
Abstract
[Fe(TPA)(N3)2](ClO4) and [Fe(TPA)Br2](ClO4), where TPA is tris-(2-pyridylmethyl)amine, crystallize in the monoclinic space group P21/c with a = 8.7029(5) Å, b = 19.168(1) Å, c = 13.5728(7) Å, β = 101.472(3)°, and a = 8.944(3) Å, b = 16.578(6) Å, c = 15.108(6) Å, β = 103.18(2)°, respectively. The structures were determined at 150 K from 3397 reflections (1426 observed) with R = 0.063 (Rw = 0.097), and at 115 K from 5617 reflections (2261 observed) with R = 0.057 (Rw = 0.065), respectively. In both cases, the iron is pseudo-octahedral with the two halide/pseudohalide ions cis. The Fe–X bond trans to the tertiary amine is shorter. The structures of [{Fe(TPA)X}2O](ClO4)2 where X = N3, Br, NCO, and two polymorphic forms of NCS, are also reported. The azide derivative [CH3CN solvate, monoclinic P21/n, a = 11.8038(11) Å, b = 22.547(2) Å, c = 17.344(2) Å, β = 106.972(4)°, determined at 100 K from 8972 reflections (4404 observed) with R = 0.087 (Rw = 0.145)] has two distinct Fe environments—the tertiary amine is cis to the oxido bridge at one site and is trans to the oxido bridge at the other site; the trans Fe–N3° distance is longer. Both the Br and NCO derivatives are monoclinic, C2/c [with a = 16.1480(17), b = 17.2036(13), c = 16.8521(12), β = 111.204(10), data collected at 293 K, 3753 reflections (2404 observed), R = 0.069 (Rw = 0.151), and a = 15.7470(9), b = 18.2270(11), c = 16.8950(8), β = 110.666(3), data collected at 90 K, 5392 reflections (3028 observed), R = 0.064 (Rw = 0.091), respectively]. Both polymorphs of the NCS derivative are monoclinic—one is P21/c and the other P21/n [a = 11.075(2), b = 15.436(2), c = 12.351(2), β = 95.528(7), data collected at 90 K, 5378 reflections (4345 observed), R = 0.068 (Rw = 0.198), and a = 12.396(2), b = 15.428(3), c = 44.505(8), β = 95.211(7), data collected at 110 K, 16,527 reflections (6540 observed), R = 0.069 (Rw = 0.105), respectively]. For the Br, NCO and NCS dimers, each iron of the [{Fe(TPA)X}2O]2+ unit is pseudo-octahedral with the halide/pseudohalide and oxide ions cis. The oxide bridge is linear, and the two halides/pseudohalides are anti. The ranking of trans influence of the ligands is O2− ≫ Br− > Cl− > N3− > NCO− ≥ NCS− > pyridyl > tertiary amine and the ranking of cis influence of the ligands is O2− ≫ N3− > NCO− > Cl− ≥ Br− > NCS−.
Graphical Abstract
The X-ray structures of two monomeric [Fe(TPA)(X)2](ClO4), where TPA is tris-(2-pyridylmethyl)amine and X = N3, and Br, and four dimeric [{Fe(TPA)Y}2O](ClO4)2, where Y =N3, Br, NCO, and NCS are presented and discussed.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tris-(2-pyridylmethyl)amine (TPA), also known as tris-(2-picolyl)amine, is a widely used tripodal ligand [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25]. It simultaneously provides four nitrogen donors (three pyridyl nitrogen atoms and a tertiary amine nitrogen) to a single metal center, thus leaving one or two available coordination sites. Because of the relative control over the metal site, iron complexes of TPA, and various substituted forms of TPA, have been used to model the metal-binding site of various metalloproteins, and have been extensively explored as catalysts for alkane functionalization [11,12,13, 26,27,28]. Many of these alkane functionalization studies have focused on compounds of the form [Fe(TPA)X2]+ where X is a halide or pseudohalide [13, 29, 30]. Given that, for octahedral complexes, one of the additional ligand atoms will be trans to a pyridine nitrogen atom and the other will be trans to the tertiary amine nitrogen atom, structural investigations are important to fully assess the trans influence of these two moieties, and to assess the extent of possible cis influences as well.
Herein we report the syntheses and structures of two mononuclear complexes ([Fe(TPA)(N3)2](ClO4) (1) and [Fe(TPA)Br2](ClO4) (2)), and several oxido-bridged dinuclear complexes ([{Fe(TPA)(N3)}2O](ClO4)2 (3), [{Fe(TPA)Br}2O](ClO4)2 (4), [{Fe(TPA)(NCO)}2O](ClO4)2 (5), [{Fe(TPA)(NCS)}2O](ClO4)2 (2 crystalline forms 6 and 7). While these studies originated in the 1990s at Duquesne University, they were only reported in three M.S. theses at that time [31,32,33]. The contact author moved from Duquesne University to the University of Louisiana at Monroe in 1997. While it might have been possible to scan the original data collection paper output and using optical character recognition software, recreate a digital version of the data collection, we decided instead to re-grow the crystals, re-collect the data, re-solve the structures, and re-refine them, as we had done for [Fe(TPA)Cl2](ClO4) and [{Fe(TPA)Cl}2O](ClO4)2 [34].
Experimental
All materials were obtained commercially and used without further purification. TPA·3HClO4 was synthesized according to literature procedures [1, 35], and used without further purification.
Synthesis
The synthesis of many of these compounds have already been reported in the literature [13, 29, 30, 36]. The following are typical preparations in our lab.
[Fe(TPA)(N3)2](ClO4) (1). A mixture of 0.2957 g of TPA·3HClO4 (0.5000 mmol) and 69.5 µL (0.500 mmol) of triethylamine in 20 mL methanol was stirred to dissolve all of the ligand. A solution of Fe(ClO4)3·10H2O (0.2670 g, 0.5000 mmol) in 2 mL methanol was added, followed by addition of 0.0650 g of NaN3 (1.00 mmol) resulting in a red–brown solution. The solution was allowed to stand at room temperature for 2 days during which time red–brown, rectangular prismatic crystals formed, which were suitable for X-ray crystallography. UV/vis: 310 nm (ε = 14.4 mM−1 cm−1), 400 nm (ε = 5.8 mM−1 cm−1), 500 nm (sh) 1H NMR (ppm) 298, 224, 144, 135, 118, 102.
