
Abstract
The design and use of innovative treatment processes are very important in preventing the possible toxic effects of organic pollutants in aquatic environments. One of these methods is the subcritical water oxidation method, which has been used recently. In the current study, the mineralization of clofibric acid (CFA) was carried out under more effective and mild conditions using persulfate (PS) as an oxidant and CoFe2O4@SiO2 catalyst by the subcritical water oxidation (sub-CWO) process. Characterization of the synthesized catalyst was performed through XRD, FTIR, TEM and SEM–EDS analyses. In the CFA oxidation with persulfate-promoted catalytic Sub-CWO process, optimum working conditions was determined as 15 mM PS, 40 min, 383 K, and 0.3 g L−1 catalyst dosage using the response surface method and Box–Behnken design. The catalyst's efficiency remained relatively stable after three cycles under optimal conditions, resulting in a 97% total organic carbon (TOC) removal. Decomposition products were determined and a degradation mechanism was proposed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although pharmaceutical compounds can degrade in biotic or abiotic processes, they persist even at low concentrations since they are constantly infused into surface waters via sewage treatment plant (STP) effluents. Due to the purpose of its design being the biological effect, all living organisms in the environment that it will encounter will be targets for it [1]. Despite all the water treatment processes, partial oxidation of clofibric acid (CFA) can be achieved [2], even though this value is stated as 51% [3], so CFA maintains its presence in the environment as a considerable threat. It is known that clofibric acid is not easily degraded by microorganisms and maintains its environmental persistence for more than 21 years [4, 5]. In 2001, one of the pharmaceuticals followed in France, Greece, Italy, and Sweden STP effluents was clofibric acid. Clofibric acid was chosen because it is an herbicide as well as being a metabolite of the lipid regulator clofibrate, that is, its widespread use. As a result, CFA was detected in the range of 0.23–0.68 μg L−1 in the Italian and Sweden STP effluent samples [1]. On the other hand, Saravan et al. [6] determined the CFA concentrations for STP effluent and surface water in Swiss lakes and German rivers as 1.6 μg L−1 and 0.551 μg L−1, respectively. The same study reported that the CFA concentrations determined for the North Sea, Greece, and the USA reached the values of 1 ng L−1, 5 ng L−1, and 0.8–2 μg L−1 on surface waters, respectively. In China, the CFA concentrations detected in Yellow River, Haihe River, and Dongjiang River by Wang et al. [7] were determined as 13.5 ng L−1, 46.4 ng L−1, and 50.6 ng L−1, respectively. The most striking result is determining the CFA concentration detected in drinking water as 270 ng L−1 [8].
Many new materials with large surface areas have been developed and tested for adsorption, some of which have the following CFA adsorption capacity: 994 mg g−1 with graphene nano-sheets [9]; 568 mg g−1 with powdered-active carbon [10]; 540 mg g−1 with bio-metal organic frameworks (MOF) derived carbon [11]. However, in the adsorption process, the contaminant is taken from the aqueous medium to the solid phase and could be recovered by enriching using a chemical eluent [12], and the adsorbent regeneration is needed. In this case, a new method will be needed to remove the organic pollutant. The failure of effective oxidation of pollutants such as CFA with STP has led to more effective advanced oxidation methods (AOPs) for detoxifying and purifying of wastewater.
The common denominator of advanced oxidation methods is the in situ generation of non-selective strong oxidizing radicals like hydroxyl radicals (•OH, Eo = 2.80 V/SCE) and/or sulfate radical (SO4•−, Eo = 2.5–3.1 V/SCE) that degrade resistant organic pollutants during these processes [13,14,15]. The oxidation of CFA was carried out using various AOPs, such as electrochemical oxidation using Fe3+/peroxymonosulfate [16], anodic oxidation [17,18,19], ozonation [20] and photocatalysis [21].
Subcritical water oxidation (Sub-CWO) is promising for the effective and short residence time oxidation of pollutants that many conventional methods cannot effectively remove and even require lengthy processing times with some AOPs [22, 23]. Subcritical water (Sub-CW) is formed in the pressure and temperature range below the water's critical temperature (374 °C) and critical pressure (22.1 MPa). Thanks to the temperature and pressure conditions applied in the Sub-CWO process, water plays a dual role as a catalyst and reaction medium [24]. Torr et al. [25] defined Sub-CW properties in specific ranges, and these are temperature (250–350 °C), pressure (5–25 MPa), density (800–600 kg m−3), static dielectric constant (27.1–14.07 F m−1), ionic product (pKw) (11.2–12.0), dynamic viscosity (0.11–0.064 mPas).
Previous studies have reported that when subcritical water oxidation is carried out with oxidant and catalyst support, refractory pollutants will degrade more effectively, eco-friendly, and quickly under mild conditions [23, 26, 27]. When oxygen is used as an oxidant in Sub-CWO [28], it is also called wet air oxidation (WAO).
In the WAO process, organic pollutants are converted into low molecular weight and biodegradable intermediates. Using catalysts allows the oxidation of substances that are difficult to convert. For this reason, the activities of catalysts such as CoAlPO4-5 [29], Ce3Cu1 [30], H4SiW12O40 and Na2HPW12O40 [31], CeO2 [32], Pt/MWNT [33], Ni/MgAlO [34], Ru/CNT [26] have been reported recently in the catalytic Sub-CWO processes. Parvas et al. [35] examined the effect of Ni/CeO2-ZrO2 catalysts with different amounts of Ni on phenol oxidation in the WAO process at 160 °C for 3 h. It was reported that Ni absent catalyst (CeO2-ZrO2) provided 11.4% phenol removal, whereas this value increased to 28.5% with 15% (wt) Ni loading. In another study, Lai et al. [36] reported that a high concentration of phenol aqueous solution (2.1 g L−1) was completely decomposed by catalytic-WAO method using Cu3-Al-500 within 2 h under mild conditions (120 °C, 1 MPa). They showed that this success was due to the promotion of hydrogen peroxide formation from oxygen during the Cu2+/Cu+ redox transition on the catalyst's surface.
Recently, the effectiveness at lower temperatures of oxidants such as hydrogen peroxide [37,38,39], persulfate [40, 41], and periodate [40] has been demonstrated even at lower temperatures. Although heat activation of persulfate can be achieved in the 40–90 °C range, increasing the oxidation efficiency requires high persulfate concentrations [42, 43]. Apart from the heat, it is possible to perform persulfate activation using many methods (UV irradiation, H2O2, O3, ultrasound, and metal ions). Among these methods, transition metal-based heterogeneous catalysts are common and effective. In the persulfate-promoted Sub-CWO process, by providing persulfate activation with a combined system supported by heat and catalyst, it may be necessary to rise to a higher temperature (> 90 °C), but more effective oxidation is provided [44, 45].
