
Abstract
The grain size of 3Y-TZP is critical because it must remain below a certain size in order to remain tetragonal during aging in the oral cavity. However, fabrication protocols regularly do not accommodate this critical size factor. Nevertheless, grain size measurements generally are not considered or undertaken. Further, when they are, they are thermally etched, which alters the material by grain growth. The didactic purpose of the present work is to provide a strategy for the application of suitable experimental methods for the examination of grain growth in 3Y-TZP, without the risk of grain growth during thermal etching. Examination of the effects of the different applicable thermal conditions is done in order to demonstrate the extents of grain growth in dental restorations following sintering, layering, and glazing. 3Y-TZP nanoscale powders were prepared by coprecipitation using aqueous solutions of zirconyl nitrate and yttrium nitrate, precipitated by ammonium hydroxide, and calcined at 800 °C. Uniaxially and cold isostatically pressed disks were bisque-fired at 800 °C for 2 h, after which they were planed and polished (0.1 μm). The 27 samples were sintered at 1400 ºC, 1500 ºC, or 1600 °C for 1, 2, or 4 h. They then were post-treated by annealing at 750 °C for 1 min under mechanical vacuum and then by post-annealing at 750 °C for 1 min in air. As the pre-polish was retained following sintering, SEM images could be used to determine the grain sizes by standard grain size measurement methods without having to impose thermal etching. These data then were implemented in a grain growth kinetics assessment. The pre-polishing procedure and SEM imaging were successful in enabling accurate grain size measurements without sample alteration. The grain growth as a function of temperature and time exhibited trends approximately logarithmic and linear, respectively. The diffusion mechanism was suggested to be of boundary control by a high-solubility solute in a doped system. Significantly, only sintering at 1400 °C for 1–2 h retained the grain size below the critical limit of ~ 300 nm for resistance to aging in the oral cavity. Also, significantly, ~ 7% grain growth, with a range of 2–14%, occurred during the modest thermal conditions used for both annealing and post-annealing. Sintering, annealing, and post-annealing reduce the number of grain boundaries and hence reduce light scattering, resulting in increasing translucency. Further, the use of the lower sintering temperature of 1400 °C still can yield a very high bulk density (≥ 99.62%), but with a relative increase in light scattering and reduced translucency, making this more suitable for masking dark stump shades. More critically, this sintering temperature for only 1–2 h is necessary in order to avoid risk of the tetragonal → monoclinic phase transformation during the aging process. Further, these conditions are sufficient to allow a limited number of layering and glazing procedures.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although the use of monolithic form of partially stabilized zirconia (PSZ) in orthopedics has been well established since the late 1980s [1], tetragonal zirconia polycrystalline (TZP) was introduced as a dental substructure only in 1995 [2]. It has the advantages of exhibiting a combination of both high strength and fracture toughness [3], although it is subject to low-temperature degradation (LTD) over time [4]. The stress-induced martensitic transformation of the metastable tetragonal to the monoclinic phase produces excellent mechanical properties [5]. In order to retain the metastable tetragonal phase at room temperature, the grain size must be held below a critical value. For zirconia powders stabilized by 3 mol% Y2O3 (3Y-TZP), this grain size must be less than approximately 800 nm to remain fully tetragonal at room temperature [6]. Critically, this critical grain size is reduced to ~ 300 nm when the ceramic must remain tetragonal during aging in the oral cavity [7]. Consequently, the grain growth kinetics during sintering/heat treatment must be controlled carefully in order to leverage the mechanical advantages deriving from the retention of the tetragonal phase.
The fabrication of TZP dental restorations often involves staining (post-annealing in air), and if the restoration is in the esthetic zone, layering and glazing (annealing in vacuum) also are required. However, there appears to be little information about the effects of annealing and post-annealing at temperatures, times, and atmospheres consistent with staining, layering, and glazing. In particular, there appears to be a complete absence of information on heat treatment under vacuum. The present work aims to examine the relevant factors of two-stage sintering temperature (1 h, 2 h, and 4 h at 1400 °C, 1500 °C, and 1600 °C) and post-sintering annealing conditions (1 min at 750 °C) commensurate with these professional processes, which are done in two atmospheric conditions (air, vacuum).
