt and Dimroth’s ET(30) [19].
These dependences can be understood by recognizing that the solvatochromism of an electronic transition of a probe molecule rests entirely on changes in the energy difference between the ground and excited state. The Franck–Condon principle tells us that the electronic transition will occur with little or no movement of the atoms of the probe and certainly no movement of the surrounding solvent molecules. Thus, the solvatochromism reflects changes in the solvation of the ground and excited states by the equilibrium solvation structure of the ground state.
Many probe molecules used to determine solvent parameters, including SA, have greater charge separation in the excited state than the ground state and interactions with the charge centres will be stronger with the excited state. The greater these interactions are, the larger the parameter will be.
In contrast, interactions that are stronger with the less polar ground state will have the opposite effect, stronger interactions leading to a reduced parameter value. Thus, the negative correlation with solvent polarization results simply from stronger interactions between the non-polar but polarizable moieties of the solvent and probe molecule, which favor the ground state rather than the excited state of the probe.
4.3 Comparison of Solvent Parameters
There is excellent consistency among both the solvent basicity parameters, SB, β and DNN and solvent acidity parameters, SA, α and ANN.
Thus, all of these solvent basicity parameters depend strongly on the partial charge on the most negative atom on the solvent molecule and on the energy of the likely electron donor orbital. Both observations are expected; thus, acid–base interactions, including hydrogen bonding, are Coulombic interactions between partially charged moieties and such interactions are stronger for larger charges. It is also well known from infra-red studies that, for example, both hydrogen bonding and interactions of metal ions at carbonyl oxygens are associated with weakening of the C=O bond as charge is drawn towards the bound positive moiety. The energy of the donor orbital is expected to influence this movement of charge, with it being greater from orbitals having higher energies so that the electrons are less strongly bound. The Abraham B parameter, which is a measure of solute hydrogen bond basicity, also depends on the partial charge on the most negative atom in the solute molecule but not on the energy of the donor orbital.
The solvent basicity parameters for alcohols and for primary and secondary amines are anomalous, in that they aren’t calculable from the properties of the isolated solvent molecules. This is consistent with the idea that the basicity of these solvents is a property of the hydrogen-bonded networks rather than simply reflecting the properties of the individual solvent molecules. This is strongly supported by the fact that Abraham B values, which are measured at low concentration of the solute, show no such anomaly.
The situation with regard to the solvent acidity parameters SA, α and ANN is similarly simple, all showing a dependence on the charge on the most positive hydrogen atom in the molecule, as does Abraham’s solute hydrogen bond acidity parameter, A. The dependence of SA on solvent polarizability seems to be an artefact of the experiment defining SA rather than a real effect.
It is striking that the acidity parameters of hydrogen-bonded solvents show no anomaly such as that found in the basicity parameters.
It isn’t immediately clear why the acidity parameters of the hydrogen-bonded solvents simply reflect the properties of the isolated molecules while their basicity parameters seem to be almost insensitive to the molecular properties.
Alcohols are very highly hydrogen bonded and, because of the 2:1 ratio of lone pairs to OH hydrogen atoms, form rings and networks of chains which are stabilized by the cooperative hydrogen bonding [31,32,33]. In these systems, the hydrogen-bonded chain can act as a base, by interacting with the solute through one of the non-hydrogen-bonded lone pairs, leaving the chain intact. In contrast, for the chain to act as a hydrogen bond donor requires breaking of the chain with the solute bonded to the OH of the alcohol at the end of the chain.
Thus, alcohols acting as bases can do so without disruption of the hydrogen-bonded structure and basicity is, logically, a property of the chain. In contrast, alcohols acting as hydrogen bond acids can only do so by breaking the hydrogen-bonded structure, with the solute interacting with the end molecule of the hydrogen-bonded chain. The results suggest that the properties of the end alcohol molecule, rather than of the chain, determine the alcohol acidity.
While this is reasonable for the alcohols, the situation with amines is different. Of course, primary and secondary amines are hydrogen-bonded liquids but the ratio of lone pairs to NH hydrogens is 0.5 and 1 in the cases of primary and secondary amines, respectively, so that the above argument isn’t applicable.
5 Conclusions
The results of the study, along with those reported previously [17,18,19,20,21,22], strongly indicate that solvent basicity and acidity parameters are largely determined by the specific properties of the individual solvent molecules. Solvent basicity reflects the partial charge on the most negative atom of the molecule and the energy of the donor orbital, while solvent acidity reflects only the partial charge on the most positive hydrogen atom in the solvent molecule. It was found that Catalan’s solvent acidity parameter, SA, contains an incidental dependence on the polarizability of the solvent molecule.