The spinel-type ferrite family (MFe2O4) is an important group of catalysts in the activation of hydrogen peroxide, persulfate, and peroxymonosulfate in AOPs [46, 47]. In the spinel-type ferrite structure, while M ions (Co, Mn, Mg, etc.) occupy the tetrahedral region, Fe3+ ions are located in the octahedral region and are mainly responsible for the catalytic activity [48]. The most significant disadvantage of transition metal-based heterogeneous catalysts is the formation of secondary pollution due to metal ion leaching [49, 50]. Silica-coated ferrite nanoparticles have active sites that are more open to catalytic reactions thanks to their dispersibility increases [51]. In addition, it is promising in long-life catalyst applications because it provides good stability and durability as a silica support material [52].
In this study, the catalytic effect of the synthesized CoFe2O4@SiO2 catalyst synthesized by the sol–gel method was examined in the sub-CWO process using persulfate oxidant for the degradation of clofibric acid. Three-level Box–Behnken design (BBD) and response surface methodology were used to optimize temperature, catalyst dosage, and persulfate concentration values in the oxidation of clofibric acid. Based on the identified degradation products, a multi-step degradation mechanism was proposed.
Experimental
Materials
Clofibric acid (C10H11ClO3), Cobalt nitrate (Co(NO3)2⋅6H2O), ferric nitrate (Fe(NO3)3⋅9H2O), citric acid, tetraethyl orthosilicate (TEOS), ammonia solution (25%), and potassium persulfate (K2S2O8) were purchased from Merck.
Synthesis of cobalt ferrite (CoFe2O4) nanoparticles
The nanocatalyst CoFe2O4 was synthesized using the sol–gel method. In summary, Co(NO3)2⋅6H2O and Fe(NO3)3⋅9H2O salts were dissolved in ultrapure water and mixed so that the molar ratio of Co:Fe was 1:2. Then, a solution of citric acid was added dropwise to this prepared mixture in such a way that the molar ratio of metal ions to citric acid was 1:1. The temperature of the solution was adjusted to 70 °C and the solution was stirred for 1 h. At the end of this period, the water was evaporated without mixing the solution and the gel structure was calcined at 600 °C for 5 h. The black-colored CoFe2O4 nanocatalyst obtained after calcination was washed with ultrapure water and ethyl alcohol and dried at 80 °C for 1 day.
Synthesis of silica coating over cobalt ferrite (CoFe2O4@SiO2) nanoparticles
For the synthesis of CoFe2O4@SiO2, 0.5 g of CoFe2O4 synthesized in the previous step was taken and mixed homogeneously in 250 mL of ethanol in an ultrasonic bath at 80 °C and 12.5 mL of a concentrated ammonia solution of 25% by mass was added. A second solution prepared by dissolving 10.8 mL of TEOS in 100 mL of ethanol was added dropwise to this mixture. Then, CoFe2O4@SiO2 was obtained by continuing the mixing process under ultrasonic for 2 h at 80 °C.
Subcritical water oxidation of clofibric acid
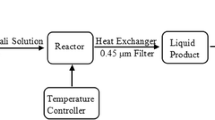
Subcritical oxidation experiments of CFA were carried out in a 200 mL stainless steel teflon-lined reactor. 100 mL of CFA solution (100 mg L−1) was used in the experiments. The reactor temperature and pressure were monitored; the solution was stirred with a magnetic stirrer throughout the oxidation. Subcritical water oxidation (Sub-CWO) experiments were performed in the presence and absence of oxidant (persulfate) and catalyst. In studies where the reuse of the catalyst was tested, the catalyst was separated by centrifugation and then washed several times with ethanol and water and dried.
Experimental design
The effects of temperature, time, oxidant concentration, and catalyst dosage on the subcritical oxidation of CFA in the presence of catalyst and oxidant under milder conditions were investigated using the response surface method and Box–Behnken design (BBD) using Design Expert 11v. The BBD design, in which 29 experiments were performed in 3 parallels, has 3 levels (− 1, 0, + 1). Experimental design and results are given in Table 1.
Analyses of mineralization efficiency
The mineralization efficiency of CFA with the Sub-CWO method was determined using a TOC analyzer (Shimadzu, Japan) for the TOC content of the initial solution (TOC0) and samples (TOCs). TOC Removal efficiency was determined using the following equation:
Characterization of catalysts
The morphological structure of the catalyst was examined with a Quanta 650 Field Emission SEM device. Elemental analysis was determined with the energy-dispersive X-ray (EDX) system. The morphology and microstructure of the CoFe2O4@SiO2 were also confirmed using a transmission electron microscope (TEM, JEM-1011, JEOL). The crystal structure of the catalyst was determined using the PANalytical EMPYREAN model XRD with the Cu-Ka X-ray source (λ = 1.5406 Å). Characterization of functional groups was examined with Jasco FT/IR-6700. The analysis of Fe, Co, and Si passing from the catalyst surface to the solution during catalytic sub-CWO process was carried out with Perkin Elmer NEXION 2000 P model ICP/MS.
Identification of intermediates
Agilent LC-QTOF-MS system with Poroshell 120-EC-C18 column (3.0 × 50 mm size 2.7 µm, Agilent) was used to analyze degradation products. A gradient system was applied using mobile phase 0.1% formic acid in water (A) and 100% methanol (B) elutions, and the steps were as follows: 0–0.5 min, 5% B; 0.5–2 min, 25% B; 2–4 min, 50% B; 4–6 min, 75% B; 6–10 min, 95% B; 10–16 min, and 5% B for equilibration of the column. Analyses were performed at 35 °C and 0.3 mL min−1 mobile phase flow rate. MS analysis was performed using an Agilent 6545 Accurate Mass QTOF-MS. Negative ion mode was used at the following conditions: drying gas flow, 14.0 L min−1; nebulizer pressure, 35 psi; gas drying temperature, 290 °C; sheath gas temperature, 400 °C; sheath gas flow, nitrogen at 12 L min−1. The scan range is from 50 to 500 m/z.
Results and Discussion
Characterization of CoFe2O4@SiO2
Figure 1a, b and c shows the SEM images of amorphous SiO2, nanoparticles CoFe2O4, and CoFe2O4@SiO2 structures, respectively. While Fig. 1b reveals the spherical structure of the CoFe2O4 nanoparticle, Fig. 1c shows that the CoFe2O4@SiO2 catalyst obtained by coating SiO2 on this core structure maintains its spherical structure after coating. The TEM image of CoFe2O4@SiO2 in Fig. 1d supports the spherical structure in the SEM images. However, the dark areas in the TEM image show the CoFe2O4 structure, which we accept as the core, while the lighter areas show the SiO2 layer, which was revealed in similar studies [53, 54].