Grain growth in ceramics
The effects of time on grain growth as a function of time under isothermal conditions and fixed atmospheres can be described by the following generalized, simplified expression [8]:
where G is the grain size at time t, n is the grain growth exponent (integral constant of values 1–4 corresponding to specific grain growth mechanisms as detailed in Table 1), G0 is the initial grain size (at time zero), and D is the proportionality constant (dependent on absolute temperature and activation energy [8]).
The grain growth exponent is derived from basic diffusion models for grain growth with these being divided into two distinct types of mechanisms for where: (1) grain boundary mobility is rate-controlled by pores located on the grain boundaries (“pore control”) or (2) where pores have detached from the grain boundaries such that grain boundary mobility is unhindered by the presence of pores and grain growth is rate-controlled at the inherent mobility of the grain boundaries (“boundary control”). For solid-state sintering in particular, pore control tends to occur during the initial and intermediate stages of sintering (up to 80–85% relative bulk density) during which the pores are present on the grain boundaries as a continuous pore network. Grain growth during these stages is significantly impeded by the presence of this porosity such that grain growth is rate-limited by the ability of the pore channels to move with the grain boundaries (This requires significant diffusion of atoms/ions around the pores by surface diffusion, lattice diffusion, or vapor diffusion, or a combination thereof).
The end of the intermediate stage of sintering is marked by the continuous pore channels along the ground boundaries closing off to give discrete pores. Compared with the continuous pore channels, these discrete pores are smaller and offer significantly less impedance to grain boundary mobility such that grain growth increases significantly during the final stage of sintering. Depending upon the mobility of the pores relative to the grain boundaries, the pores will either stay on, and move with, the grain boundaries during grain growth or detach from the grain boundaries to become enclosed within the grains. Grain growth during the final stage is usually characterized by boundary control in which the grain boundaries move at whatever rate governed by their own inherent diffusion rate possibly impeded to some extent by the rate of diffusion of secondary phases (these can include discrete pores) located on the grain boundaries as well as by solute drag.
More recent work has suggested that high-temperature plastic flow in Y-TZP is influenced by the grain boundary segregation of Y3+ ions and other dopant cations [11,12,13,14,15]. Above 1300 °C, tetragonal grains with segregated Y3+ form intergrowths with cubic grains. The implied redissolution of the Y3+ triggers the tetragonal to cubic phase transformation, ultimately resulting in cubic grains only. However, this process must be localized as the XRD data confirm the presence of both polymorphs. This new diffusive transformation mechanism was named grain boundary segregation-induced phase transformation (GBSIPT). Further, with increasing soak times up to 50 h, continuing Y3+ segregation, inward progression of the tetragonal grains, and Y3+ redissolution to cubic phase transformation resulted in negligible grain growth. This mechanism explains how the tetragonal to cubic phase transformation in the absence of grain growth can occur. Unfortunately, the authors did not discuss the contradiction between Y3+ segregation (outward diffusion) and redissolution (inward diffusion), which requires a reversal of the chemical gradient. If the proposed mechanism is credible, then this reversal would need to be considered in terms of the effects of stress. That is, it is well known that segregation from the grain volume to the grain boundary is caused by stress. Consequently, the growth and impingement of the tetragonal intergrowth on the cubic matrix potentially could establish a reversed stress state, thereby facilitating redissolution.
Derivation of grain growth rate expressions based on “pore control” and “boundary control,” and their respective inherent diffusional mechanisms, gives Eq. (1) and the grain growth integer exponents are summarized in Table 1. These are based on simplified diffusion models, and although possibly simplistic and more than one diffusion mechanism can be operating at the one time, they give an indication on the diffusion of the nature of the grain growth that is occurring [8].
The proportionality constant D of Eq. (1) is specific to the particular grain growth mechanism and is derived from the underlying diffusion mechanism which, in turn, is dependent on the absolute temperature and activation energy for diffusion as expressed by the fundamental Arrhenius equation:
where
D = Diffusion rate (ions/cm2 s).
D0 = Pre-exponent constant.
Q = Activation energy for diffusion.
R = Gas constant (8.314 J/K mol).
T = Absolute temperature.
The activation energy is specific to the particular diffusion mechanism including the effect of atmosphere. Rearrangement of the above Arrhenius equation to Eq. (3) enables D0 and Q to be determined graphically from a plot of ln D versus \(\frac{1}{T}\), which yields a straight line of slope \(\frac{ - Q}{R}\), with ordinate intercept ln D0:
This linear relation can be simplified as follows:
where x is the abscissa, y is the ordinate, a is the slope, and b is the ordinate intercept. Equation (3) indicates that this method is valid only when the dependent variables are diffusion and temperature, viz. the diffusion mechanism is dependent solely on the temperature.
