
Abstract
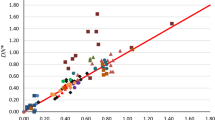
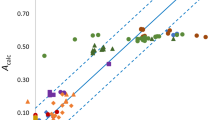
Abraham’s measure of the solute hydrogen bond basicity, \(\beta_{2}^{{\text{H}}}\), is analyzed in terms of molecular properties derived from computational chemistry. It is found that the solute basicity is largely determined by the partial charges on negative centers on the solute molecule. For \(\beta_{2}^{{\text{H}}}\) < 0.5 the basicity is determined by the partial charge on the most negative atom of the solute molecule but for multifunctional solutes with higher \(\beta_{2}^{{\text{H}}}\) values there are contributions from the basicity of other basic centers. The \(\beta_{2}^{{\text{H}}}\) values of amines are not reproduced by the equation defined by correlation of the other solutes considered.


Similar content being viewed by others

Notes
This is, of course, well known and was the basis of Kamlet and Taft’s scale of hydrogen bond acidities [6 and references therein].
The quadrupolar amplitude is calculated as \(A = \sqrt {\sum {q_{ij} q_{ij} } } \, \, i = x,y,z \, {\text{and}} \, j = x,y,z\) where the qij are the components of the traceless quadrupole.
Complex charge distributions, such as those of polyatomic molecules, are commonly represented by a series of superimposed point objects. The first is a point charge (the net charge), which is a scalar quantity, the second is the dipole, which is a vector and the third is the quadrupole, which is a tensor. Just as the dipole has no net charge, the quadrupole has no net moment. The dipole moment and quadrupolar amplitude are used here simply as quantitative measures of the scale of charge centers imbedded in the bulk solvent, the “intensity” of embedded charges. The simplest way to see the necessity for both the dipolar and quadrupolar contributions is to consider CO2, which, despite having partial charges on the O and C atoms has a zero dipole moment, but a non-zero quadrupolar amplitude.
For clarity, the convention is adopted that \(\beta_{2}^{{\text{H}}}\) refers to the experimentally determined basicity parameter, \(\beta_{2}^{{\text{H*}}}\) represents the basicity parameter calculated using Eq. 4 and the coefficients listed in Table 1. Where hydrogen bonding to more than one site on the solute molecule is considered, \(\beta_{2i}^{{\text{H*}}}\) represents the calculated basicity parameter for site i on the solute molecule and \(\sum {\beta_{2}^{{\text{*H}}} }\) is the sum of the calculated \(\beta_{2i}^{{\text{H*}}}\) for the various sites on the solute molecule.
The values for the methyl and ethyl acids and esters are slightly lower.
References
Abraham, M.H.: Scales of solute hydrogen-bonding: their construction and application to physicochemical and biochemical processes. Chem. Soc. Rev. 22, 73–83 (1993)
Abraham, M.H., Grellier, P.L., Prior, D.V., Morris, J.J., Taylor, P.J.: Hydrogen bondint. Part 10. A scale of solute hydrogen-bond basicity using log K values for complexation in tetrachloromethane. J. Chem. Soc. Perkin Trans 2 (1990). https://doi.org/10.1039/p29900000521
Waghorne, W.E., O’Farrell, C.: Solvent basicity, a study of Kamlet-Taft β and Gutmann DN values using computationally derived molecular properties. J. Solution Chem. 47, 1609–1625 (2018)
Waghorne, W.E.: A study of Kamlet-Taft β and π* scales of solvent basicity and polarity/polarizability using computationally derived molecular properties. J. Solution Chem. 49, 466–485 (2020). https://doi.org/10.1007/s10953-020-00979-z
Waghorne, W.E.: A study of the Reichardt parameter using solvent molecular properties derived from computational chemistry and consideration of the Kamlet and Taft α scale of solvent solvent hydrogen bond acidities. J. Solution Chem. 49, 1360–1372 (2020). https://doi.org/10.1007/s10953-020-01002-1
Waghorne, W.E.: Using computational chemistry to explore experimental solvent parameters—solvent basicity, acidity and polarity/polarizability. Pure Appl. Chem. 92, 1539–1551 (2020)