SEM images of a SiO2, b CoFe2O4, and c CoFe2O4@SiO2, and d TEM image of CoFe2O4@SiO2.
Figure 2 shows the EDS spectra of amorphous SiO2 and CoFe2O4@SiO2 catalysts, respectively. Figure 2b conclusively confirmed the presence of all the elements that the CoFe2O4@SiO2 catalyst in the core–shell structure obtained after coating the CoFe2O4 core with a SiO2 layer [55].

EDS spectra of a SiO2 and b CoFe2O4@SiO2.
The phase purity and crystal structure of the prepared CoFe2O4, SiO2, and composite CoFe2O4@SiO2 structures were examined by X-ray diffraction analysis and the results are given in Fig. 3 a. The XRD diffraction pattern for CoFe2O4 is consistent with the standard JPDS card (No: 22–1086), and the sharp diffraction peaks describe good crystallization [53]. A broad peak at 2θ = 22° is characteristic of pure SiO2 [56], and it can be detected in the composite CoFe2O4@SiO2 structure. When the FTIR spectra of spinel ferrite CoFe2O4 and CoFe2O4@SiO2 structures are examined, the tetrahedral Fe–O band is observed at 550 cm−1, while the peak belonging to the Co–O band is observed at 460 cm−1 in both spectra (Fig. 3b). Moreover, bands of symmetrical and asymmetrical stretching vibrations of the Si–O–Si bond are observed at 810 and 1072 cm−1, respectively [51, 57]

XRD patterns of a SiO2, CoFe2O4 and CoFe2O4@SiO2 and b FTIR spectra of CoFe2O4 and CoFe2O4@SiO2.
Clofibric acid oxidation by sub-CWO
The effect of only the applied temperature on the oxidation of clofibric acid with the Sub-CWO method was tested at 353, 373, and 393 K, and no mineralization was observed at all three temperatures after 60 min of treatment (Fig. 4). It appears that for effective oxidation of clofibric acid needs high temperature. When 5 mM PS promotes oxidation, it is seen that mineralization increases. Sulfate radical and other active species formed due to thermal activation of PS carry out mineralization. At 393 K, the PS-promoted Sub-CWO method achieved 75.02 ± 1.40% TOC removal. Apart from the heat activation of persulfate, its catalytic activation with metal ions is also possible. For this purpose, when the effect of CoFe2O4 and CoFe2O4@SiO2 catalysts on CFA oxidation with 5 mM PS was examined, influential mineralization thanks to both catalysts even at 353 K. Prepared CoFe2O4 and CoFe2O4@SiO2 catalysts provided 79.36 ± 1.22% and 82.95 ± 1.32% mineralization, respectively, at 353 K under the same conditions. In addition, the behavior of the catalyst alone (CoFe2O4@SiO2) showed that it does not have the property of adsorption of CFA.

The effect of heat, oxidant and catalyst on oxidation of clofibric acid ([PS] = 5 mM, [Catalyst] = 0.4 g L−1).
The oxidation of various organic pollutants can be effectively carried out thanks to the high reaction rate, improved organic matter solubility, low viscosity, and excellent mass transfer provided by the PS-promoted subcritical water environment. And also, the application of a catalyst plays a crucial role in increasing the oxidation efficiency by increasing the rate of chemical reactions in relatively mild conditions [58].
Box–Behnken design for optimization of clofibric acid oxidation
Temperature, time, oxidant concentration and catalyst dosage in the mineralization of CFA with CoFe2O4@SiO2 catalyst and PS-supported Sub-CWO method were investigated using the response surface method and Box–Behnken design. The ANOVA results of the reduced quadratic model (Table 2) by removing the ineffective terms (with high p-value) in the proposed quadratic model. The F-value (187.82), p-value (< 0.0001), and R2 (0.9905) values of the proposed quadratic model indicate that the model has high fitting. Based on ANOVA results, the independent variables have a more linear and subsequently quadratic effect on the response. On the other hand, the interactive effects of the independent variables on the response were quite low.
The polyfunctional coded Eq. (2) used to generate the estimated results according to the quadratic model is as follows.
The results given in Table 1 show that the TOC removal is between 72.36% and 95.65%, depending on the Sub-CWO conditions. The effect of all factors for 90% CFA mineralization can be summarized in Fig. 5. For the targeted 90% TOC removal, the temperature of 384 K, the treatment time of 40 min, 10 mM PS, and 0.3 g L−1 catalyst will be sufficient. The effects of factors on the response will also be examined in more detail in 3D graphics.

Effects of factors for 90% TOC removal efficiency.
Effects of operating parameters on TOC removal rate
The effects of temperature, PS concentration, catalyst dosage, and time on the oxidation of clofibric acid with Sub-CWO were examined with BBD, and the effects of the factors are given in 3D graphics in Fig. 6. Temperature is the most important factor of the Sub-CWO method. It was determined in Sect. "Clofibric acid oxidation by sub-CWO" that temperature alone did not affect CFA oxidation at least between 353–393 K. One of the activation methods of PS is thermal activation so that PS can homolytically decompose to sulfate radicals (SO4•−) in the range of 323–403 K according to Eq. (3) with second-order rate constant [59, 60].

The effects of a time and temperature (Cox = 10 mM, Catalyst = 0, 3 g L−1), b) PS.concentration and temperature (time = 40 min, Catalyst = 0.3 g L−1), and c catalyst dosage and temperature (time = 40 min, Cox = 10 mM) on CFA mineralization.
Although keeping the temperature below 373 K does not seem sufficient for CFA mineralization, it can be said that when the temperature rises above 384 K, a significant increase in mineralization efficiency is not observed and even the mineralization rate slows down (Fig. 6a). By increasing the temperature from 353 to 373 K, the TOC removal rate enhanced from 68.76% to 84.45%, but by increasing the temperature from 384 to 393 K, an increase of only 1% (from 90.03 to 91.36%) was achieved. High temperature facilitates the breaking of the peroxide bond (O–O) of PS and supports the formation of more sulfate radicals [61].