[Fe(TPA)Br2](ClO4) (2). A mixture of 0.1480 g of TPA·3HClO4 (0.2500 mmol) and 35.0 µL (0.251 mmol) of triethylamine in 20 mL methanol was warmed (to ~ 60 °C) and stirred to dissolve all of the ligand. After the solution cooled to room temperature, 0.0605 g of NaBr (0.500 mmol) and 0.1336 g of Fe(ClO4)3·10H2O (0.2500 mmol) were added resulting in a dark red solution. A white powdery precipitate formed in a few minutes. After stirring for 30 min, the mixture was filtered, and the red filtrate was allowed to stand at room temperature overnight during which time dark red, needle-like crystals formed, which were suitable for X-ray crystallography. UV/vis: 282 nm (sh), 318 nm (ε = 7.0 mM−1 cm−1), 395 nm (ε = 4.1 mM−1 cm−1) 1H NMR (ppm) 170, 144, 115, 112, 98.
[{Fe(TPA)(N3)}2O](ClO4)2 (3). A mixture of 0.5945 g of TPA·3HClO4 (1.010 mmol) and 700 µL (5.00 mmol) of triethylamine in 40 mL methanol was stirred to dissolve all of the ligand. A solution of Fe(ClO4)3·10H2O (0.5427 g, 1.020 mmol) in 4 mL methanol was added, followed by addition of 0.0758 g of NaN3 (1.17 mmol) resulting in a red–brown mixture. This mixture was allowed to stand at room temperature for 2 days during which time small red crystals formed which proved to be unsuitable for X-ray crystallography. The solids were dissolved in acetonitrile, and the solution was placed in a desiccator containing ethyl acetate. After 2 days orange needle-shaped crystals formed which were suitable for X-ray crystallography and proved to be 3·CH3CN. UV/vis: 312 nm (sh), 430 nm (ε = 4.2 mM−1 cm−1) 1H NMR (ppm) 34, 31, 24.6, 23.2, 19.2, 17.9, 16.9, 16.5, 13.8, 12.6, 7.1, 6.4.
[{Fe(TPA)Br}2O](ClO4)2 (4). A mixture of 0.3908 g of TPA·HClO4 (1.000 mmol) and 280 µL (2.009 mmol) of triethylamine in 60 mL acetonitrile was warmed (to ~ 60 °C) and stirred to dissolve all of the ligand. After the solution cooled to room temperature, 0.5344 g of Fe(ClO4)3·10H2O (1.000 mmol) was added and dissolved, followed by 0.1030 g of NaBr (1.000 mmol) resulting in a dark red solution. This solution was allowed to stand at room temperature for 5 days (the volume reduced to ~ 40 mL), and 8 mL of methanol was added. This solution was allowed to stand at room temperature, and within 3 days red–brown crystals formed, which were suitable for X-ray crystallography. UV/vis: 320 nm (ε = 7.1 mM−1 cm−1), 382 nm (ε = 4.2 mM−1 cm−1), 444 nm (sh), 558 nm (sh) 1H NMR (ppm) 34, 29, 20.8, 18, 16.5, 15.8, 13.8, 12.8, 11.3 7.6, 7.1, 6.4.
[{Fe(TPA)(NCO)}2O](ClO4)2 (5). A mixture of 0.5925 g of TPA·3HClO4 (1.000 mmol) and 630 µL (4.50 mmol) of triethylamine in 40 mL methanol was stirred to dissolve all of the ligand. A solution of Fe(ClO4)3·10H2O (0.5345 g, 1.000 mmol) in 2 mL methanol was added, followed by addition of 0.0811 g of KOCN (1.00 mmol) resulting in a brown mixture. This mixture was stirred for 40 min, gravity filtered and allowed to stand at room temperature for several days during which time golden–brown plate-like crystals formed which were suitable for X-ray crystallography, and red crystals. The red crystals were either [{Fe(TPA)Cl}2O](ClO4)2 or [{Fe(TPA)(OCN)}2O](ClO4)2. UV/vis: 310 nm (ε = 10.5 mM−1 cm−1), 370 nm (ε = 7.8 mM−1 cm−1), 450 nm (sh), 500 nm (sh), 640 nm (ε = 0.14 mM−1 cm−1) 1H NMR (ppm) 34, 31, 24.8, 19, 17.8, 16.8, 16.1, 13.7, 12.3, 6.8, 6.2.
[{Fe(TPA)(NCS)}2O](ClO4)2 (6). A mixture of 0.1490 g of TPA·3HClO4 (0.2518 mmol), 160 µL (1.12 mmol) of triethylamine, and 0.0245 g NaSCN (0.302 mmol) in 30 mL methanol was stirred to dissolve all of the ligand. A solution of Fe(ClO4)3·10H2O (0.1408 g, 0.2635 mmol) in 2 mL methanol was added, resulting in an orange–red mixture, which was gravity filtered and the red filtrate was allowed to stand at room temperature for 3 days during which small red crystals formed. These were collected, dissolved in a minimal amount of acetonitrile, and the solution was placed in a desiccator containing ethyl acetate. Red rectangular prisms formed over several days which were suitable for X-ray crystallography. UV/vis: 316 nm (ε = 12.8 mM−1 cm−1), 378 nm (ε = 11.6 mM−1 cm−1), 424 nm (ε = 6.7 mM−1 cm−1) 1H NMR (ppm) 34, 31, 24, 20, 18, 17, 16, 14, 13, 12, 9, 8, 7, 4.
[{Fe(TPA)(NCS)}2O](ClO4)2 (7). In a procedure very similar to that used for 6 above, the red filtrate produced X-ray quality crystals.
All the perchlorate salts included in this work proved to be relatively stable for small scale routine synthesis and purification procedures. However, caution should be observed because perchlorate salts of metal complexes with organic ligands are potentially explosive.