Cho, C.W., Stolte, S., Yun, Y.-S., Krossing, I., Thöming, J.: In silico prediction of linear free energy relationship descriptors for neutral and ionic compounds. RSC Adv. 5, 80634 (2015). https://doi.org/10.1039/c5ra13595h
Devereux, M., Popelier, P.L.A., McLay, I.M.: A refined model for prediction of hydrogen bond acidity and basicity parameters from quantum chemical molecular descriptors. Phys. Chem. Chem. Phys. 11, 1595–1603 (2009). https://doi.org/10.1039/b816321a
Green, A.J., Popelier, P.L.A.: Theoretical prediction of hydrogen-bond basicity pKBHX using quantum chemical topology descriptors. J. Chem. Inf. Model. 14, 553–561 (2014)
Tortorella, S., Corosati, E., Sorbi, G., Bocci, G., Cross, S., Cruciani, G., Storchi, L.: Combining machine learning and quantum mechanics yields more chemically aware molecular descriptors for medicinal chemistry applications. J. Comput. Chem. 42, 2068–2078 (2021). https://doi.org/10.1002/jcc.26737
Schwöbel, J., Ebert, R.-U., Kühne, R., Schüürmann, G.: Prediction of intrinsic hydrogen bond acceptor strength of organic compounds by local molecular properties. J. Chem. Inf. Model. 49, 956–962 (2009). https://doi.org/10.1021/ci900040z
Schwöbel, J., Ebert, R.-U., Kühne, R., Schüürmann, G.: Prediction of the intrinsic hydrogen bond acceptor strength of chemical substances from molecular structure. J. Phys. Chem. A 113, 10104–10112 (2009)
Rahaman, O., Doren, D.J., Di Toro, D.M.: Quantum mechanical estimation of Abraham hydrogen bond parameters using 1:1 donor–acceptor complexes. J. Phys. Org. Chem. 27, 783–793 (2014). https://doi.org/10.1002/poc.3337
Hirshfeld, F.L.: Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 44, 129–138 (1977)
Marenich, A.V., Jerome, S.V., Craner, C.J., Truhlar, D.G.: Charge Model 5: An extension of Hirshfeld population analysis for the accurate description of molecular interactions in gaseous and condensed phases. J. Chem. Theory Comput. 6, 527–541 (2012)
Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J.A., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, E.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, O., Foresman, J.B., Ortiz, J.V., Cioslowski, J., Fox, D.J., Gaussian 09 ed., Gaussian, Inc.: Wallingford CT (2009)
Sekine, T., Usayama, M., Yamaguchi, S., Moriya, H.: The formation of mixed dimers with two carboxylic acids in carbon tetrachloride. Bull. Chem. Soc. Japan 40, 27–32 (1967)
Barrow, G.M., Yerger, E.A.: The dimerization of acetic acid in carbon tetrachloride and chloroform. J. Am. Chem. Soc. 76, 5248–5249 (1954)
Earon, G., Symons, M.C.R.: Spectroscopic studies of the solvation of N,N dimethyl amides in pure and mixed solvents. J. Chem. Soc Faraday Trans. I 84, 3459–3473 (1988)
Eaton, G., Symons, M.C.R., Rastogi, P.P.: Spectroscopic studies of the solvation of amides with N–H groups Part 1. The carbonyl group. J. Chem. Soc. Faraday Trans. I 85, 3257–3271 (1989)
Eaton, G., Symons, M.C.R., Rastogi, P.P., O’Cuinn, C., Waghorne, W.E.: Solvation of some amides in mixed solvent systems Comparison of the results of infrared spectroscopic and calorimetric measurements. J. Chem. Soc. Faraday Trans. 88, 1137–1142 (1992)
Cox, B.G.: A nuclear magnetic resonance study of the rates of protonation of dimethylacetamide and dimethylbenzamide in concentrated acid solutions. J. Chem. Soc. (1970). https://doi.org/10.1039/j29700001780
Waghorne, W.E., Ward, A.J.I., Clune, T.G., Cox, B.G.: Effect of different cations on the N-CO rotational barrier of N,N-dimethylacetamide. Variable temperatre proton magnetic resonance study. J. Chem. Soc. Faraday Trans. I 76, 1131–1137 (1980)
Waghorne, W.E., Rubalcava, H.: Infrared spectroscopic study of the effects of different cations on N,N-dimethylacetamide and fully deuterated N,N-dimethylformamide. J. Chem. Soc Faraday Trans. I 78, 1199–1207 (1982)
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Waghorne, W.E. A Study of the Abraham Effective Solute Hydrogen Bond Basicity Parameter Using Computationally Derived Molecular Properties. J Solution Chem 51, 1133–1147 (2022). https://doi.org/10.1007/s10953-022-01168-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10953-022-01168-w