The effect of oxidant (PS) concentration and temperature was depicted in Fig. 6b when the catalyst dosage is 0.3 g L−1 and the time is kept constant at 40 min. Figure 6b finds out that adequate oxidation cannot be achieved below 10 mM PS concentration. Effective mineralization is achieved when the PS concentration is 10–15 mM, especially when the temperature is kept above 373 K. Increasing the PS concentration from 5 to 10 mM at 383 K improved mineralization from 85.30% to 91.05%. However, if the PS concentration was 15 mM, the mineralization efficiency slowly increased, and the yield reached 94.10%. This result indicates that higher PS concentration will cause less improvement in mineralization efficiency. Zhou et al. [41] observed that PS concentration above 11.1 mM did not positively affect COD removal in the oxidation of benzothiazole by the PS-promoted WAO process. Interestingly, in the aforementioned study, it was reported that polymerization also took place apart from oxidation. While more polymers were produced in the 373–413 K range, the polymerization decreased when the temperature increased to 453 K in persulfate-promoted WAO. The sulfate radical formed by persulfate activation also leads to the formation of hydroxyl radicals according to Eq. (4) [59]. Both radicals contributed to CFA mineralization. However, if the PS concentration is above the optimum conditions, it will lead to the formation of undesirable side reactions, which are given in Eqs. (5–7). Therefore, the decrease in the concentration of active radical species in the aqueous medium will cause a decrease in the mineralization efficiency [13, 62].
To better understand the effects of catalyst dosage and temperature at 10 mM PS on the mineralization of CFA, a 3D graph is shown in Fig. 6c. In this study, the catalyst dosage was examined in the range of 0.2–0.4 g L−1, it is evident that the change in the catalyst dosage does not have an improving effect on the mineralization efficiency at each temperature applied. The effect of catalyst dosage is more dependent on PS concentration. For example, while the catalyst dosage was 0.3 g L−1, the mineralization increased from 91.05% to 94.10% by increasing the PS concentration from 10 to 15 mM at 383 K. As the increase in the catalyst dosage will increase the large surface area to volume ratio, it facilitates the formation of the active sites required for PS activation.
Persulfate activation occurs on the surface of heterogeneous catalysts such as CoFe2O4@SiO2 by different mechanisms. First of all, SO4•− radicals are formed as a result of the electron transfer of metals on the catalyst surface to PS (Eqs. (8) and (9)) [63]. Moreover, the Co(III)/Co(II) cycle on the catalyst surface can occur according to Eq. (10) and as a result of its reaction hydroxyl radicals as well as are produced [64].
Sulfate radical formation is also supported by the reaction between persulfate and the Fe(III) on the catalyst by following Eqs. (11 and 12). The higher redox potential of Co3+/Co2+ (E° = + 1.81 V) compared to Fe3+/Fe2+ (E° = + 0.77 V) facilitates the reduction cycle of Fe(III) to Fe(II) on the heterogeneous catalyst surface [65].
Considering the above reactions (Eqs. 8–12), the mechanism shown in Fig. 7 was proposed for the radicalic pathway. This possible mechanism shows the degradation of CFA molecules by producing SO4•− and •OH radicals in the CoFe2O4@SiO2 + PS system.

Illustration of the possible activation mechanism of PS by the CoFe2O4@SiO2 catalyst.
Table 3 compares the results obtained in this study for clofibric acid mineralization with the results of studies based on different oxidation methods. The clofibric acid concentration differs in previous studies (Table 3). Lin et al. [16] used the electrochemical oxidation (electro/Fe3+)/PMS) process based on peroxymonosulfate (PMS, 20 mM) activation with Fe(III) (2 mM) ions for oxidation CFA (0.23 mM). The degradation and TOC removal efficiency were reported to be 80% for 60 min and 65% for 3 h, respectively. By the anodic oxidation process, more effective TOC removal efficiencies were achieved, especially using a Boron Doped Diamond (BDD) anode, for example, 100% (6 h) with BDD-carbon PTFE couple [18] and 91% (7 h) with BDD-stainless steel couple [17] for 179 mg L−1 CFA. With the Pt-carbon PTFE anode–cathode pair, 96% TOC removal was achieved in 6 h in the photoelectro-Fenton method [19], while 100% TOC removal was achieved in 6 h in the electro-Fenton method with the BDD-O2 diffusion anode–cathode pair [18] for 179 mg L−1 CFA. In the above-mentioned anodic oxidation, electro-Fenton and photoelectro-Fenton methods, the solution pH value was adjusted to pH 3.0, where optimum efficiency was obtained. Sable et al. [20] achieved 40% mineralization of CFA (25 mg L−1) with the ozone process alone, while the mineralization was enhanced up to 73–81% in 6 h by catalytic ozonation using hydrotalcite-derived catalysts (Mg3Fe0.5Al1 and Cu0.75Mg0.25Al2O4). In CFA (50 mg L−1) oxidation with Zn3La400 photocatalyst at 365 nm, 80% degradation and 29.5% TOC removal were reported in 180 min [21].
Obviously, using catalysts in photolysis and ozonation methods has significantly improved CFA mineralization. The electrochemical advanced oxidation processes such as electro-Fenton, photoelectro-Fenton, anodic oxidation, and EC/Fe3+/PMS for CFA mineralization seem to be very effective but long processing time methods. As a result, CFA mineralization can be achieved through promising and effective processes such as PS-promoted Sub-CWO and PS-promoted catalytic Sub-CWO processes. These methods can be carried out in mild conditions and within a limited duration. This study achieved 75.02% mineralization efficiency at 393 K in 1 h with the PS (5 mM) supported Sub-CWO process without using a catalyst. The CFA mineralization efficiency achieved under the same conditions using the CoFe2O4@SiO2 catalyst reached 89.05%. In optimum conditions, with the effect of the catalyst, both lower temperature (383 K) was applied and higher (97.71%) TOC removal was achieved.
It is a very important advantage that the CoFe2O4@SiO2 structure can be collected and reused with an external magnet. When the CoFe2O4@SiO2 catalyst's reusability is repeated in three cycles, it is seen that there is no significant decrease in the mineralization efficiency, and 97% TOC removal is achieved at the end of the three cycles (Table 4). Since the stability and recovery of the catalyst will positively reduce the cost of the method, the highly efficient catalytic Sub-CWO method can be considered a promising method applied in mild conditions. The amounts of Fe, Co, and Si transferred to the solution during the Sub-CWO process at pH 3.44 were determined as 3.73, 3.63, and 2.60 mg L−1 by ICP-OES, respectively. Based on the total mass of the catalyst used, it can be said that the mass loss in the catalyst is approximately ~ 3% based on these species alone. Since CFA oxidation is carried out in an acidic medium, the use of the CoFe2O4@SiO2 catalyst in a near-neutral medium will significantly reduce the release of these ions.