X-Ray Structure Determinations
Some details of the crystal and data collections are collected in Table 1. X-ray diffraction data were collected primarily at Louisiana State University on a Nonius KappaCCD diffractometer using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) and an Oxford Cryostream cryostat. Data were collected with ω and ϕ scans. Data reduction included corrections for background, Lorentz, polarization and absorption effects [by the multi-scan method (HKL Scalepack)] [37]. Most structures were solved using direct methods [38], expanded using Fourier techniques [39], and calculations were performed using the teXsan for Windows [40] crystallographic software package. Data for 4 were collected at Sam Houston State University on a Rigaku XtaLAB Mini diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å) with ω scans. Using Olex2 [41], the structure was solved with ShelXT using intrinsic phasing [42] and refined with ShelXL [43]. Full-matrix least-squares refinement with anisotropic thermal parameters for all the non-hydrogen atoms converged. The function minimized in refinement was ∑w(Fo2 − Fc2)2 where w = 1/[σ2(Fo) + (p)2Fo2/4]. The hydrogen atoms were placed in idealized positions (C–H 0.95 Å), with Uiso = 1.2 Ueq of the attached atom. Neutral atom scattering factors were taken from Cromer and Waber [44]. Anomalous dispersion effects were included in Fc [45], and the values for ∆f′ and ∆f″ were those of Creagh and McAuley [46]. The values for the mass attenuation coefficients were those of Creagh and Hubbell [47].
For 4 and 5 the bridging O atom is located on a twofold axis, and for 6 the bridging oxide is located on an inversion center. For 7, there are multiple diiron dimers; in two cases the bridging O atom is located on an inversion center, and there is no crystallographically imposed symmetry for the other dimer. For 1, 2 and 3, there is no crystallographically imposed symmetry. For 1, the perchlorate is disordered about the Cl1-O1 axis and was modelled with two sets of oxygen atoms with occupancies of 0.447 and 0.553. For 3, one of the perchlorate ions is disordered about the Cl2-O21 axis and was modelled with two sets of oxygen atoms with half occupancy. Selected relevant bond distances and angles can be found in Table 2. Tables of positional and thermal parameters for all reported compounds can be found in the Supplementary Material.
Spectroscopy
Electronic spectra were recorded either on a HP 8452A (a single beam, diode-array spectrophotometer) at Duquesne University, a Shimadzu UV1601-PC at the University of Louisiana at Monroe (also a single beam, diode-array spectrophotometer), or a Jasco V-550 (a double-beam spectrophotometer) at Sam Houston State University. HPLC grade acetonitrile (99.9%) and 1 cm path length cuvettes were used.
NMR spectra were recorded at ambient temperature on a Bruker ACP300 at Duquesne University or a JEOL Eclipse + 300 FT-NMR at Sam Houston State University. Both were operating at 300 MHz and 5 mm tubes were used. The acquisition conditions were 5.0 µsec pulses, 8192 data points, a scan width of 100,000 Hz, 0.0 relaxation delay, acquisition time 0.041 s in the former case, and 5.4 µsec, 8192 data points, 1 s relaxation time, x-offset 100 ppm, and x-sweep 200 ppm in the latter case. The number of scans ranged from 20,000 to 100,000. CD3CN (99.9%) was used as solvent and the residual proton signal of the solvent was assigned a chemical shift δ value of 1.94 ppm.
Results and Discussion
Synthesis and Spectroscopy
The units [Fe(TPA)X2]+ and [{Fe(TPA)X}2O]2+ where X is a halide or pseudohalide, are appealing targets for catalysts for alkane hydroxylation and for structural analysis. The mononuclear systems must either be prepared under relatively acidic conditions, or under rigorously dry conditions given the high affinity of Fe(III) for oxide. More often than not, dinuclear oxido-bridged products are obtained. Paramagnetically shifted NMR spectroscopy is an extremely useful tool for understanding these Fe(III) compounds in solution [13]. Spectra for 1, 2, and 4 have been reported and assigned previously [13]. For mononuclear complexes, signals are observed with chemical shifts in the 200–100 ppm range, while dinuclear compounds exhibit signals with chemical shifts in the 50–10 ppm range due to the strong antiferromagnetic coupling mediated by the bridging oxide [13]. For the dinuclear systems, the intensity pattern of the signals in the 20–10 ppm range indicates if the TPA ligands on the two Fe(III) centers are symmetric or are asymmetric [11, 12]. An asymmetric arrangement refers to having the tertiary amine nitrogen on one Fe(III) center cis to the oxido-bridge while the other tertiary amine nitrogen on the other Fe(III) center is trans to the oxido-bridge. A symmetric arrangement refers to having the tertiary amine nitrogens with the same relative position on both Fe(III) centers (cis to the oxido-bridge), which produces a pattern of intensities of 4:4:2:2. Electronic spectroscopy is also quite useful for the study of the solution structure of the oxido-bridged dinuclear complexes. The absorption band in the 550–700 nm range is sensitive to the Fe–O–Fe angle. This band appears as a shoulder near 550 nm for linear bridges (the compounds are red), and red-shifts as the Fe–O–Fe unit bends.
X-Ray Structures
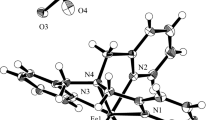
The structure of [Fe(TPA)(N3)2](ClO4) (1) is shown in Fig. 1 and selected relevant bond distances and angles can be found in Table 2. The complex cation has an octahedral Fe(III) center coordinated to four nitrogen atoms of TPA and two cis nitrogen atoms of azide, and is remarkably similar to [Fe(TPA)Cl2](ClO4). Two of the pyridyls are trans. One of the azides is trans to the tertiary amine and the other is trans to pyridine. The Fe(III)–Naz of the former is 1.932(6) Å and the latter is 1.959(7) Å, indicating that pyridine has a stronger trans influence than the tertiary amine. The Fe(III)–Npy distance trans to azide [2.174(6) Å] is long compared to the two Fe(III)–Npy distances that are mutually trans [2.142(6) and 2.119(5) Å]. The Fe(III)–N3° distance [2.197(6) Å] is the longest of the Fe–N distances in 1.

