
Abstract
We report three new cases of a germline heterozygous gain-of-function missense (p.(Met1141Lys)) mutation in the C2 domain of phospholipase C gamma 2 (PLCG2) associated with symptoms consistent with previously described auto-inflammation and phospholipase Cγ2 (PLCγ2)-associated antibody deficiency and immune dysregulation (APLAID) syndrome and pediatric common variable immunodeficiency (CVID). Functional evaluation showed platelet hyper-reactivity, increased B cell receptor-triggered calcium influx and ERK phosphorylation. Expression of the altered p.(Met1141Lys) variant in a PLCγ2-knockout DT40 cell line showed clearly enhanced BCR-triggered influx of external calcium when compared to control-transfected cells. Our results further expand the molecular basis of pediatric CVID and phenotypic spectrum of PLCγ2-related defects.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
The PLCG2 gene encodes phospholipase Cγ2 (PLCγ2), a transmembrane signaling enzyme that catalyzes the production of second messenger molecules, diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3), that utilizes calcium as a cofactor and propagates downstream signals in a variety of hematopoietic cells. IP3 increases intracellular calcium levels by inducing the release of endoplasmic reticulum (ER) calcium stores. The released intracellular Ca2+ leads to the translocation of additional PLCγ2 to the plasma membrane, where its activation leads to the sustained influx of extracellular Ca2+ across the plasma membrane [1].
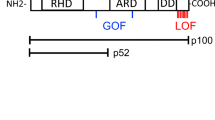
Recently, heterozygous germline mutations in human PLCG2 were linked to two clinical phenotypes with some overlapping features—PLCγ2-associated antibody deficiency and immune dysregulation syndrome (PLAID) and autoinflammation, antibody deficiency, and immune dysregulation syndrome (APLAID) [2,3,4]. The majority of these mutations occur in the autoinhibitory C-terminal SH2 domain, conferring gain-of-function activity (Fig. 2a). These PLCG2 variants have also been described as somatic mutations causing clonal expansion in Ibrutinib-resistant chronic lymphocytic leukemia (CLL) [5]. Another hotspot region in the PLCγ2 protein associated with Ibrutinib resistance mutations is amino acids 1139 to 1141 within the C2 domain which is involved in calcium binding and membrane anchoring of PLCγ2 [6].
To our knowledge, there has never been a report of a germline mutation affecting the C2 domain of PLCγ2. Here we report 3 patients from 2 families with a novel gain-of-function heterozygous missense mutation in the C2 domain of PLCG2 (c. 3422 T>A) [p.Met1141Lys], which are associated with clinical symptoms that expand the phenotype of PLCG2 mutations.
Materials and Methods
Patient Samples
Written informed consent for genetic testing and participation for all three patients was provided by the parent and/or for their children in accordance with the Declaration of Helsinki. Blood was collected from healthy pediatric sex-matched donors after consent at BC Children’s Hospital or through the BC Children’s Hospital BioBank. This research study was approved by the University of British Columbia institutional review board.
Whole-Exome Sequencing of Patient
Whole-exome sequencing (WES) was performed at Canada’s Michael Smith Genome Sciences Centre. Exome library was constructed using the Agilent All Exon V4+UTR capture kit and sequenced on Illumina Hiseq2000. Reads were aligned to the human genome (GRCh37-lite) using Burrows–Wheeler Aligner and duplicate marked with Picard [7, 8]. Variants were called using mpileup (SAMtools), subsequently filtered with varFilter, and annotated using an in-house pipeline that combines SnpEff [9], Ensembl variant database (including pathogenicity prediction), dbSNP [10], NHLBI exome sequencing database (https://esp.gs.washington.edu/drupal/), COSMIC [11], and an in-house human variation database [12]. Annotated variants were then filtered: (1) according to the inheritance mode, (2) ExAC Browser (exac.broadinstitute.org) and (3) previous reported pathogenicity with a phenotype of B cell defects in the Online Mendelian Inheritance in Man (OMIM) and ClinVar databases. There were 193 unique variants identified of which PLCG2 was considered to be the most suitable for further evaluation based on in silico modeling and its known role in B cell development. The heterozygous variant was identified in 50 of 96 reads.
Cell Lines and Reagents
Human CD19+ B cells were incubated in RPMI medium (Gibco®, Waltham, USA), 10% fetal bovine serum, 1% L-glutamine, and 1% penicillin/streptomycin. The wild-type and PLCγ2 knockout DT40 chicken B cell lines were a kind gift from Dr. Michael Gold (University of British Columbia) and grown in RPMI medium (Gibco®), 10% fetal bovine serum, 1% chicken serum, 1% L-glutamine, and 1% penicillin/streptomycin.