Identification of degradation products
In order to understand the CFA degradation mechanism, samples were taken at 15 and 30 min processing times and it was observed that CFA (− m/z = 213) was completely degraded within 15 min. Based on the 10 degradation products obtained from LC-QTOF-MS analysis, a degradation mechanism for CFA oxidation by the PS promoted-catalytic Sub-CWO method was proposed in Fig. 8. Activation of PS with heat and CoFe2O4 catalyst promotes the formation of sulfate radicals. In addition, there is also the formation of hydroxyl radicals in the bulk solution depending on Eq. (4) and Eq. (10). It has been reported that reactive oxygen species (ROS) such as superoxide anion radicals (O2•−) and hydroperoxyl ions (HO2−) are present in the environment due to some reactions given by PS (Eqs. (11 and 12)) [68].

Proposed degradation pathway of CFA by PS-promoted catalytic Sub-CWO.
It is known that both radicals are successful in the overall mineralization of many organic pollutants toward CO2, H2O, and inorganic ions depending on the heteroatom type in their structure. The hydroxyl radical preferentially gives an electrophilic addition reaction to electron-rich unsaturated bonds. Specifically, the sulfate radical provides electrons to attack these bonds [69]. Hydrogen abstraction from the benzene ring is also the main degradation pathway by the sulfate radical [70]. Also, the two radicals behave differently in their reactions with aliphatic carboxylic groups. The sulfate radicals prefer to abstract an electron from the oxygen atom in the carboxylic group. In contrast, the hydroxyl radicals capture a hydrogen atom from the aliphatic chain's alpha position [71].
Based on the identified degradation products, the CFA structure can be hydroxylated to form a P1 (2-(4-chloro-2-hydroxyphenoxy)-2-methylpropanoic acid. In the possible degradation pathway, the P1 structure is turned into the P5 structure (2-(3,4-dihydroxy-phenoxy)-2-methyl propanoic acid) by dechlorination by advanced •OH attacks and might be followed by the ring cleavage reaction to form the P6 structure (2-methyl-2-[4-oxo-1-(2-oxo-ethylidene)-but-2-enoxy]-propionic acid). P6-like degradation products have also been suggested in previous studies [67, 72]. Also, P2 (2-phenoxy-isobutyric acid) is formed by eliminating the chloride ion due to electron transfer to the ring, and then P7 structure (2-(4-hydroxy-phenoxy)-isobutyric acid) is formed by hydroxylation. In another degradation pathway, the CFA structure can be transformed into the P3 (4-chlorophenol) intermediate by •OH attack on the aryloxy-carbon bond [72]. The formation of the P12 (2-hydroxyisobutyric acid) degradation product as a result of this reaction is possible. After decarboxylation from the aliphatic group in the CFA structure, the P4 (1-chloro-4-isopropenyloxy-benzene) structure is formed at another degradation pathway. Elimination of Cl− in the P4 degradation product allows the formation of the P8 (isopropenyloxy-benzene) structure. Other degradation products detected are P9 (phenol), P10 (chloro-benzoic acid), and P11 (hydroquinone) are given in the proposed degradation pathways.
Conclusion
The oxidation efficiency increases due to the increase in the probability of adequate high-frequency collisions thanks to the increase in the rate of movement of oxidative species and organic molecules formed in subcritical water conditions. In addition, while problems such as high energy and operating cost requirements, instrumental corrosion, and salt accumulation in the reactor occur in supercritical water conditions, these negative effects are not observed in the sub-CWO process. The use of a heterogeneous catalyst and oxidant allows sub-CWO to be carried out under milder conditions.
In this study, 97.67% TOC removal was achieved after 1 h of sub-CWO using 15 mM persulfate and 0.3 g L−1 catalyst dosage at 383 K, which was determined as the optimum condition for 100 mg L−1 clofibric acid solution. The distribution of nanosized CoFe2O4 in the silica-coated magnetic heterogeneous catalyst structure resulted in more active sites with catalytic properties. The results obtained also support this claim. The catalyst's magnetic feature contributes to its high recovery and reusability. In the persulfate-assisted catalytic sub-CWO of clofibric acid, low ion leaching values were measured even under acidic conditions (pH 3.44). This approach has reduced secondary pollution sources to some extent, presenting a more environmentally friendly method.
Data availability
All the data supporting this study's findings are available in this manuscript.
References
Ferrari B, Paxéus N, Giudice RL, Pollio A, Garric J (2003) Ecotoxicological impact of pharmaceuticals found in treated wastewaters: study of carbamazepine, clofibric acid, and diclofenac. Ecotoxicol Environ Saf 55:359–370. https://doi.org/10.1016/S0147-6513(02)00082-9
Tixier C, Singer HP, Oellers S, Müller SR (2003) Occurrence and fate of carbamazepine, clofibric acid, diclofenac, ibuprofen, ketoprofen, and naproxen in surface waters. Environ Sci Technol 37:1061–1068. https://doi.org/10.1021/es025834r
Ternes TA (1998) Occurrence of drugs in German sewage treatment plants and river. Water Res 32:3245–3260. https://doi.org/10.1016/S0043-1354(98)00099-2
Emblidge JP, DeLorenzo ME (2006) Preliminary risk assessment of the lipid-regulating pharmaceutical clofibric acid, for three estuarine species. Environ Res 100:216–226. https://doi.org/10.1016/j.envres.2005.03.014
Winkler M, Lawrence JR, Neu TR (2001) Selective degradation of ibuprofen and clofibric acid in two model river biofilm systems. Water Res 35:3197–3205. https://doi.org/10.1016/S0043-1354(01)00026-4