Thermal ellipsoid plot of [Fe(TPA)(N3)2]ClO4 (1) with selected atom labels
The structure of dark red [Fe(TPA)Br2](ClO4) (2) is shown in Fig. 2 (selected relevant bond distances and angles can be found in Table 2), and exhibits the same general pattern as 1 and as [Fe(TPA)Cl2](ClO4): two of the pyridyls are trans, one of the bromides is trans to the tertiary amine and the other is trans to pyridine. This latter bromide exhibits the longer of the two Fe(III)–Br distances [2.449(1) Å vs. 2.389(1) Å] again indicating that pyridine has a stronger trans influence than the tertiary amine. The corresponding Fe(III)–Npy distance is longer [2.222(6) Å] than the two Fe(III)–Npy distances that are mutually trans [2.133(5) and 2.121(5) Å]. While the Fe–N3° distance would appear to be shorter than the Fe–Npy trans to bromide, the distances are not distinguishable at the 3σ limit [2.207(5) vs. 2.222(6) Å]. The Fe–N3° distance in 2 [2.207(5) Å] is somewhat longer than the analogous distance in 1 [2.197(6) Å] suggesting that azide exhibits a weaker trans influence than bromide.

Thermal ellipsoid plot of [Fe(TPA)Br2]ClO4 (2) with selected atom labels
The Fe(II) compound analogous to 2 ([Fe(TPA)Br2]) has been structurally characterized [48], and the general pattern of distances and angles are observed—the Fe–Br distance trans to the tertiary amine is shorter as are the mutually trans Fe-pyridyl distances, and the Npy–Fe–Npy angle is significantly less than 180°. What is remarkable is that all of the iron-ligand distances are significantly shorter in 2 compared to the corresponding distances in the Fe(II) compounds. For example, the mutually trans Fe-pyridyl distances of 2 (2.127 Å) as compared to the Fe(II) compounds (2.188 Å), or the shorter Fe–Br distance (2.389 Å vs 2.5342 Å). We reported the same pattern comparing [Fe(TPA)Cl2]+ and [Fe(TPA)Cl2] [34]. In the chloride case, the average Fe(III) ligand bond distance was reported to be 2.196 Å and the average Fe(II) ligand bond distance was reported to be 2.283 Å, a difference of 0.087 Å. In the current bromide cases, the average Fe(III) ligand bond distance is 2.254 Å and the average Fe(II) ligand bond distance is 2.337 Å, a difference of 0.083 Å. Both differences are considerably smaller than the difference of 0.135 Å in effective ionic radii. As we noted previously, since the effective ionic radii values of Shannon [49] are based on coordination primarily by oxygen [seven oxygen structures and one fluorine structure for Fe(III), and five strictly oxygen structures and one structure with F/OH disorder for Fe(II)], one might assume that π donation from the ligands is included in the effective ionic radius, so the difference in size is due to two factors: the increase in π antibonding electron count and the difference in charge on the metal center (the greater the charge, the smaller the radius). However, the nitrogen atoms of TPA are not π donors, thus the difference in size would appear to be primarily due to the difference in charge.
If we compare the average iron to ligand distance in 1 and 2, naturally 2 is larger due to the presence of two Fe–Br bonds (2.253 Å vs 2.087 Å, respectively). If we confine ourselves to comparison of the Fe-TPA distances, the average for 2 is 2.171 Å, while it is 2.158 Å for 1. The difference is entirely due to the greater trans influence of Br versus N3. Excluding the Fe–N value for the pyridine trans to the halide/pseudohalide, the average Fe-TPA distance for 2 is 2.154 Å and for 1 it is 2.153 Å. For comparison, the analogous distances for [Fe(TPA)Cl2](ClO4) are 2.201 Å for the overall coordination sphere, 2.168 Å for the average Fe-TPA distances, and 2.156 Å excluding the Fe–N value for the pyridine trans to Cl [34].
Figure 3 shows the cationic portion of [{Fe(TPA)(N3)}2O](ClO4)2·CH3CN (3·CH3CN), which consists of an oxido-bridged diiron(III) core with the tertiary amine of TPA cis to the oxido bridge on Fe1 and the tertiary amine of TPA trans to the oxido bridge on Fe2, thus an asymmetric dinuclear center. Selected relevant bond distances and angles can be found in Table 2. The Fe–O distance to Fe1 is longer than the Fe–O distance to Fe2 [1.807(4) Å vs. 1.788(4) Å] reflecting the stronger trans influence of pyridine as compared to the tertiary amine. Each iron is octahedral (four nitrogen atoms from TPA, the oxygen atom of the oxido-bridge, and a nitrogen atom from azide). The azides on each iron center are cis to the oxido-bridge, so consequently the azide on Fe1 is trans to the tertiary amine and the azide on Fe2 is trans to pyridine. The Fe–Naz distance on Fe1 [1.969(5) Å] is considerably shorter than the distance on Fe2 [2.023(5) Å] once again reflecting the stronger trans influence of pyridine as compared to the tertiary amine. Each of these distances are significantly longer the average Fe–Naz distance in 1 (1.946 Å); further discussed below. As expected, the bonds trans to oxygen [the tertiary amine at 2.264(5) Å and the pyridine at 2.230(5) Å] are elongated, reflecting the strong trans influence of oxide—the two Fe–N3° distances are 2.264(5) Å and 2.232(5) Å, and the average of the four Fe–Npy distances which are trans to pyridine is 2.166 Å. The Fe–O–Fe angle is 173.9(2)°, a significant departure from 180°. The structure of [{Fe(TPA)(N3)}2O](ClO4)2 (not a solvate) has been reported previously [36] and exhibits the same rough structure (the asymmetric arrangement) and pattern of distances. These are included in Table 2 for comparison.