Isolation of Peripheral Blood Mononuclear Cells
Peripheral blood was collected via a peripheral blood draw into blood collection tubes containing ACD (trisodium citrate, citric acid, and dextrose) (BD Vacutainer®). Human peripheral blood mononuclear cells (PBMCs) were isolated from fresh blood samples of patient #3 and healthy controls by Ficoll-Paque (GE Healthcare Life Sciences) density centrifugation method.
Isolation of Human CD19+ B Cells
Human B cells were purified after isolation of PBMCs as described above by negative selection using the StemSep® Human B Cell Enrichment kit according to the manufacturer’s instructions (StemCell Technologies, Vancouver, Canada). The purity of the CD19+ fraction was > 95% as determined by CD19 expression by FACS on an LSRII flow cytometer (BD Biosciences).
Cell Lysis, Immunoprecipitation, SDS-PAGE, and Western Blotting
Cell lysates of isolated primary CD19+ B cells from patient #3 and healthy controls were prepared using cell lysis buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin; Cell Signaling) in the presence of protease inhibitor cocktails (Roche, Germany). Equal amounts of protein were separated by 10% SDS-PAGE gel, transferred to PVDF membrane, and blocked with 2% BSA. PLCγ2 protein was detected by rabbit anti-PLCγ2 (Q-20) polyclonal antibody (Santa Cruz Biotechnology, Dallas, USA). After washing, bound antibody was detected with HRP-conjugated anti-rabbit secondary antibody and Novex ECL chemiluminescent substrate (Invitrogen/ThermoFisher Scientific, MA, USA) or with anti-rabbit 680RD secondary antibody on the LiCor Phosphorimager Odyssey (LiCor, USA).
B Cell Immunophenotyping
Immunophenotyping on isolated PBMCs from patient #3 and healthy controls was performed using combinations of Brilliant Violet 605™-CD19 (SJ25C1; BioLegend), PE-Cy7-CD19 (HIB19; BioLegend), Pacific Blue™-CD19 (HIB19; BioLegend), PerCP-CD24 (ML5; BioLegend), Brilliant Violet 421™-CD38 (HIT2; BioLegend), V450-CD38 (HIT2; BD Biosciences), PE-CD38 (HIT2, BioLegend), PE-CD27 (M-T271; BioLegend), FITC-IgD (IA6-2; BioLegend), FITC-CD21 (Bu32; BioLegend), PE-Cy5-CD21 (B-ly4; BD Pharmingen), Alexa Fluor® 700-CD10 (CB-CALLA; eBioscience), APC-CD10 (HI10a; BioLegend), and APC-IgM (MHM-88; BioLegend). Intracellular staining was done with a combination of BD Cytofix™ incubation on ice for 15 min and BD FACS Permeabilizing Solution 2 incubation at room temperature for 10 min. Cells were analyzed using a LSR II flow cytometer (BD Biosciences) and data was analyzed using FlowJo (Tree Star, Ashland, OR, USA).
Calcium Flux Assay
PBMCs from patient #3 and healthy controls were washed once in HBSS (no Ca2+, no Mg2+, Life Technologies) + 1% FBS (Gibco®) and resuspended in dye-loading buffer consisting of 4 μM FLUO-4AM (Molecular Probes) and Probenecid (Life Technologies) in HBSS+1% FBS for 45 min at 37 °C at a concentration of 1 × 106 PBMCs/mL. Cells were washed again with HBSS+1%FBS and then incubated on ice for 20 min with 5 μL of Pacific Blue™-CD19 (HIB19; BioLegend) followed by addition of 1 mL HBSS+1%FBS. Samples were warmed again to 37 °C and within 10 min baseline fluorescence was detected with FITC filter on LSR II flow cytometer in the CD19+ positive fraction. Intracellular calcium flux was induced by BCR stimulation with 10 μg/mL anti-IgM antibody (Jackson Immunoresearch) followed by the addition of extracellular Ca2+ to measure external flux.