Saravanan M, Karthika S, Malarvizhi A, Ramesh M (2011) Ecotoxicological impacts of clofibric acid and diclofenac in common carp (Cyprinuscarpio) fingerlings :hematological, biochemical, ionoregulatory and enzymological responses. J Hazard Mater 195:188–194. https://doi.org/10.1016/j.jhazmat.2011.08.029
Wang L, Ying GG, Zhao JL, Yang XB, Chen F, Tao R, Liu S, Zhou LJ (2010) Occurrence and risk assessment of acidic pharmaceuticals in the Yellow River, Hai River and Liao River of north China. Sci Total Environ 408:3139–3147. https://doi.org/10.1016/j.scitotenv.2010.04.047
Shi W, Ji S, Xu Q, Duan X, Song Z, Xu G (2019) Treatment of pharmaceutical wastewater containing clofibric acid by electron beam irradiation. J Radioanal Nucl Chem 322:407–414. https://doi.org/10.1007/s10967-019-06701-8
Zhang YL, Liu YJ, Dai CM, Zhou XF, Liu SG (2014) Adsorption of clofibric acid from aqueous solution by graphene oxide and the effect of environmental factors. Water Air Soil Pollut 225:2064. https://doi.org/10.1007/s11270-0142064-0
Marques SC, Marcuzzo JM, Baldan MR, Mestre AS, Carvalho AP (2017) Pharmaceuticals removal by activated carbons: role of morphology on cyclic thermal regeneration. Chem Eng J 321:233–244. https://doi.org/10.1016/j.cej.2017.03.101
Bhadra BN, Jhung SH (2018) Adsorptive removal of wide range of pharmaceuticals and personal care products from water using bio-MOF-1 derived porous carbon. Microporous Mesoporous Mater 270:102–108. https://doi.org/10.1016/j.micromeso.2018.05.005
Ighalo JO, Ajala OJ, Umenweke G, Ogunniyi S, Adeyanju CA, Igwegbe CA, Adeniyi AG (2020) Mitigation of clofibric acid pollution by adsorption: A review of recent developments. J Environ Chem Eng 8:104264. https://doi.org/10.1016/j.jece.2020.104264
Brillas E (2023) Removal of insecticides from waters and soils by sulfate radical-based advanced oxidation processes. Appl Res. https://doi.org/10.1002/appl.202300055
Giwa A, Yusuf A, Balogun HA, Sambudi NS, Bilad MR, Adeyemi I, Chakraborty S, Curcio S (2021) Recent advances in advanced oxidation processes for removal of contaminants from water: a comprehensive review. Process Saf Protect 146:220–256. https://doi.org/10.1016/j.psep.2020.08.015
Rueda-Márquez JJ, Levchuk I, Manzano M, Sillanpää M (2020) Toxicity reduction of industrial and municipal wastewater by advanced oxidation processes (photo-Fenton, UVC/H2O2, electro-Fenton and galvanic Fenton): a review. Catalysts 10:612. https://doi.org/10.3390/catal10060612
Lin H, Wu J, Zhang H (2014) Degradation of clofibric acid in aqueous solution by an EC/Fe3+/PMS process. Chem Eng J 244:514–521. https://doi.org/10.1016/j.cej.2014.01.099
Sirés I, Cabot PL, Centellas F, Garrido JA, Rodríguez RM, Arias C, Brillas E (2006) Electrochemical degradation of clofibric acid in water by anodic oxidation: comparative study with platinum and boron-doped diamond electrodes. Electrochim Acta 52:75–85. https://doi.org/10.1016/j.electacta.2006.03.075
Sirés I, Centellas F, Garrido JA, Rodríguez RM, Arias C, Cabot PL, Brillas E (2007) Mineralization of clofibric acid by electrochemical advanced oxidation processes using a boron-doped diamond anode and Fe2+ and UVA light as catalysts. Appl Catal B 72:373–381. https://doi.org/10.1016/j.apcatb.2006.12.002
Sirés I, Arias C, Cabot PL, Centellas F, Garrido JA, Rodríguez RM, Brillas E (2007) Degradation of clofibric acid in acidic aqueous medium by electro-Fenton and photoelectro-Fenton. Chemosphere 66:1660–1669. https://doi.org/10.1016/j.chemosphere.2006.07.039
Sable SS, Medina F, Contreras S (2014) Clofibric acid degradation by catalytic ozonation using hydrotalcite-derived catalysts. Appl Catal B 150–151:30–36. https://doi.org/10.1016/j.apcatb.2013.11.042
Sescu AM, Harja M, Favier L, Berthou LO, de Castro CG, Pui A, Lutic D (2020) Zn/La mixed oxides prepared by coprecipitation: synthesis, characterization and photocatalytic studies. Materials 13:4916–4928. https://doi.org/10.3390/ma13214916
Du X, Zhang R, Gan Z, Bi J (2013) Treatment of high strength coking wastewater by supercritical water oxidation. Fuel 104:77–82. https://doi.org/10.1016/j.fuel.2010.09.018
Javaid R, Qazi UY, Ikhlaq A, Zahid M, Alazmi A (2021) Subcritical and supercritical water oxidation for dye decomposition. J Environ Manage 290:112605. https://doi.org/10.1016/j.jenvman.2021.112605
Reddy SN, Nanda S, Dalai AK, Kozinski JA (2014) Supercritical water gasification of biomass for hydrogen production. Int J Hydrog Energy 39:6912–6926. https://doi.org/10.1016/j.ijhydene.2014.02.125
Toor SS, Rosendahl L, Rudolf A (2011) Hydrothermal liquefaction of biomass: a review of subcritical water technologies. Energy 36:2328–2342. https://doi.org/10.1016/j.energy.2011.03.013
Barge AS, Vaidya PD (2019) Ruthenium-decorated carbon nanotubes as catalyst for wet air oxidation. J Environ Chem Eng 7:102914. https://doi.org/10.1016/j.jece.2019.102914
Ghasemi MN, Esmaeilzadeh F, Mowla D, Elhambakhsh A (2022) Treatment of methyldiethanolamine wastewater using subcritical and supercritical water oxidation: parameters study, process optimization and degradation mechanism. Environ Sci Pollut Res 29:57688–57702. https://doi.org/10.1007/s11356-022-19910-8
Fang L (2016) Catalytic wet oxidation of waste drilling fluid. Oxid Commun 39:2728–2732
Lin SS, Chang DJ, Chen MT, Chen CC (2001) Wet air oxidation of a direct dye solution catalyzed by CoAlPO4-5. Performance assessment and kinetic study. J Environ Sci Health Part A 36:2055–2068. https://doi.org/10.1081/ESE-100107448
Zhu W, Bin Y, Li Z, Jiang Z, Yin T (2002) Application of catalytic wet air oxidation for the treatment of H-acid manufacturing process wastewater. Water Res 36:1947–1954. https://doi.org/10.1016/S0043-1354(01)00419-5