Thermal ellipsoid plot of the [{Fe(TPA)(N3)}2O]2+ unit of [{Fe(TPA)(N3)}2O](ClO4)2·CH3CN (3) with selected atom labels, and hydrogen atoms omitted
The Fe–Naz distances for both 1 and 3 have shorter distances trans to the tertiary amine and longer distances trans to pyridine indicative that pyridine has a stronger trans influence than the tertiary amine. Further, the distances in 3 are longer than the corresponding distances in 1. The major influence is the presence of the oxido-bridge which clearly results in longer Fe–Naz distances [1.969(5) Å for 3 vs 1.932(6) Å for 1 for Fe–Naz trans to the tertiary amine and 2.023(5) Å for 3 vs 1.959(7) Å for 1 for Fe–Naz trans to pyridine]. The strong π donation of oxide to the Fe centers in 3 increases the electron density on Fe which results in longer Fe–Naz bonds, suggesting that the azide is less able to participate in π donation to Fe. This is a cis effect. Except for the shortest Fe–Naz distance in 1, the Fe–Naz distances are typical [36, 50,51,52,53,54,55,56,57,58]. Typically, the two N–N distances of azide are distinctly different for metal complexes with terminal (i.e. non-bridging) azides, and normally the coordinated N has the longer N–N distance. This is not what is observed for the azide trans to pyridine in 1—the distances are indistinguishable at the 3σ level [1.079(9) Å vs 1.094(11) Å] but the shorter distance is to the bound N. The Fe–N–N angles for 1 and 3 are typical, ranging from 132.2° to 134.0° [36, 50,51,52,53,54,55,56,57,58].
Figure 4 shows the cationic portion of [{Fe(TPA)Br}2O](ClO4)2·H2O (4·H2O), and exhibits the same general pattern as the various polymorphs of [{Fe(TPA)Cl}2O](ClO4)2 [13,14,15, 34]: a symmetric oxido-bridged diiron(III) core with the tertiary amines cis to the oxido-bridge. There are two hydrogen bonds between H2O, ClO4−, and a symmetry related ClO4−. Selected relevant bond distances and angles can be found in Table 2. The Fe centers are six-coordinate, similar to Fe1 in 3. The tertiary amines (and the bromides which are trans to the tertiary amines) on each Fe are anti. The bridging oxide sits on a twofold axis. Coincidentally, one of the polymorphs of [{Fe(TPA)Cl}2O](ClO4)2 has similar cell parameters, the same space group, and shows a similar pattern of distances and angles [14]. The Fe–O–Fe angle of 174.6(5)° is significantly different from linearity. The Fe–O distance of 1.7844(11) Å is quite short, indicating a strong bond. The Fe–Npy distance trans to oxygen is quite long 2.237(7) Å indicative of the strong trans influence of oxygen, and this is the longest Fe–N distance in the structure. The Fe–N3° distance is the second longest Fe–N in the coordination sphere of 4 at 2.220(6) Å. The average of the Fe–Npy distances is 2.144 Å. The Fe–Br distance of 2.4477(13) Å is longer than the average of the Fe–Br distances in 2 (2.419 Å), but quite similar to the Fe–Br distance in 2 for the bromide trans to pyridine [2.449(1) Å]. The short Fe–Br distance in 2 [2.389(1) Å] is the shortest reported Fe–Br distance among six-coordinate Fe(III) centers with pyridyl and amine donors [59,60,61]. The longest Fe–Br distances among this group are found in an anion with trans bromides [61] (average Fe–Br 2.5316 Å). The remaining Fe–Br distances in this group range from 2.4046(8) to 2.4525(7) Å, with an average of 2.438 Å.

Thermal ellipsoid plot of the [{Fe(TPA)Br}2O]2+ unit of [{Fe(TPA)Br}2O](ClO4)2·H2O (4) with selected atom labels
If we compare the Fe–Br distance in 4 [2.4477(13) Å] to the analogous (trans to the tertiary amine) distance in 2 [2.389(1) Å], and the Fe–Naz distance trans to the tertiary amine in 3 [1.969(5) Å] and the analogous Fe–Naz distance in 1 [1.932(6) Å] we see a significant lengthening of the distances for the oxido-bridged structures. We suggest that this is due to the strong multiple bond nature of the Fe–O bond. Oxide is a strong π donor, and the increased π electron density on Fe weakens the ability of bromide to serve as a π donor, resulting in a longer and weaker bond. This is a cis effect.
If we compare the average iron to ligand distance in 4 and the Fe1 site of 3 (which is structurally similar to 4) as we did with the monomers 1 and 2, naturally 4 is larger due to the presence of the Fe–Br bond (2.160 Å vs 2.096 Å, respectively). If we confine ourselves to comparison of the Fe-TPA distances, the average for 4 is 2.186 Å, while it is 2.199 Å for the Fe1 site of 3. This is reversed compared to 1 and 2, and we suggest that this difference is due to the inability of the bromide to donate in a π sense as discussed above. Excluding the Fe–N value for the pyridine trans to oxide, the average Fe-TPA distance for 4 is 2.169 Å and for the Fe1 site of 3 it is 2.191 Å. These average distances are both longer than those observed for 1 and 2. This is a cis effect of oxide. Another way to state this is that the Fe centers in 1 and 2 are more Lewis acidic than the Fe centers in 3 and 4.
Figure 5 shows the asymmetric unit of [{Fe(TPA)(NCO)}2O](ClO4)2·2CH3OH (5·2CH3OH), while Fig. 6 shows the complete formula unit. Selected relevant bond distances and angles can be found in Table 2. There is a hydrogen bond between CH3OH and ClO4−. The dimer is symmetric with the bridging oxide sitting on a twofold axis. The Fe–O–Fe angle is 169.0(2)°, significantly deviating from linearity. The Fe(III) center is octahedral with the tertiary amine cis to the oxido-bridge and a pyridine ring trans to the oxido-bridge. The Fe–Npy distance of the pyridine trans to oxide is the longest metal–ligand bond in the structure [2.231(3) Å], indicative of the strong trans influence of oxide. The Fe–O distance is typically short [1.7925(6) Å], and the Fe–N3° is typically long [2.219(3) Å] as compared to the Fe–Npy distances of the mutually trans pyridine rings (the average value is 2.152 Å). The isocyanate is trans to the tertiary amine nitrogen and the Fe–N distance of the isocyanate is 1.993(3) Å, which is typical [55,56,57, 62], as are the distances within the isocyanate and the Fe–N–C angle.