Intracellular ERK Phosphorylation
Freshly isolated PBMCs from patient #3 and healthy controls were stimulated with 10 μg/mL soluble anti-IgM antibody (Jackson Immunoresearch Labs, West Grove, USA) at 37 °C. At indicated time-points, cells were fixed with BD Phosflow™ Fix Buffer I for 10 min at 37 °C followed by permeabilization with BD FACS Permeabilizing Solution 2 for 10 min at room temperature. Cells were then stained with V450-CD20 (L27, BD Bioscience), Alexa Fluor 488 – phospho-PLCγ2 (pY759) (K86-689.37; BD Biosciences) and PerCP-eFluor® 710 phospho-ERK1/2 (T202/Y204) (MILAN8R, eBioscience) for 20 min at RT and analyzed on a BD LSR II flow cytometer.
Collagen Stimulation of Platelets
Whole blood was collected in a BD Vacutainer® Plus plastic citrate tube (0.109 Molar/3.2% sodium citrate) from patient #3 and healthy controls after a 3 mL discard. A total of 90 μL of whole blood was then immediately mixed with 10 μL of PBS containing soluble native collagen fibrils (type I) (Chrono-log Corporation) at indicated concentration for 15 min. Subsequently, 5 μL of this mixture was incubated with a flow cytometry antibody mix of 3 μL of FITC-CD61 (VI-PL2; BioLegend) and APC-CD62P (P-Selectin) (AK4; BioLegend) each for 15 min on ice in the dark. At the end of the incubation, 1 mL of 1% paraformaldehyde solution was added and the final mixture was acquired on a LSRII cytometer (BD Biosciences).
Cloning and Transfection Studies
Plasmids used for transfection studies contained full-length wild-type PLCG2 cloned into pCMV6-Entry with a C-terminal Myc-DDK (FLAG) tag (Origene, Rockville, MD, USA) or an empty vector. A mutant PLCG2 plasmid corresponding to our patient’s genetic variant was constructed by site-directed mutagenesis (Q5 Site-Directed Mutagenesis Kit, New England BioLabs Inc., MA, USA) as per manufacturer’s instructions. For transfection studies, 1 × 106 PLCγ2−/− DT40 cells were transfected with 0.5 μg of plasmid DNA in Opti-MEM (Gibco®) by using the B-023 program in an Amaxa Nucleofector II device (Lonza, Allendale, NJ, USA). After overnight incubation, cells were stimulated with 10 μg/mL of anti-chicken IgM (#8300-01, Southern Biotech) in HBSS (no Ca2+, no Mg2+, Life Technologies) + 1% FBS (Gibco®) and calcium flux analyzed as described.
Statistical Analysis
Descriptive statistics were generated on all data using Prism version 6 for Mac (GraphPad Software, San Diego, CA, USA). Significance of observed changes was determined using paired and unpaired Student t tests and two-tailed Mann-Whitney test.
Results
Cases #1 and #2
A 21-month-old female presented with a history of recurrent rashes since birth and scattered atrophic scarring on physical examination. At 2 days of life, she developed vesiculobullous eruptions positive for Serratia marcescens. She continued to develop recurrent episodes of 1–2 mm erythematous, non-folliculocentric papules that evolved into urticarial lesions or flaccid vesicles on an erythematous base. Lesions appeared in migrating crops and resolved spontaneously over 1–2 weeks with rare atrophic scarring (Fig. 1). The eruptions were exacerbated by heat and moisture. In addition to cutaneous findings, she also had recurrent episodes of bloody stools and was treated for multiple respiratory infections, including RSV, adenovirus, and coronavirus. There were no ocular symptoms. Immune cell subset abnormalities included a reduced percentage of CD19+CD27+IgM+IgD− B cells of total CD19+ B cells (0.1%; normal range 0.2–0.3%), a reduced IgM level (0.23 g/L; normal range 0.4–1.9 g/L), and elevated soluble B cell–activating factor (BAFF) level (2382 pg/mL). There were no documented autoantibodies. Her antibody responses to tetanus, diphtheria, and pneumococcal vaccinations were normal.