An additional contribution to the grain growth of 3Y-TZP arises from the tetragonal to monoclinic (tet → mono) phase transformation during cooling of a fraction of the tetragonal grains. This phase transformation is diffusionless [16], and when it occurs, it results in small but significant volumetric increase of ~ 3–5 vol% in the size of the grains. This gives an added contribution to the effective grain growth in which case Eq. (2) is modified to the form [17]:
where
m′ = rate of the tet → mono transformation (= mono/(tet + mono)/t
m = monoclinic fraction of phase assemblage (= mono/(tet + mono).
Accurate measurement of grain size
Accurate measurement of the grain sizes of zirconia is problematic because each of the commonly used techniques, for sample preparation to reveal the microstructure, risks alteration of the sample and, in particular, the actual grain size. Similarly, the imaging techniques used to capture the microstructural images have inherent limitations and can lead to measurement artifacts. Lastly, the grain size measurement used to determine the grain size is subject to error and bias. The methods used for each of these main steps in microstructural characterization are examined briefly below with particular mention of their issues/limitations.
Sample preparation for avoidance of tet → mono phase transformation
Polishing
Examination of the microstructure of bulk samples (for example, by optical microscopy or scanning electron microscopy (SEM)) necessitates the preparation of highly polished, flat surface on the sample. This is done typically by a series of grinding and polishing steps down to ~ 0.05 µm final polish. However, for TZP materials especially where the final polish is coarser than ~ 0.05 µm, this can result in residual stress-induced tet → mono transformation at the surface [18], thereby giving an artifact of a slightly larger grain size at the surface.
Acid etching
This is involves immersing the polished surface in acid at room temperature [19, 20], or elevated temperatures [21] to remove material preferentially from the grain boundaries to highlight the grain structure. In the case of zirconia, this technique uses employs HF as the etchant and this can also chemically induce the tet → mono transformation with the associated grain expansion altering the grain size [22].
Thermal etching
Similar to acid etching, this is done to reveal the surface microstructure, but uses heat to etch the surface. This method involves heating the polished sample for a short time (~ 15–30 min) at a temperature approximately 150–200 °C below the temperature used to initially sinter the TZP [23, 24]. This results in additional diffusion and, in particular, the movement of material from the grain boundaries to highlight the grain structure. However, these conditions can be sufficient to cause additional grain growth, thereby affecting the subsequent measured grain size. Further, cooling from the thermal etching temperature risks causing the thermally induced tet → mono transformation and associated grain expansion, although this is unlikely since the critical grain size would not be likely to be exceeded.
Polishing/thinning
Samples to be examined by transmission electron microscopy (TEM) are prepared by thinning down to a thickness of ~ 50 nm, typically by ultrathin mechanical polishing, focused ion beam milling (FIB), or argon ion milling (commonly known as precision ion polishing, PIP). Similar to polishing of SEM samples, this can result in the stress-induced tet → mono transformation and associated grain expansion at the sample surface [18]. Also, FIB can introduce Ga contamination which, although not a problem for grain size measurements, may affect compositional analysis.
Microstructural/structural examination for grain size measurement
Scanning electron microscopy (SEM)
SEM involves direct topographical imaging of the surface microstructure (using secondary electron mode) and has the advantages that it the sample surface can be examined as-sintered, and the low-energy electron beam is unlikely to alter the sample. However, this method has the disadvantage that, even with a highly polished surface, the grain boundaries generally cannot be detected. This can be overcome with chemical or thermal etching, but this risks sample alteration. This method also requires the surface to be perpendicular to the incident beam to produce an undistorted (non-oblique) image.
Electron backscattered diffraction (EBSD)
EBSD complements SEM by detecting backscattered electrons to give a compositional image of the microstructure based on variations in elemental composition and/or atomic density to reveal grain orientations and grain boundaries. EBSD has the disadvantages that the material must be nearly fully dense and requires a scratch-free, highly polished surface.
Transmission electron microscopy (TEM)
This method has the same advantages as SEM, but the high magnification limits the number of grains that can be examined in a given field of view. Also, it has the disadvantage that the high-energy electron beam can induce the tet → mono transformation, thus slightly increasing the measured grain size [22].