Thermal ellipsoid plot of the asymmetric unit of [{Fe(TPA)(NCO)}2O](ClO4)2·2CH3OH (5) with selected atom labels

Thermal ellipsoid plot of [{Fe(TPA)(NCO)}2O](ClO4)2·2CH3OH (5)
Figure 7 shows the cationic portion of [{Fe(TPA)(NCS)}2O](ClO4)2 (6), and selected relevant bond distances and angles can be found in Table 2. As with 5, the dimer is symmetric with the bridging oxide sitting on a special position, but, in this instance, it is a center of symmetry rather than a twofold axis, thus the Fe–O–Fe angle is symmetrically required to be 180°. The Fe(III) center is octahedral with the tertiary amine cis to the oxido-bridge and a pyridine is trans. The Fe–Npy distance of the pyridine trans to the oxide is the longest metal–ligand bond in the structure [2.213(2) Å], again indicative of the strong trans influence of oxide. The Fe–O distance is typically short [1.7921(4) Å], and the Fe–N3° is typically long [2.208(3) Å] as compared to the Fe–Npy distances of the mutually trans pyridine rings (the average value is 2.146 Å). The isothiocyanate is trans to the tertiary amine nitrogen and the Fe–N distance of the isothiocyanate is 2.046(3) Å.

Thermal ellipsoid plot of the [{Fe(TPA)(NCS)}2O]2+ unit of [{Fe(TPA)(NCS)}2O](ClO4)2 (6) with selected atom labels
These details are again observed for 7, as pictured in Fig. 8 which shows the independent atoms which include one complete dimeric unit with no imposed symmetry, and two centrosymmetric units (as for 6). Closer views of the centrosymmetric units are shown in Figs. 9 and 10, while Fig. 11 shows the dimeric unit with no imposed symmetry. The details for these units are all similar to 6. Selected relevant bond distances and angles can be found in Table 2. All contain octahedral Fe(III) centers with bridging oxides. For the unit shown in Fig. 11, the Fe–O–Fe angle is 177.6(3)°. For the other centers, the angle is required to be 180°. In each case, the tertiary amine is cis to the oxido-bridge and a pyridine is trans to the oxido-bridge. The Fe–Npy distances of the pyridines trans to the oxide are the longest metal–ligand bonds for each iron center [the average distance is 2.212(2) Å], again indicative of the strong trans influence of oxide. The Fe–O distances are typically short [the average distance is 1.795(3) Å], and the Fe–N3° is typically long [the average distance is 2.210(5) Å] as compared to the Fe–Npy distances of the mutually trans pyridine rings [the average value is 2.142(3) Å]. The isothiocyanate is trans to the tertiary amine nitrogen and the average Fe–N distance of the isocyanate is 2.011(7) Å.

Thermal ellipsoid plot of the three unique units of [{Fe(TPA)(NCS)}2O](ClO4)2 (7)

Thermal ellipsoid plot of the asymmetric unit of one of the centrosymmetric units of [{Fe(TPA)(NCS)}2O]2+ of (7) with selected atom labels

Thermal ellipsoid plot of the asymmetric unit of the other centrosymmetric unit of [{Fe(TPA)(NCS)}2O]2+ of (7) with selected atom labels

Thermal ellipsoid plot of the asymmetric unit of the diiron center without imposed symmetry of [{Fe(TPA)(NCS)}2O]2+ of (7) with selected atom labels
Generally, isothiocyanates coordinated to Fe(III) bind in a bent fashion (at least in cases where the other donors includes aliphatic and aromatic amines), and 6 and 7 are no exception with an average Fe–N–C angle of 163.1° and a range of 157.0° to 169.2° [56,57,58, 63, 64]. The C–S bonds for 6 and 7 are shorter than what is normally observed (1.602 Å vs 1.629 Å) while the N–C distances are longer by roughly the same amount (1.162 Å vs 1.141 Å).
The [{Fe(TPA)(NCS)}2O]2+ unit is similar to [{Cr(TPA)(NCS)}2O]2+ [65] with the same relative arrangement of atoms (the oxide sits on an inversion center for the Cr(III) complex). The M–O distance is shorter for Fe(III) (the Fe–O average is 1.795 Å for 5 instances and the Cr–O distance is 1.800 Å) and the M–Ntrans distance is longer for Fe(III) (the Fe–Ntrans average is 2.212 Å for 5 instances and the Cr–Ntrans is 2.121 Å) suggesting a stronger trans influence for oxide for Fe(III) as compared to Cr(III). While octahedral high spin Fe(III) has a larger effective ionic radius than octahedral Cr(III) (0.645 Å vs. 0.615 Å, respectively, a difference of 0.030 Å) [49], the average metal ligand bond distance for all of the [Fe(TPA)(NCS)}2O]2+ units (2.087 Å) is significantly longer than the corresponding [Cr(TPA)(NCS)}2O]2+ unit (2.024 Å), a difference of 0.063 Å. As mentioned above, the effective ionic radii values of Shannon are based on coordination primarily by oxygen—mostly oxygen for Fe(III) (seven oxygen structures and one fluorine structure) and two fluoride structures, one oxide structure, one hydroxide structure and one acetylacetonate structure for Cr(III), so one might assume that π donation from the ligands is included in the effective ionic radius. This implies that the longer Fe–N distances are σ effects, reflecting the two σ antibonding electrons in Fe(III) as compared to Cr(III) which is d3.