Cutaneous manifestations of PLCG2 mutation. Various polymorphic cutaneous manifestations including a vesiculobullous (case 1) b urticarial (case 1) and c, d nonspecific erythematous, polymorphic eruptions (case 2)
Family history was significant for a similar history of relapsing-and-remitting polymorphic eruptions in her mother occurring since infancy resulting in extensive atrophic scarring (Fig. 1). Her mother also had a history of significant respiratory infections and chronic obstructive pulmonary disease. Additionally, she reported episodes of episcleritis, arthritis, and recurrent urinary tract infections. B cell immunophenotyping showed an increased percentage of naïve B cell populations in the total CD19+ B cell population: CD19+CD27− (89%, normal range 40–80%), CD19+CD27−CD21+ (86%, normal range 35–76%), and CD19+CD27−IgM+IgD+ (87%, normal range 33–76%). There was a decreased percentage of CD19+CD27+IgM+IgD-cells of total CD19+ B cells (0.7%; normal range 1.5–6.2%) similar to her daughter. She also had a reduced IgM level (0.32 g/L; normal range 0.5–3.7 g/L). She has been treated with regular intravenous immunoglobulin (IVIG) infusions, high-dose steroids, omalizumab, and, most recently, anakinra resulting in improved skin manifestations. A fever/autoinflammatory disorders next-generation sequencing panel (Fulgent Genetics, CA, USA) performed on the mother only reported two potential variations of significance in PLCG2 and CARD14. Targeted sequencing for those two genes was performed in the daughter, identifying the missense mutation in PLCG2, c.3422 T>A [p.Met1141Lys] that was common to both affected individuals (Fig. 2a).

Structure of PLCγ2 and multiple sequence alignment. a Schematic representation of the protein domains of PLCγ2. Annotated are previous cases of APLAID, PLAID, and somatic cancer-associated mutations linked to the Met1141 site. The yellow star refers to the mutation identified in the current report. b The region surrounding the p.Met1141Lys substitution site was aligned to the corresponding regions in a variety of organisms. Indicated in red is the substitution site. Asterisks indicate perfect conservation. PLCγ2, phospholipase C gamma 2; PH, pleckstrin homology domain; EF, EF hand; SH2, Src Homology 2 domain; SH3, Src Homology 3 domain; C2, C2 domain; APLAID, autoinflammation and phospholipase Cγ2 (PLCγ2)-associated antibody deficiency, and immune dysregulation (APLAID) syndrome; PLAID, PLCG2-associated antibody deficiency, and immune dysregulation syndrome
Case #3
A 6-year-old girl presented with a history of recurrent viral (parainfluenza virus type 2, respiratory syncytial virus (RSV) and bacterial (Haemophilus influenzae) respiratory infections resulting in restrictive lung disease. There was no history of ocular, dermatologic, or inflammatory symptoms. Her father died at the age of 51 years due to emphysema.
She met laboratory criteria for CVID with undetectable IgA (< 0.04 g/L), IgM (< 0.03 g/L), and IgG (< 0.27 g/L) levels, and normal total T (CD3+, CD3+CD4+, CD3+CD8+), CD19+ B, and CD3−CD56+ NK absolute cell counts. Lymphocyte proliferation in response to phytohemagglutinin (PHA), pokeweed (PWM), and Staphylococcus aureus Cowan I (SAC) was normal. Even though her blood type is Group O, anti-A and anti-B isohemagglutinins were not detectable. Furthermore, despite documented hepatitis B, vaccination the anti-HBs titer was non-reactive. This was not tested upon boost vaccination. B cell immunophenotyping showed (i) increased fraction of transitional B cells (Fig. 3a, b, e), (ii) lack of memory B cells (Fig. 3c, d, e), and (iii) lack of marginal zone B cells (Fig. 3c). She is currently treated with monthly IVIG infusions.

B cell subset immunophenotyping of CD19+ B cells from patient (PLCγ2M1141K) and healthy pediatric donor. a CD21+/lowCD10+ transitional B cells. b CD38highCD10+ transitional B cells. c CD27+IgD+IgM+ marginal zone B cells, CD27+IgD-IgM-switched memory B cells and CD27-IgD+IgM-naïve B cells. d IgG+IgM+, IgG-IgM-, and IgG-IgM+ B cells. e CD24highCD38high transitional B cells, CD24highCD38low memory B cells and CD24lowCD38low naïve B cells. 7AAD was used to exclude dead cells. Each of these plots represents three different experiments
Whole-exome sequencing identified a heterozygous single nucleotide variant in PLCG2, c.3422 T>A, resulting in a missense protein variant [p.Met1141Lys] (Fig. 2a). All rare variants identified, unique to the patient, are available in a supplementary file.