X-ray diffraction (XRD)
This method is based on the line broadening of XRD peaks and correlation of this to crystallite size [25, 26]. It is meaningful only if the grains are single crystals rather than polycrystalline. It has the limitations that it assumes approximately equiaxed particles and the results can depend on which XRD peaks are used for calculation.
Grain size measurement
Standardized grain counting methods
The accurate measurement of the grain sizes of zirconia has limitation because there is no perfect way to determine the actual grain size in a densified material. Standardized methods of the measurement of average grain size include comparison procedure, the intercept procedures (lineal and circular), and the planimetric (or Jeffries) procedure [27]. These test methods are used to determine the average grain size of specimens’ grain areas, diameters, or intercept lengths, assuming they have approximately log-normal unimodal distribution of equiaxed grain sizes, but they do not characterize the nature of this distribution. These test methods deal only with determination of planar grain size, that is, characterization of the two-dimensional grain sections as revealed by the sectioning plane (this planar grain size being numerically equivalent to the grain size in a three-dimensional volume if the material is isotropic) [28].
The comparison procedure [27] involves visual comparison of the sample grain structure to a series of graded equiaxed grain structure images and choosing the image which best matches visually (taking into account the magnification). These charts were constructed to reflect the typical log-normal distribution of grain sizes that result when a plane is passed through a three-dimensional array of equiaxed grains and show a distribution of grain dimensions, ranging from very small to very large, depending on the relationship of the planar section and the three-dimensional array of grains. For specimens consisting of equiaxed grains, the method of comparing the specimen with a standard chart is quick and convenient and gives an indication of an approximate relative grain size as used in, for example, commercial quality control purposes. A general bias in the comparison grain size ratings is that the estimated grain size is somewhat coarser than its actual size. It is not sufficiently accurate for quantitative grain size measurements.
The intercept methods [27] involve the random application of a test line of known length (LT) on the microstructural image and the counting of the number (n) of either grains that intercept the test line or grain boundary intersections that lie on the test line. The line used is either a straight line (Heyn lineal intercept procedure) or a circle (Hilliard single-circle procedure), the latter used if there is some directional variation of the grain size. In counting the number of grain intercepts, whole grains are counted as 1 and grains in which the line terminates are counted as ½. In counting the number of grain intersections, each grain boundary intersection is counted as 1, tangential intersections with a grain boundary are counted as 1, grain boundary triple point intersections are counted as 1½, and end points of line are not counted unless they lie exactly on a grain boundary in which case they are counted as ½. The grain size is taken to be the “intercept length” (L) which numerically is the length of the line (taking into account image magnification M) divided by the number of grain intercepts (the “grain intercept count”) or grain boundary intersections (the “grain boundary intersection count”) that lie on the line (Eq. 8).
where
L = Intercept length (length of test line/number of grain intercepts)
LT = Test line of known length
n = Number of grains along test line
M = Magnification.
The accuracy and precision of a single intercept measurement are reliant on an optimal number of grains being counted (typically at least 50) this being a balance between counting enough grains to give statistical reliability versus not counting too much leading to counting errors. Also, it is recommended that grain intercepts or grain boundary intersections be marked off as they are counted to ensure reliability of the counting. An average grain size for the sample is then obtained by making measurements on typically 3 to 5 microstructural fields (more if necessary to obtain required precision as specified in the standard method [27]).
The planimetric method (also called Jeffries’ procedure) involves the application circle of known area (A) randomly on the microstructural image and the counting of the number (n) of grains that lie within it [27]. Grains lying entirely within the area are counted as 1 and grains cut by the area perimeter are counted as ½. Numerically, the number of grains counted per area (taking into account the image magnification (M)) is reciprocated to give the area per grain, then the square root taken to give the length per grain (Eq. 9). This latter quantity is taken to be the measured grain size.
Like the intercept method, it is recommended that grains be marked off to ensure reliability of the counting and that an optimal number of grains (typically ~ 50) be counted to ensure reliability of the individual measurement, and an appropriate number of microstructural fields of view be measured to give an average grain size of acceptable precision [27].