Arslan-Alaton I, Ferry JL (2002) Application of polyoxotungstates as environmental catalysts: wet air oxidation of acid dye Orange II. Dyes Pigm 54:25–36. https://doi.org/10.1016/S0143-7208(02)00031-1
Chang DJ, Chen IP, Chen MT, Lin SS (2003) Wet air oxidation of a reactive dye solution using CoAlPO4-5 and CeO2 catalysts. Chemosphere 52:943–949. https://doi.org/10.1016/S0045-6535(03)00302-3
Garcia J, Gomes HT, Serp P, Kalck P, Figueiredo JL, Faria JL (2005) Platinum catalysts supported on MWNT for catalytic wet air oxidation of nitrogen containing compounds. Catal Today 102–103:101–109. https://doi.org/10.1016/j.cattod.2005.02.013
Ovejero G, Rodríguez A, Vallet A, García J (2013) Catalytic wet air oxidation of a non-azo dye with Ni/MgAlO catalyst. Chem Eng J 215–216:168–173. https://doi.org/10.1016/j.cej.2012.11.028
Parvas M, Haghighi M, Allahyari S (2019) Catalytic wet air oxidation of phenol over ultrasound-assisted synthesized Ni/CeO2–ZrO2 nanocatalyst used in wastewater treatment. Arab J Chem 12:1298–1307. https://doi.org/10.1016/j.arabjc.2014.10.043
Lai C, He T, Li X, Chen F, Yue L, Hou Z (2019) Catalytic wet air oxidation of phenols over porous plate Cu-based catalysts. Appl Clay Sci 181:10525. https://doi.org/10.1016/j.clay.2019.105253
Kayan B, Akay S, Kulaksız E, Gözmen B, Kalderis D (2017) Acid Red 1 and Acid Red 114 decolorization in H2O2-modified subcritical water: process optimization and application on a textile wastewater. Desalin Water Treat 59:248–261. https://doi.org/10.5004/dwt.2017.0552
Javaid R, Qazi UY, Kawasaki SI (2015) Efficient and continuous decomposition of hydrogen peroxide using a silica capillary coated with a thin palladium or platinum layer. Bull Chem Soc Jpn 88:976–980. https://doi.org/10.1246/bcsj.20150052
Görmez Ö, Çalhan SD, Gözmen B (2022) Degradation of isoniazid by anodic oxidation and subcritical water oxidation methods: application of Box-Behnken design. J Environ Sci Health C Toxicol 40:1–26. https://doi.org/10.1080/26896583.2022.2026192
Kayan B, Gözmen B, Demirel M, Gizir AM (2010) Degradation of acid red 97 dye in aqueous medium using wet oxidation and electro-Fenton techniques. J Hazard Mater 177:95–102. https://doi.org/10.1016/j.jhazmat.2009.11.076
Zhou L, Xie Y, Cao H, Guo Z, Wen J, Shi Y (2019) Enhanced removal of benzothiazole in persulfate promoted wet air oxidation via degradation and synchronous polymerization. Chem Eng J 370:208–217. https://doi.org/10.1016/j.cej.2019.03.201
Ghauch A, Tuqan AM, Kibbi N (2012) Ibuprofen removal by heated persulfate in aqueous solution: a kinetics study. Chem Eng J 197:483–492. https://doi.org/10.1016/j.cej.2012.05.051
Ji YF, Xie WP, Fan Y, Shi YY, Kong DY, Lu JH (2016) Degradation of trimethoprim by thermo-activated persulfate oxidation: reaction kinetics and transformation mechanisms. Chem Eng J 286:16–24. https://doi.org/10.1016/j.cej.2015.10.050
Xu XY, Zeng GM, Peng YR, Zeng Z (2012) Potassium persulfate promoted catalytic wet oxidation of fulvic acid as a model organic compound in landfill leachate with activated carbon. Chem Eng J 200–202:25–31. https://doi.org/10.1016/j.cej.2012.06.029
Yuan XZ, Guan RP, Wu ZB, Jiang LB, Li YF, Chen XH, Zeng GM (2018) Effective treatment of oily scum via catalytic wet persulfate oxidation process activated by Fe2+. J Environ Manage 217:411–415. https://doi.org/10.1016/j.jenvman.2018.03.129
Zhu Z, Ma C, Yu K, Lu Z, Liu Z, Yan Y, Tang X, Huo P (2020) Fabrication of CoFe2O4-modified and HNTs-supported g-C3N4 heterojunction photocatalysts for enhancing MBT degradation activity under visible light. J Mater Sci 55:4358–4371. https://doi.org/10.1007/s10853-019-04170-8
Görmez Ö, Yakar E, Gözmen B, Kayan B, Khataee A (2022) CoFe2O4 nanoparticles decorated onto graphene oxide and graphitic carbon nitride layers as a separable catalyst for ultrasound-assisted photocatalytic degradation of Bisphenol-A. Chemosphere 288:132663. https://doi.org/10.1016/j.chemosphere.2021.132663
Liang X, He Z, Zhong Y, Tan W, He H, Yuan P, Zhu J, Zhang J (2013) The effect of transition metal substitution on the catalytic activity of magnetite in heterogeneous Fenton reaction: in interfacial view. Colloids Surf A: Physicochem Eng Aspects 435:28–35. https://doi.org/10.1016/j.colsurfa.2012.12.038
Duan XG, Sun HQ, Kang J, Wang YX, Indrawirawan S, Wang SB (2015) Insights into heterogeneous catalysis of persulfate activation on dimensional-structured nanocarbons. ACS Catal 5:4629–4636. https://doi.org/10.1021/acscatal.5b00774
Yang Q, Choi H, Al-Abed SR, Dionysiou DD (2009) Iron–cobalt mixed oxide nanocatalysts: heterogeneous peroxymonosulfate activation, cobalt leaching, and ferromagnetic properties for environmental applications. Appl Catal B 88:462–469. https://doi.org/10.1016/j.apcatb.2008.10.013
Singh C, Goyal A, Bansal S, Singh S (2017) SiO2@MFe2O4 core-shell nanostructures: efficient photocatalysts with excellent dispersion properties. Mater Res Bull 85:109–120. https://doi.org/10.1016/j.materresbull.2016.09.010
Šuligoj A, Trendafilova I, Maver K, Pintar A, Ristić A, Draži G, Abdelraheem WHM, Jagličić Z et al (2023) Multicomponent Cu-Mn-Fe silica supported catalysts to stimulate photo-Fenton-like water treatment under sunlight. J Environ Chem Eng 11:110369. https://doi.org/10.1016/j.jece.2023.110369
Yu M, Feng Z, Huang Y, Wang K, Liu L (2019) CoFe2O4 nanoparticles directly grown on carbon nanotube with coralline structure as anodes for lithium ion battery. J Mater Sci: Mater Electron 30:4174–4183. https://doi.org/10.1007/s10854-019-00709-2
Li Z, Wang J, Liu M, Chen T, Chen J, Ge W, Fu Z, Peng R, Zhai X, Lu Y (2018) Core-shelled mesoporous CoFe2O4–SiO2 material with good adsorption and high-temperature magnetic recycling capabilities. J Phys Chem Solids 115:300–306. https://doi.org/10.1016/j.jpcs.2017.12.056