Characterization of the PLCG2, c.3422 T>A [p.Met1141Lys] Variant
A missense variant in PLCG2, c.3422 T>A [p.Met1141Lys] in all three affected individuals. This variant was not found in any population databases that aggregate next-generation sequencing data, including Exome Aggregation Consortium (ExAC) [13], Genome Aggregation Database (gnomAD) (https://doi.org/10.1101/531210), 1000 Genomes Project [14], ClinVar [15], dbSNP [10], and Exome Variant Server (https://evs.gs.washington.edu/EVS; accessed May 24, 2019) (Supplementary Table 1). However, it was found in the Catalogue of Somatic Mutations in Cancer [11], associated with chronic lymphocytic leukemia originally [6], which suggests potential gain-of-function activity. The region surrounding the p.Met1141Lys substitution was also found to be highly conserved in evolution as evidenced by sequence alignment (Fig. 2b) and in silico conservation metrics (PhyloP = 2.192, fitCons = 0.745) (Supplementary Table 2). Of note, a variety of in silico pathogenicity prediction tools predicted this PLCG2 variant to be pathogenic (Supplementary Table 3), including Combined Annotation Dependent Depletion (CADD = 29) [16], Deleterious Annotation of genetic variants using Neural Networks (DANN = 0.989) [17], Rare Exome Variant Ensemble Learner (REVEL = 0.711) [18], Variant Effect Scoring Tool (VEST4 = 0.838), MetaLR = 0.334 and MetaSVM = − 0.378 [19], Mendelian Clinically Applicable Pathogenicity score (M-CAP = 0.04) [20], GenoCanyon (damaging) [21], Polyphen-2 (damaging) [22], MutationTaster2 (disease causing) [23], SIFT [24], Functional Analysis through Hidden Markov Models (FATHMM = − 0.28) [25], and Protein Variation Effect Analyzer (PROVEAN = − 5.09) [26].
Hematological Cells Manifest Evidence of Altered Function of PLCG2
To investigate the relevance of the PLCγ2 variant, we performed functional assays on hematopoietic cells from patient #3 assessing calcium flux, downstream signaling, and platelet activation related to PLCγ2 function.
We measured intracellular and plasma Ca2+ flux after B cell receptor (BCR) stimulation in the patient’s primary B cells by flow cytometry. We found that BCR stimulation induced increased external calcium entry in the patient’s primary B cells relative to controls (Fig. 4a). Adding to the significance of this observation, the patient’s B cell population was enriched in transitional B cells, B cells that typically exhibit a reduced calcium response compared with naïve and IgM+CD27+ B cells normally [27]. Altogether, these findings suggest that the patient’s mutation leads to PLCγ2 gain-of-function.

Evidence of altered BCR signaling. a Calcium flux in primary CD19+ B cells after BCR stimulation. Fresh PBMCs from patient (PLCγ2M1141K) and healthy control were incubated in Ca2+-free media and stimulated via BCR followed by addition of exogenous Ca2+. Representative of three independent blood collections including one prior to initiation of IVIG therapy with three different sex-matched pediatric controls. b, c ERK1/2 phosphorylation after BCR stimulation. Primary CD19+ B cells from patient (PLCγ2M1141K) and healthy control were stimulated for the indicated times and the phosphorylation of PLCγ2 (b) and ERK1/2 (c) was measured by intracellular flow cytometry. These experiments were repeated with three blood draws from the patient. Each data point is presented as a ratio of MFI to baseline MFI at start of incubation. Values represent means ± SEM. d Collagen-induced platelet alpha granule release. Fresh blood was collected without use of a tourniquet from the patient (PLCγ2M1141K) and healthy controls into sodium citrate tubes with a discard tube first and stimulated with collagen. Alpha granule release from platelets was assessed by measuring the surface expression of CD62P on CD61+ platelets by flow cytometry. Each column represents data from 3 individual samples. e Equal amounts of PLCγ2 protein in about 1 million isolated CD19+ B cells was confirmed by immunoblotting. Representative of two independent collections
The increased production of DAG could activate downstream effectors such as extracellular signal-regulated kinase (ERK). We evaluated ERK phosphorylation after BCR stimulation by flow cytometry analysis. We found that despite equivalent levels of PLCγ2 phosphorylation between samples, there was more prolonged ERK phosphorylation in the affected patient B cells relative to controls (Fig. 4b, d). This gain-of-function activity was not the result of increased PLCγ2 protein expression as total PLCγ2 expression in primary CD19+ B cell lysates from a patient and healthy control was comparable as detected by immunoblot (Fig. 4e).
In murine models of PLCγ2 gain-of-function mutations, platelets showed enhanced α-granule secretion upon stimulation with collagen [28]. We evaluated secretion of α-granules using surface expression of CD62P (P-selectin) on freshly isolated human platelets after collagen stimulation. Platelet activation results in the flipping of CD62P on the inner membrane to the outside of the cell as granules release their contents. We found that the patient’s platelets have enhanced α-granule release after stimulation with collagen in vitro (Fig. 4c). These data provide support for a gain-of-function phenotype.