The basicities of hydrogen-bonded solvents are anomalous, in that they don’t reflect the above dependence on the molecular properties; this anomaly is not found for Abraham’s B, which is a measure of the basicity of the isolated solute molecule.
Notes
Kamlet and Taft [3] originally defined their β scale in terms of the average of values determined from a range of experiments, including the differences in the solvatochromism of 4-nitroaniline and N,N-diethyl-4-nitroaniline and of 4-nitrophenol and 4-nitroanisole, the log10 of the formation constant for the hydrogen-bonded complex formed with 4-fluorophenol, the limiting 19F NMR shift of for the hydrogen-bonded complex with 4 fluorophenol and the log10 of the formation constant for the hydrogen-bonded complex with phenol. Subsequent measurements have tended to be based on single measurements, commonly of the difference in the solvatochromism 4-nitroaniline(1) and N,N-diethyl-4-nitroaniline(2) \(\left( {\beta = - {{\left( {\Delta \nu_{1} - \Delta \nu_{2} } \right)} \mathord{\left/ {\vphantom {{\left( {\Delta \nu_{1} - \Delta \nu_{2} } \right)} {2.80}}} \right. \kern-0pt} {2.80}}} \right)\) where ν is expressed in kK, [3] or something similar.
References
Kosower, E.M.: The effect of solvent on spectra. I. A new empirical measure of solvent polarity: Z-values. J. Am. Chem. Soc. 80, 3253–3260 (1958)
Karl Dimroth, K., Reichardt, C., Siepmann, T., Bohlmann, F.: Über pyridinium-N-phenol-betaine und ihre verwendung zur charakterisierung der polarität von lösungsmitteln. Justus Liebigs Ann. Chem. 661, 1–37 (1963)
Kamlet, M.J., Taft, R.W.: The solvatochromic comparison method. I. The β-scale of solvent hydrogen-bond acceptor (HBA) basicities. J. Am. Chem. Soc. 98, 377–383 (1976)
Taft, R.W., Kamlet, M.J.: The solvatochromic comparison method. 2. The α-scale of solvent hydrogen-bond donor (HBD) acidities. J. Am. Chem. Soc. 98, 2886–2894 (1976)
Gutmann, V.: Empirical parameters for donor and acceptor properties of solvents. Electrochim. Acta 21, 661–670 (1976)
Kamlet, M.J., Abboud, J.L., Taft, R.W.: The solvatochromic comparison method. 6. The π* scale of solvent polarities. J. Am. Chem. Soc. 99, 6027–6038 (1977)
Taft, R.W., Abboud, J.L., Kamlet, M.J.: Linear solvation energy relationships. 12. The dδ term in the solvatochromic equations. J. Am. Chem. Soc. 103, 1080–1086 (1981)
Gritzner, G.: The softness parameter (SP), a measure of the soft donor properties of solvents. Z. Phys. Chem. 158, 99–107 (1988)
Abraham, M.H.: Hydrogen bonding. Part 7. A scale of solute hydrogen-bond acidity based on log k values for complexation in tetrachloromethane. J. Chem. Soc. Perkin Trans. 699, 711 (1989)
Abraham, M.H., Grellier, P.L., Prior, D.V., Morris, J.J., Taylor, P.J.: Hydrogen bonding. Part 10. A scale of solute hydrogen-bond basicity using log K values for complexation in tetrachloromethane. J. Chem. Soc Perkin Trans. 2, 521–529 (1990)
Abraham, M.H.: Scales of solute hydrogen-bonding:their construction and application to physicochemical and biochemical processes. Chem. Soc. Rev. 22, 73–83 (1993)
Reichardt, C.: Solvatochromic dyes as solvent polarity indicators. Chem. Rev. 94, 2319–2358 (1994)
Catalán, J., Lopéz, V., Pérez, P., Martin-Villami, R., Rodríguez, J.-G.: Progress towards a generalized solvent polarity scale: the solvatochromism-7-nitrofluorene and its homomorph 2-fluoro-7-nitrofluorene. Liebigs Ann. 1995, 241–252 (1995)
Catalán, J., Diaz, C., Lopéz, V., Pérez, P., de Paz, J.-L.G., Rodríguez, J.-G.: A generalized solvent basicity scale: the solvatochromism of 5-nitroindoline and its homomorph 1-methyl-5-nitroindoline. Liebigs Ann. 1996, 1785–1794 (1996)
Catalán, J., Diaz, C.: A generalized solvent acidity scale: the solvatochromism of o-tert-butylstilbazolium betaine dye and its homomorph o, o’-di-tert-butylstilbazolium betaine dye. Liebigs Ann. Recueil 1997, 1941–1949 (1997)