For the five independent examples of [{Fe(TPA)(NCS)}2O]2+, the average Fe–O distance is 1.795(3) Å, the average Fe–N3° distance is 2.210(2) Å, the average Fe–Npy distance trans to the oxide is 2.212(2) Å, and the average Fe–Npy distances of the mutually trans pyridine rings is 2.142(3) Å. For four independent examples of [{Fe(TPA)Cl}2O]2+, the average Fe–O distance is 1.788(2) Å, the average Fe–N3° distance is 2.220(6) Å, the average Fe–Npy distance trans to the oxide is 2.250(14) Å, and the average Fe–Npy distances of the mutually trans pyridine rings is 2.145(4) Å. It is important to point out that these two units (and all of the oxido-bridged units described in this paper except 3) contain an essentially linear oxido-bridge with a pyridine group trans to the bridge, two mutually trans pyridine groups, a tertiary amine cis to the oxido-bridge, and a halide or pseudohalide trans to the tertiary amine. Also, the orientations of the tertiary amine nitrogen atoms on either side of the oxido-bridge are anti. Thus, the only significant change is in the identity of the halide or pseudohalide. The biggest impact of this change is on the Fe–N3° distance. Since the diiron unit is asymmetric for 3, the opportunity to compare the trans influence of oxide and azide is provided. The Fe–N3° distance trans to oxide is 2.264(5) Å and trans to azide is 2.232(5) Å, demonstrating that O2− > N3− in trans influence. The corresponding distances (Fe–N3°) are 2.219(3) Å and 2.210(2) Å for NCO− and NCS−, respectively. As mentioned above, the Fe–N3° distance is 2.220(6) Å when Cl− is trans. What remains is to rank Br−. This can be done by comparing the Fe–N3° distances for 1 & 2 [2.197(6) Å and 2.207(5) Å, respectively], as well as comparing the Fe–Npy distances of the trans pyridine [2.174(6) Å and 2.222(6) Å, respectively], clearly demonstrating that Br− has a stronger trans influence than N3−. For [Fe(TPA)X2]+, the Fe–Npy distances of the trans pyridine when X = Br− is 2.222(6) Å and when X = Cl− is 2.204(2) Å [34], clearly demonstrating that Br− has a stronger trans influence than Cl−. Analysis of the Fe–N3° distances for these compounds implies that Br− has a stronger trans influence, but the data is less compelling [2.207(5) Å for Br− and 2.204(2) Å for Cl−], falling short of a 3σ difference. Altogether, comparison of the Fe–N3° distances, the Fe–Npy distances of the trans pyridine, and the Fe–X distances in the monomeric complexes allow the general ranking the trans influence of these ligands as O2− ≫ Br− > Cl− > N3− > NCO− ≥ NCS− > pyridyl > tertiary amine.
If we locally designate the Fe–O vector as the z axis, the Fe \({\text{d}}_{\text{z}^2}\) orbital is oriented for a σ interaction with the oxide (and with the pyridine trans to oxide). If we locally designate the Fe–N3° vector as the x axis, the Fe \({\text{d}}_{{\text{x}^2}-{\text{y}^2}}\) orbital is oriented for a σ interaction with the tertiary amine (and with the halide/pseudohalide). Further, the Fe dxz orbital is oriented for a π interaction with both the oxide px orbital and the halide/pseudohalide π symmetry orbital in the xz plane. Oxide is a good π donor, so this orientation is expected to synergistically enhance π acidity of the pseudohalide and suppress π basicity of the halide—these are cis influences. Evidence for cis influences can be observed by comparing mono-iron cases to diiron cases—so [Fe(TPA)(N3)2]+ to [{Fe(TPA)(N3)}2O]3+, [Fe(TPA)Br2]+ to [{Fe(TPA)Br}2O]3+, and [Fe(TPA)Cl2]+ to [{Fe(TPA)Cl}2O]3+—and examination of the bond trans to the tertiary amine. For the azide mono-iron case the distance is 1.932 Å and for the diiron case there is elongation to 1.969 Å, for the bromide mono-iron case the distance is 2.389 Å and for the diiron case there is elongation to 2.4477 Å, and for the chloride mono-iron case the distance is 2.2425 Å and for the diiron cases the corresponding distance is longer in every case (ranging from 2.296 to 2.3145 Å), thus the presence of the cis oxide results in a weakening of the bond to the halide/pseudohalide in all three cases. A similar lengthening of the tertiary amine distance (2.197 Å for the mono-iron azide, 2.232 Å for the diiron azide, 2.207 Å for the mono-iron bromide, 2.220 Å for the diiron bromide, 2.204 Å for the mono-iron chloride, 2.220 Å for the diiron chloride) is observed. If we rank the average Fe–Npy distances that are mutually trans for the oxido-bridged systems, we find 2.166(6) Å (N3) > 2.152(3) Å (NCO) > 2.145(4) Å (Cl) ≥ 2.144(7) Br > 2.142(3) Å (NCS), and if we do the same for 1 & 2 we find 2.130(5) Å (N3) > 2.127(5) Å (Br). For these series, the trans influences are the same (since these distances are all for mutually trans pyridine rings). Any observable differences must be cis influences; thus the cis influence ranking is O2− ≫ N3− > NCO− > Cl− ≥ Br− > NCS−.
References
Anderegg G, Wenk F (1967) Helv Chim Acta 50:2330–2332
Tajika Y, Tsuge K, Sasaki Y (2005). Dalton Trans. https://doi.org/10.1039/B414532A
Robertson NJ, Carney MJ, Halfen JA (2003) Inorg Chem 42:6876–6885
Hodgson DJ, Zietlow MH, Pederson E, Toftlund H (1988) Inorg Chim Acta 149:111–117
Gafford BG, O’Rear C, Zhang JH, O’Connor CJ, Holwerda RA (1988) Inorg Chem 28:1720–1726
Gultneh Y, Farooq A, Karlin KD, Liu S, Zubieta J (1993) Inorg Chim Acta 211:171–175
Towle DK, Botsford CA, Hodgson DJ (1988) Inorg Chim Acta 141:167–168