PLCG2 (p.(Met1141Lys)) Mutation is Characterized by the Ability to Amplify BCR-Triggered External Calcium Flux
To experimentally confirm the gain-of-function activity of the PLCG2M1141K genetic variant, we transiently transfected a PLCγ2 knockout (KO) DT40 cell line with a wild-type and mutant PLCG2 construct and assessed calcium flux after BCR stimulation. BCR stimulation of PLCγ2 KO DT40 cells transfected with the mutant PLCG2 (p.(Met1141Lys)) construct resulted in increased external calcium flux compared with a wild-type PLCG2 construct (Fig. 5a). The difference in area under the curve for calcium flux starting at 60 s between wild-type and mutant PLCG2 was significant (463 ± 25, 414–512 versus 937 ± 40, 858–1017, p = 0.02; (mean ± SEM, 95% CI)). There was no significant difference in transfection efficiency between wild-type and mutant plasmid (27.13 ± 1.04% versus 28.23 ± 0.20%, p = NS) and equal expression was confirmed by Western blotting.

BCR stimulation of PLCγ2 KO DT40 cells transfected with PLCγ2 constructs. a PLCγ2 KO DT40 cells were transiently transfected with empty plasmid, normal, and mutant PLCγ2M1141K construct corresponding to our patients’ genetic variant. After overnight incubation, the transfected DT40 cells were incubated in Ca2+-free media and stimulated via BCR followed by addition of exogenous Ca2+. Each data point is representative of three independent experiments with measurements shown in 15-s intervals. Viable cells were analyzed for 60 s prior to BCR stimulation in order to establish an average baseline by which the relative fold increase in fluorescence is measured. b Western blot showing equal transfection expression of PLCγ2 constructs
Discussion
We have identified a germline heterozygous gain-of-function mutation in the C2 domain of PLCγ2, which was previously identified as a somatic mutation in CLL, thus further expanding the phenotype of PLCG2-related inherited immunodeficiencies. Our experimental studies confirm that the genetic variant was associated with increased BCR-triggered external calcium flux in primary B cells, increased and prolonged BCR-triggered ERK phosphorylation in primary B cells and platelet hyper-reactivity. A potentially causative relationship between the candidate genotype and the clinical phenotype was suggested by transfecting a PLCγ2 knockout DT40 B cell line with mutant PLCG2 (c.3422 T>A [p.Met1141Lys]) which resulted in increased external calcium flux after BCR stimulation similar to the patient’s primary B cells.
In antigen-stimulated B cells, the first transient Ca2+ flux is caused by inositol 1,4,5-trisphosphate (IP3) binding to IP3 receptors (IP3R), which triggers rapid Ca2+ release from finite internal Ca2+ stores in the endoplasmic reticulum (ER). This initial phase of receptor-activated Ca2+ signaling is followed by a sustained influx of extracellular Ca2+ across the plasma membrane [1, 29]. The C2 domain of PLCγ2, in which this mutation resides, has previously been shown to play two roles in this second phase. Firstly, the initial Ca2+ flux via IP3R promotes lipase-independent translocation of additional PLCγ2 from the cytosol to the plasma membrane leading to more production of IP3 and DAG after binding to the adapter protein BLNK [1]. DAG in turn activates more influx of Ca2+ by directly activating transient receptor potential cation (TRPC) Ca2+ channels in the plasma membrane [30]. The C2 domain is critical for this Ca2+ entry–dependent translocation of PLCγ2 as deletion of one of the expected Ca2+ binding sites in the C2 domain of PLCγ2 (Δ1138E-1140D) results in the absence of this second sustained phase [1]. The proximity of the amino acid substitution in the patient’s PLCγ2 (M1141K) to this putative calcium-binding site may result in the formation of a higher affinity Ca2+ binding site, leading to increased IP3 and DAG production. Secondly, binding of the PLCγ2 SH2 domain to BLNK is stabilized by the PLCγ2 C2 domain in a Ca2+-regulated manner [31]. Interestingly, total deletion of the C2 domain in PLCγ2 ablated both intracellular and extracellular BCR-triggered Ca2+ responses [1]. The additional requirement of the C2 domain for the initial BCR-mediated Ca2+ flux could be explained by the universal role of C2 domains in promoting binding to phospholipid bilayers. A mutation within the C2 domain has the potential to affect the association of PLCγ2 with the plasma membrane. For example, mutants that increase the positive surface charge of the C2 domain in p110α, a catalytic subunit of PI3K involved in tonic BCR signaling, lead to a gain-of-function [32]. This is analogous to our variant as methionine is substituted with positively charged lysine at position 1141.