Catalán, J.: Toward a generalized treatment of the solvent effect based on four empirical scales: dipolarity (SdP, a new scale), polarizability (SP), acidity (SA), and basicity (SB) of the medium. J. Phys. Chem. B 113, 5951–5960 (2009)
Waghorne, W.E., O’Farrell, C.: Solvent basicity, a study of Kamlet-Taft β and Gutmann DN values using computationally derived molecular properties. J. Solution Chem. 47, 1609–1625 (2018)
Waghorne, W.E.: A study of Kamlet-Taft β and π* scales of solvent basicity and polarity/polarizability using computationally derived molecular properties. J. Solution Chem. 49, 466–485 (2020). https://doi.org/10.1007/s10953-020-00979-z
Waghorne, W.E.: A study of the Reichardt parameter using solvent molecular properties derived from computational chemistry and consideration of the Kamlet and Taft α scale of solvent hydrogen bond acidities. J. Solution Chem. 49, 1360–1372 (2020). https://doi.org/10.1007/s10953-020-01002-1
Waghorne, W.E.: Using computational chemistry to explore experimental solvent parameters—solvent basicity, acidity and polarity/polarizability. Pure Appl. Chem. 92, 1539–1551 (2020). https://doi.org/10.1515/pac-2020-0108
Waghorne, W.E.: A study of the Abraham effective solute hydrogen bond basicity parameter using computationally derived molecular properties. J. Solution Chem. 51, 1133–1147 (2023)
Waghorne, W.E.: A study of Abraham’s effective hydrogen bond acidity and polarity/polarizability parameters, A and S, using computationally derived molecular properties. J. Solution Chem. (2023). https://doi.org/10.1007/s10953-023-01269-0
Waghorne, W.E.: Solvent polarity/polarizability parameters: A study of Catalan’s SPPN, using computationally derived molecular properties, and comparison with π* and ET(30). Liquids 4, 163–170 (2024)
Hirshfeld, F.L.: Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 44, 129–138 (1977)
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J.A., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, E.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, O., Foresman, J.B., Ortiz, J.V., Cioslowski, J., Fox, D.J.: Gaussian. Gaussian Inc., Wallingford (2009)
Catalan, J., Palomar, J., Diaz, G., de Paz, J.L.G.: On solvent basicity: analysis of the SB scale. J. Phys. Chem. 101, 5183–5189 (1997)
Gordon, J.E.: Dependence of the acidity and basicity of water on the extent of its hydrogen-bonded structure. J. Am. Chem. Soc. 94, 650–651 (1972)
Duboc, C.: Etude par spectrophotométrie iI.R. de l’autoassociation de quelques alcools aliphatiques en solution dans le tétrachlorure de carbone—I. Détermination de la concentration en monomères alcooliques. Spectrochim. Acta 30, 431–439 (1974)
Erlich, R.H., Popov, A.I.: Spectroscopic studies of ionic solvation. X. A study of the solvation of sodium ions in nonaqueous solvents by 23Na nuclear magnetic resonance. J. Am. Chem. Soc. 93, 5620–5623 (2002)
Marcus, Y.: The properties of organic liquids that are relevant to their use as solvating solvents. Chem. Soc. Rev. 22, 409–416 (1993)
Robinson, H.L., Symons, M.C.R.: Infrared spectroscopic studies of the solvation of aprotic solvents and ions in methanol. J. Chem. Soc Faraday Trans. I 81, 2131–2144 (1985)
Symons, M.C.R., Thomas, V.K.: Structure of methanol + inert solvent systems. J. Chem. Soc Faraday Trans. I 77, 1883–1890 (1981)
Symons, M.C.R., Thomas, V.K.: Solvation of anions by protic solvents. Nuclear magnetic resonance and infrared correlations. J. Chem. Soc Faraday Trans. I 77, 1891–1897 (1981)
Funding
Open Access funding provided by the IReL Consortium.
Author information
Authors and Affiliations
Contributions
WEW carried out the research and wrote the manuscript
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Waghorne, W.E. Solvent Acidity and Basicity Scales: Analysis of Catalan’s SB and SA Scales and Gutmann’s Acceptor Number and Comparison with Kamlet and Taft’s β and α Solvent Scales, Gutmann’s Donor Number and Abraham’s B and A Solute Scales. J Solution Chem 53, 747–760 (2024). https://doi.org/10.1007/s10953-024-01382-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10953-024-01382-8