Kim M, Kim Y-U, Han J (2007) Polyhedron 26:4003–4008
Mandon D, Machkour A, Goetz S, Welter R (2002) Inorg Chem 41:5364–5372
Davies CJ, Solan GA, Fawcett J (2004) Polyhedron 23:3105–3114
Norman RE, Yan S, Que L Jr, Backes G, Ling L, Sanders-Loehr J, Zhang JH, O’Connor CJ (1990) J Am Chem Soc 112:1554–1562
Norman RE, Holz RC, Ménage S, O’Connor CJ, Zhang JH, Que L Jr. (1990) Inorg Chem 29:4629–4637
Kojima T, Leising RA, Yan S, Que L Jr. (1993) J Am Chem Soc 115:11328–11335
Hazell A, Jensen KB, McKenzie CJ, Toftlund H (1994) Inorg Chem 33:3127–3134
Seth SK, Mandal PC, Kar T, Mukhopadhyay S (2011) J Mol Struct 994:109–116
Eckenhoff WT, Biernesser AB, Pintauer T (2012) Inorg Chim Acta 382:84–95
Mandel JB, Maricondi C, Douglas BE (1988) Inorg Chem 27:2990–2996
Tong B, Norman RE, Chang S-C (1999) Acta Crystallogr Sect C Cryst Struct Commun C55:1236–1238
Tong B, Chang S-C, Carpenter EE, O’Connor CJ, Lay JO Jr., Norman RE (2000) Inorg Chim Acta 300–302:855–861
Jacobson RR, Tyeklár Z, Farooq A, Karlin KD, Zubieta J (1988) J Am Chem Soc 110:3690–3692
Tyeklár Z, Jacobson RR, Wei N, Murthy NN, Zubieta J, Karlin KD (1993) J Am Chem Soc 115:2677–2689
Oshio H, Ichida H (1995) J Phys Chem 99:3294–3302
Nobutoshi K, Hirotaka N, Yoshinori K, Gin-ya A, Masatatsu S, Akira U, Koji T (1995) Bull Chem Soc Jpn 68:581–589
Murthy NN, Karlin KD (1993). J Chem Soc Chem Commun. https://doi.org/10.1039/c39930001236
Adams H, Bailey NA, Fenton DE, He Q-Y (1995). J Chem Soc Dalton Trans. https://doi.org/10.1039/dt9950000697
Kal S, Xu S, Que L Jr (2020) Angew Chem Int Ed 59:7332–7349
Puri M, Que L Jr (2015) Acc Chem Res 48:2443–2452
He C, Mishina Y (2004) Curr Opin Chem Biol 8:201–208
Leising RA, Norman RE, Que L Jr (1990) Inorg Chem 29:2553–2555
Leising RA, Zang Y, Que L Jr (1991) J Am Chem Soc 113:8555–8557
Xue J (1996) M. S. Thesis, Duquesne University
Hangun Y (1996) M. S. Thesis, Duquesne University
Mullaney M (1996) M. S. Thesis, Duquesne University
Xue J, Xie M, Nadir S, Lewis JC, Jayaratna NB, Arachchilage HJ, Norman RE (2021) J Chem Cryst 51:483–490. https://doi.org/10.1007/s10870-020-00872-z
Gafford BG, Holwerda RA (1989) Inorg Chem 28:60–66
Shin JW, Rowthu SK, Lee JE, Lee HI, Min KS (2012) Polyhedron 33:25–32
Otwinowski Z, Minor W (1997) Methods Enzymol 276:307–326
Altomare A, Cascarano G, Giacovazzo C, Guagliardi A (1993) J Appl Crystallogr 27:343–350
Beurskens PT, Admiraal G, Beurskens G, Bosman WP, De Gelder R, Isreal R, Smith JMM (1994) Technical report of the Crystallography Laboratory. University of Nijmegen, Nijmegen
Molecular Structure Corporation (1997) TeXsan for windows: crystal structure analysis package. Molecular Structure Corporation, The Woodlands
Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H (2009) J Appl Crystallogr 42:339–341
Sheldrick GM (2015) Acta Crystallogr A 71:3–8
Sheldrick GM (2015) Acta Crystallogr C 71:3–8
Cromer DT, Waber JT (1974) International tables for X-ray crystallography. Birmingham, Kynoch
Ibers JA, Hamilton WC (1964) Acta Crystallogr 17:781–782
Creagh DC, McAuley WJ (1992). In: Wilson AJC (ed) International tables for X-ray crystallography. Kluwer, Boston, pp 219–222
Creagh DC, Hubbell JH (1992). In: Wilson AJC (ed) International tables for X-ray crystallography. Kluwer, Boston, pp 200–206
Rana S, Bag S, Patra T, Maiti D (2014) Adv Synth Catal 356:2453–2458
Shannon RD (1976) Acta Crystallogr Sect A A32:751–767
Dori Z, Ziolo RF (1973) Chem Rev 73:247–254
Wang S, Wang L, Wang X, Luo Q (1997) Inorg Chim Acta 254:71–77
Dutta SK, Beckmann U, Bill E, Weyhermüller T, Wieghardt K (2000) Inorg Chem 39:3355–3364
Beckmann U, Bill E, Weyhermüller T, Wieghardt K (2003) Inorg Chem 42:1045–1056
Shin JW, Rowthu SR, Hyun MY, Song YJ, Kim C, Kim BG, Min KS (2011) Dalton Trans 40:5762–5773
Pogány L, Moncol J, Pavlik J, Šalitroš I (2017) New J Chem 41:5904–5915
Krüger C, Augustín P, Nemec I, Trávníček Z, Oshio H, Boča R, Renz F (2013) Eur J Inorg Chem 2013:902–915
Natke D, Preiss A, Klimke S, Shiga T, Boca R, Ohba M, Oshio H, Renz F (2021) Eur J Inorg Chem 2021:1498–1504
Mizoguchi TJ, Lippard SJ (1997) Inorg Chem 36:4526–4533
Merkel M, Schnieders D, Baldeau SM, Krebs B (2004) Eur J Inorg Chem 2004:783–790
Merkel M, Pascaly M, Krebs B, Astner J, Foxon SP, Schindler S (2005) Inorg Chem 44:7582–7589
Afshar RK, Eroy-Reveles AA, Olmstead MM, Mascharak PK (2006) Inorg Chem 45:10347–10354
Nemec I, Herchel R, Boča R, Trávníček Z, Svoboda I, Fuess H, Linert W (2011) Dalton Trans 40:10090–10099
Setifi F, Golhen S, Ouahab L, Turner SS, Day P (2002) Cryst Eng Comm 4:1–6
Setifi F, Ota A, Ouahab L, Golhen S, Yamochi H, Saito G (2002) J Solid State Chem 168:450–456
Gafford BG, Holwerda RA, Schugar HJ, Potenza JA (1988) Inorg Chem 27:1126–1128
Acknowledgements
We thank Drs. Shih-Chi Chang and Frank Fronczek for their assistance and expertise with X-ray data collection. Financial support of the Louisiana Board of Regents Support Fund and the Welch Foundation (x-0011) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xue, J., Hangun-Balkir, Y., Mullaney, M. et al. Syntheses and Structures of [Fe(TPA)X2](ClO4) and [{Fe(TPA)Y}2O](ClO4)2 Where TPA = Tris-(2-pyridylmethyl)amine, X = N3, or Br, and Y = N3, Br, NCO, or NCS. J Chem Crystallogr 53, 50–65 (2023). https://doi.org/10.1007/s10870-022-00941-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-022-00941-5