Hemmat K, Nasseri MA, Allahresani A, Ghiami S (2019) CoFe2O4@SiO2: a magnetically recyclable heterogeneous catalyst for the synthesis of spirooxindole derivatives. J Organomet Chem 903:120996. https://doi.org/10.1016/j.jorganchem.2019.120996
Liang Y, Ouyang J, Wang H, Wang W, Chui P, Sun K (2012) Synthesis and characterization of core–shell structured SiO2@YVO4:Yb3+, Er3+ microspheres. Appl Surf Sci 258:3689–3694. https://doi.org/10.1016/j.apsusc.2011.12.006
Joseph AM, Thangaraj B, Gomathi RS, Adaikalam AAR (2017) Synthesıs and characterızatıon of cobalt ferrıte magnetıc nanopartıcles coated wıth polyethylene glycol. Adv Nano Bio M&D 1: 71–77 https://www.researchgate.net/publication/329170292
Fu J, Kyzas GZ (2014) Wet air oxidation for the decolorization of dye wastewater: an overview of the last two decades. Chinese J Catal 35:1–7. https://doi.org/10.1016/S1872-2067(12)60724-4
Ganiyu SO, Zhou M, Martínez-Huitle CA (2018) Heterogeneous electro-Fenton and photoelectro-Fenton processes: a critical review of fundamental principles and application for water/wastewater treatment. Appl Catal B 235:103–129. https://doi.org/10.1016/j.apcatb.2018.04.044
Aktaş Y, Gözmen B, Oturan MA (2022) Degradation of phthalic acid by anodic oxidation in acidic aqueous solutions with high chromium content using boron-doped diamond anode. Sep Purif Technol 293:121098. https://doi.org/10.1016/j.seppur.2022.121098
Hussain I, Li M, Zhang Y, Li Y, Huang S, Du X, Liu G, Hayat W et al (2017) Insights into the mechanism of persulfate activation with nZVI/BC nanocomposite for the degradation of nonylphenol. Chem Eng J 311:163–172. https://doi.org/10.1016/j.cej.2016.11.085
Ren W, Huang X, Wang L, Liu X, Zhou Z, Wang Y, Lin C, He M, Ouyang W (2021) Degradation of simazine by heat-activated peroxydisulfate process: a coherent study on kinetics, radicals and models. Chem Eng J 426:131876. https://doi.org/10.1016/j.cej.2021.131876
Yan J, Chen Y, Qian L, Gao W, Ouyang D, Chen M (2017) Heterogeneously catalyzed persulfate with a CuMgFe layered double hydroxide for the degradation of ethylbenzene. J Hazard Mater 338:372–380. https://doi.org/10.1016/j.jhazmat.2017.05.007
Pham AN, Xing G, Miller CJ, Waite TD (2013) Fenton-like copper redox chemistry revisited: Hydrogen peroxide and superoxide mediation of copper-catalyzed oxidant production. J Catal 301:54–64. https://doi.org/10.1016/j.jcat.2013.01.025
Liu H, Bruton TA, Li W, Buren JV, Prasse C, Doyle FM, Sedlak DL (2016) Oxidation of benzene by persulfate in the presence of Fe(III)- and Mn(IV)-containing oxides: stoichiometric efficiency and transformation products. Environ Sci Technol 50:890–898. https://doi.org/10.1021/acs.est.5b04815
Doll TE, Frimmel FH (2004) Kinetic study of photocatalytic degradation of carbamazepine, Clofibric acid, Iomeprol and Iopromide assisted by different TiO2 materials—determination of intermediates and reaction pathways. Water Res 38:955–964. https://doi.org/10.1016/j.watres.2003.11.009
Rosal R, Gonzalo MS, Boltes K, Letón P, Vaquero JJ, García-Calvo E (2009) Identification of intermediates and assessment of ecotoxicity in the oxidation products generated during the ozonation of clofibric acid. J Hazard Mater 172:1061–1068. https://doi.org/10.1016/j.jhazmat.2009.07.110
Yan Y, Wei Z, Duan X, Long M, Spinney R, Dionysiou DD, Xiao R, Alvarez PJJ (2023) Merits and limitations of radical vs. nonradical pathways in persulfate-based advanced oxidation processes. Environ Sci Technol 57:12153–12179. https://doi.org/10.1021/acs.est.3c05153
Qi F, Chu W, Xu B (2013) Catalytic degradation of caffeine in aqueous solutions by cobalt-MCM41 activation of peroxymonosulfate. Appl Catal B 134–135:324–332. https://doi.org/10.1016/j.apcatb.2013.01.038
Zhao L, Ji Y, Kong D, Lu J, Zhou Q, Yin X (2016) Simultaneous removal of bisphenol A and phosphate in zero-valent iron activated persulfate oxidation process. Chem Eng J 303:458–466. https://doi.org/10.1016/j.cej.2016.06.016
Xia X, Zhu F, Li J, Yang H, Wei L, Li Q, Jiang J, Zhang G, Zhao Q (2020) A review study on sulfate-radical-based advanced oxidation processes for domestic/industrial wastewater treatment: degradation, efficiency, and mechanism. Front Chem 8:592056. https://doi.org/10.3389/fchem.2020.592056
Zhang X, Liu Z, Kong Q, Liu G, Lv W, Li F, Lin X (2018) Aquatic photodegradation of clofibric acid under simulated sunlight irradiation: kinetics and mechanism analysis. RSC Adv 8:27796–27804. https://doi.org/10.1039/C8RA03140A
Acknowledgement
I would like to thank Prof. Dr. Ahmet Murat GİZİR and Prof. Dr. Belgin GÖZMEN for their support. This work was supported by Mersin University Research Fund (Project No: BAP 2019-2-AP1-3605).
Funding
Open access funding provided by the Scientific and Technological Research Council of Türkiye (TÜBİTAK). Open access funding provided by the Scientific and Technological Research Council of Türkiye (TÜBİTAK).
Author information
Authors and Affiliations
Contributions
The design of the research, experimental studies, data evaluation, visualization, conceptualization, and writing-original draft process carried out within the scope of this article were carried out by Özkan Görmez.
Corresponding author
Ethics declarations
Conflict of interest
The author declares that he has no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary information
Not applicable.
Ethical approval
Not applicable.
Additional information
Handling Editor: Zhao Shen.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Görmez, Ö. Mineralization of clofibric acid by persulfate-promoted catalytic subcritical water oxidation process using CoFe2O4@SiO2 catalyst. J Mater Sci (2024). https://doi.org/10.1007/s10853-024-09913-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10853-024-09913-w