Statistics of grain size determined by image analysis
The grain sizes can be assessed statistically using image analysis software (e.g., ImageJ, National Institutes of Health, Maryland, USA), in which the spatial parameters of individual grains contained within the microstructural image are measured/calculated as defined geometric quantities (such as equivalent spherical diameter, aspect ratio, and roundness). In particular, image analysis has the advantage that it can provide a spherical equivalent of a non-circular grain by averaged circularization, but its accuracy depends on the number of grains examined owing to the location of the surface plane relative to the grain centers. Histograms or other visual approaches are used to demonstrate the distributions in the measured grain size quantities. Assuming a normal distribution of grain sizes (as is the case typically for equiaxed grain structures), the following standard log-normal Gaussian function is commonly applied to the distribution data in order to determine average grain size [29]:
where
f(D′) = Grain size distribution function.
A′ = Equivalent circular area of a grain.
D′ = Equivalent diameter of a grain.
σ = Skewness (≥ 1).
\(D_{x}^{\prime }\) = Average grain diameter.
D and A are the measured grain diameter and equivalent circular area of each grain, Dx is the average grain diameter, and \(\sigma\) is the skewness value (≥ 1), which is the deviation from a Gaussian distribution, as determined by curve fitting.
The methods described above give a linear representative dimension of the grains which, although commonly taken as a measure of the grain size of the material, is not actually the true grain size (in whatever way this is defined for a non-spherical shape). Both methods underestimate the grain size because, if the true grain size is taken as some “diametral” dimension through the center of the grain then, owing to the plane of section through the material passing at a random height through each grain, the grains viewed on the plane of section will be either maximal size (if cut through the grain “diameter”) or less than this (if cut “off-diameter”). Hence, the average measured grain size for the collection of grains on the plane of section has to be numerically smaller than the true grain size. In addition, for the intercept methods, the line drawn on the microstructure will not necessarily pass through the center of each grain area present on the plane of section such that there is further underestimation of the grain size. A correction factor of 1.56 has been derived to account for the underestimation (i.e., true grain size = 1.56 measured grain size) based on the assumption of a log-normal statistical distribution of where the grains are sectioned through the grain and a log-normal statistical distribution of the true sizes of the actual grains [30]. However, published grain sizes are usually reported as the as-measured sizes rather than corrected values hence are underestimations of the actual true size.
Objectives of work
In the present work, the grain sizes of 3Y-TZP samples sintered at different temperatures (1400 °C, 1500 °C, and 1600 °C) for different times (1, 2, and 4 h) are determined by the principal approaches of intercept method, planimetric method, and grain size statistics by image analysis. The results are contextualized by preliminary consideration of the preceding grain size measurement techniques in order to apply the most representative method. The quantified effects on grain size of annealing under vacuum and post-annealing in air for conditions typical of low-temperature glazing (1 min at 750 °C) of the sintered starting 3Y-TZP dental materials are then reported. These results aim to elucidate the effects of glazing of dental restorations, where there is a risk of exceeding the critical size effect for the tet → mono phase transformation [22] in 3Y-TZP, which is used regularly for dental restorations.
Materials and methods
Sample fabrication
An ultrafine 3Y-TZP starting powder (~ 40 nm particle size) was synthesized in this work by a hydroxide coprecipitation method typical of that used to prepare commercial zirconia powders. Aqueous solutions of zirconium nitrate hydrate (ZrO (NO3)2·1.71H2O) and yttrium (III) nitrate hexahydrate (Y(NO3)3·6H2O) were mixed (in stoichiometric ratio to produce nominally 3 mol% Y-doped zirconia) by magnetic stirring for 10 h. The mixture was precipitated using an aqueous solution of ammonium hydroxide (NH4OH). The precipitates were filtered, washed with deionized water five times, and dried at 110 °C for 48 h. The powders were lightly crushed by hand using an aluminous porcelain mortar and pestle and calcined at 800 °C for 15 min (30 °C/min heating rate) in an Al2O3 crucible. The particle size of the calcined powder was reduced by high-energy planetary ball milling (Emax, Retsch, Germany) at 2000 rpm for 40 min; the mill and media consisted of 2Y-TZP. The milled powders were dried for 48 h at 110 °C, after which they were dry-pressed by double-ended compaction at 220 MPa in a hardened steel cylindrical die. Each of the triplicate samples (13 mm diameter and ~ 2 mm thickness) then was vacuum-sealed in a non-lubricated latex sheath and cold isostatically pressed at 200 MPa. The samples were bisque-fired at 800 °C for 2 h (heating rate 11 °C/h), after which CAD/CAM (Amann Girrbach AG, Vorarlberg, Austria) was used to machine the bisque-fired blanks using a five-axis milling system equipped with a tungsten carbide cylindrical burr. The finish was achieved using progressively smaller burrs of diameters 1.0, 0.6, and 0.3 mm to produce highly flat and smooth surfaces satisfactory for SEM imaging (i.e., the surface is effectively scratch-free at the magnifications used). The samples were placed on a zirconia refractory surface and two-stage sintered at 1400 °C, 1500 °C, or 1600 °C by heating at a rate of 11 °C/h to 950 °C, soaking for 2 h, heating at a rate of 100 °C/h to the sintering temperature, soaking for 1, 2, or 4 h, and nature cooling. The three sintering temperatures and three soak times gave a total of nine sample permutations. Following characterization of the sintered samples, they were annealed by preheating a furnace at 400 °C, sample insertion at this temperature, soaking for 6 min, heating at 50 °C/min to 750 °C with application of vacuum (0.426 kPa) at 450 °C, soaking for at 750 °C for 1 min, opening the door and cooling to 400 °C over 5 min, and removing. Finally, a post-annealing heat treatment was done using the same procedure except that no vacuum was applied. Figure 1 shows the schematic of the overall experimental program and includes characterization using analytical methods after sintering and polishing.