The clinical findings in the first two cases share some of the clinical features previously reported in patients with APLAID, including polymorphous rashes, lack of cold-induced urticaria, colitis, arthritis, episcleritis, and immunodeficiency. There are notable differences in the dermatological presentation as patients #1 and #2 appear to have a broader spectrum of cutaneous manifestations including urticarial, granulomatous, and vesiculobullous rashes, while patient #3 has no significant cutaneous findings to date. One of the limitations of our study is that we were unable to obtain a more detailed functional profile of hematopoietic cells in these two patients as was done with patient #3.
Our third patient does not, as of yet, have any evidence of severe inflammation and the presentation was more consistent with pediatric CVID. The association between PLCG2 mutations and CVID is further supported by a recent study of 185 CVID cases, which revealed 12 missense variants of PLCG2 [33]. Overexpression studies in a PLCG2 deficient DT40 B cell line, as done in our study, showed enhanced ERK phosphorylation and enhanced activation following IgM stimulation.
Interestingly, the B cell defects of the third patient more closely resembled those observed in PLCγ2-deficient mice despite completely opposing effects on BCR-stimulated ERK phosphorylation and calcium flux [34]. These mice were viable but had decreased mature B cells, a block in pro-B cell differentiation, and B1 B cell deficiency. IgM receptor-induced Ca2+ flux and proliferation in response to B cell mitogens was absent. Ig levels were reduced and T cell–independent antibody production was absent. FcγR signaling was also defective, resulting in a loss of collagen-induced platelet aggregation, mast cell FcεR function, and NK cell FcγRIII and 2B4 function [35]. Targeted deletion of Plcγ2 in germinal center B cells demonstrated an additional essential role in the maintenance of memory B cells [36]. To date, there are no reported cases of humans with PLCγ2 deficiency. This suggests that PLCγ2 activity may be tightly regulated in normal B cell development; a decrease in PLCγ2 activity results in diminished BCR signaling that strongly impairs development of transitional B cells [37], while BCR hyper-reactivity causes B cell apoptosis. Either way, there is a defect in B cell development.
In this study, we report the same PLCG2 mutation in three patients with different phenotypes. The reasons for this may include variable expressivity of the genotype in various hematopoietic cell subsets leading to overdominance of the gain-of-function allele in certain cells. For example, the B cell lineage in our third case may have had increased mutant allele transcription and gene activity resulting in a more pronounced effect on BCR signaling in early B cell development. Further, yet unknown modifier genes and environmental factors may affect this phenotype.
Our study highlights the utility of the whole genome/exome sequencing approach in elucidating genetic causes of autoinflammatory syndromes and CVID and provide further insight into the mechanism of BCR signaling in human B cells. Moreover, this work emphasizes the ongoing critical need for functional studies to link genotype to phenotype. Our results also expand the clinical spectrum of diseases resulting from PLCG2 mutations, while observing similarities such as impaired B cell memory and antibody production.
References
Nishida M, Sugimoto K, Hara Y, et al. Amplification of receptor signaling by Ca2+ entry-mediated translocation and activation of PLCγ2 in B lymphocytes. EMBO J. 2003;22(18):4677–88.
Ombrello MJ, Remmers EF, Sun G, et al. Cold urticarial, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med. 2012;366(4):330–8.
Zhou Q, Lee GS, Brady J, et al. A hypermorphic missense mutation in PLCG2, encoding phospholipase Cγ2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am J Hum Genet. 2012;91(4):713–20.
Neves JF, Doffinger R, Barcena-Morales G, et al. Novel PLCG2 mutation in a patient with APLAID and cutis laxa. Front Immunol. 2018;9:2863.
Maddocks KJ, Ruppert AS, Lozanski G, et al. Etiology of Ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol. 2015 Apr;1(1):80–7.
Burger JA, Landau DA, Taylor-Weiner A, et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun. 2016 May 20;7:11589. https://doi.org/10.1038/ncomms11589.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;26(5):589–95.
Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
Cingolani P, Platts A, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6(2):80–92.
Sherry ST. Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11.
Forbes SA, Bindal N, Bamford S, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39(Database issue):D945–50.
Fejes AP, Khodabakhshi AH, Birol I, et al. Human variation database: an open-source database template for genomic discovery. Bioinformatics. 2011;27(8):1155–6.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91.
1000 Genomes Project Consortium, Auton A, Brooks LD, et al. Nature. 2015;526(7571):68–74.
Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–8.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–94.
Quang D. Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. 2015;31(5):761–3.
Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99(4):877–85.
Dong C, Wei P, Jian X, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. 2015;24(8):2125–37.
Jagadeesh KA, Wenger AM, Berger MJ, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48(12):1581–6.
Lu Q, Hu Y, Sun J, et al. A statistical framework to predict functional non-coding regions in the human genome through integrated analysis of annotation data. Sci Rep. 2015;5:10576.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Schwarz JM, Cooper DN, Schuelke M, et al. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–2.
Sim NL, Kumar P, Hu J, et al. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40(Web Server issue):W452–7.
Shihab HA, Gough J, Mort M, et al. Ranking non-synonymous single nucleotide polymorphisms based on disease concepts. Hum Genomics. 2014;8:11. https://doi.org/10.1186/1479-7364-8-11.
Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31(16):2745–7.
Foerster C, Voelxen N, Rakhmanov M, et al. B cell receptor-mediated calcium signaling is impaired in B lymphocytes of type Ia patients with common variable immunodeficiency. J Immunol. 2010;184(12):7305–13.
Elvers M, Pozgaj R, Pleines I, et al. Platelet hyperreactivity and a prothrombotic phenotype in mice with a gain-of-function mutation in phospholipase Cgamma2. J Thromb Haemost. 2010;8(6):1353–63.
Feske S. Calcium signaling in lymphocyte activation and disease. Nat Rev Immunol. 2007;7(9):690–702.
Numaga T, Nishida M, Kiyonaka S, et al. Ca2+ influx and protein scaffolding via TRPC3 sustain PKCbeta and ERK activation in B cells. J Cell Sci. 2010;123(Pt 6):927–38.
Engelke M, Oellerich T, Dittmann K, et al. Cutting edge: feed-forward activation of phospholipase Cγ2 via C2 domain-mediated binding to SLP65. J Immunol. 2013;191(11):5354–8.
Gymnopoulos M, Elsliger MA, Vogt PK. Rare cancer-specific mutations in PIK3CA show gain of function. PNAS. 2007;104(13):5569–74.
Szymanski AM, Baysac K, Marcy H, et al. PLCG2 variants influence CVID susceptibility: expanding the spectrum of PLCG2-associated immune dysregulation. 20218 ACR/ARHP Annual Meeting. Abstract Number: 1971.
Bell SE, Vigorito E, McAdams S, et al. PLCgamma2 regulates Bcl-2 levels and is required for survival rather than differentiation of marginal zone and follicular B cells. Eur J Immunol. 2004;34(8):2237–47.
Wang D, Feng J, Marine JC, et al. Phospholipase Cgamma2 is essential in the functions of B cell and several Fc receptors. Immunity. 2000;13(1):25–35.
Hikida M, Casola S, Takahashi N, et al. PLC-gamma2 is essential for formation and maintenance of memory B cells. J Exp Med. 2009;206(3):681–9.
Rowland SL, DePersis CL, Torres RM, Pelanda R. Ras activation of Erk restores impaired tonic BCR signaling and rescues immature B cell differentiation. J Exp Med. 2010;207(3):607–21.
Acknowledgements
We would like to acknowledge Dr. Jacob Bleesing at Cincinnati Children’s Hospital for providing lymphocyte phenotyping data for patients 1 and 2.
Funding
This work was supported by the Rare Disease Foundation and Canadian Child Health Clinician Scientist Program.
Author information
Authors and Affiliations
Contributions
All authors contributed substantially to this work and there are no conflicts of interest to disclose.
Corresponding author
Ethics declarations
Written informed consent for genetic testing and participation for all three patients was provided by the parent and/or for their children in accordance with the Declaration of Helsinki. Blood was collected from healthy pediatric sex-matched donors after consent at BC Children’s Hospital or through the BC Children’s Hospital BioBank. This research study was approved by the University of British Columbia institutional review board.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
A hypermorphic missense mutation in the C2 domain of PLCG2 is associated with pediatric CVID
Rights and permissions
About this article

Cite this article
Novice, T., Kariminia, A., Del Bel, K.L. et al. A Germline Mutation in the C2 Domain of PLCγ2 Associated with Gain-of-Function Expands the Phenotype for PLCG2-Related Diseases. J Clin Immunol 40, 267–276 (2020). https://doi.org/10.1007/s10875-019-00731-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-019-00731-3