Experimental program
The critical difference between the above methodology and that of others is the stage of pre-polishing, which was done before sintering. This resulted in the ability to visualize the grains without risk of grain growth (as could be the case if thermal etching was used) or the tet → mono phase transformation (which would give a ~ 3–5 vol% increase in grain size) induced by grinding since the precision mechanically machined surface was retained upon sintering. Consequently, reliable grain sizes at the sample surface without the above artifacts could be determined.
Characterization of physical properties
Relative bulk density, open porosity, and closed porosity of heat-treated samples were measured by hydrostatic weighing according to AS1774.5 [31]. The phase assemblage of samples after the various heat treatments were analyzed by X-ray diffraction (XRD; X’pert Pro MPD, Philips, CuKα radiation, 45 kV, 40 mA, scan rate 0.13° 2θ/s, step size 0.026° 2θ). Only the surfaces of samples were measured by XRD since converting them to powders for traditional powder diffraction is not applicable to densified TZP materials because it causes extensive tet → mono transformation.
Microstructural visualization
Usually, grain size measurements are taken on the sections taken through the bulk of the material so as to obtain values representative of the overall material. However, in dental materials, surface microstructure is of importance in the esthetic appearance, and so, grain size at the surface is an important characterization. In this work, the grain size of the heat-treated samples was determined on both the surface (i.e., the precision-machined surface) and in the bulk (on intergranular fracture surface obtained by controlled diametral breakage) of each sample. To ensure consistency of grain size measurement, and also to give relevancy to dental restoration fabrication in which samples are heat-treated typically by mounting them on posts such that the entire surface is exposed to the same heat treatment, microstructural visualization and grain size measurement in this work were taken on the exposed upper surface of each sample (for surface measurements) and at the center of the diametral fracture surface (for bulk measurements). Samples were mounted on stubs, carbon-coated (Leica ACE600 sputter coater), and microstructures examined by SEM (FEI Nova NanoSEM 450 FE-SEM, 5.0 kV accelerating voltage).
Grain size measurement
Grain size was measured on SEM microstructural images by three methods: intercept method [27], planimetric method [27], and grain size statistics by image analysis with each method being performed on each individual microstructural image. For the intercept method, counting lines at arbitrary orientations of horizontal, vertical, and oblique (45° and 135°) were applied in random positions on each microstructural image to count the number of grains (according to the counting rules) and the intercept length calculated for each line using [Eq. (8)]. The linear intercept lengths from the four lines were averaged to obtain the average intercept length for the sample. Typically ~ 30–50 grains were counted for each line. The planimetric method [20] was performed on SEM microstructural images (each corresponding to a 13.8 µm × 7.8 μm field on the sample surface) and involved counting (as per the counting rules) typically at least ~ 70 grains in this field and the grain size calculated using Eq. (9). At least 2 fields of view were used per sample to obtain the average grain size of the sample. Grain size statistics by image analysis was the third method to measure the grain size of samples. A publicly available software package (Image J, National Institutes of Health, Maryland, USA) was used and measurement involved a sequence of calibrating the image size to the actual microstructure size, optimizing brightness and contrast of the microstructural image, thresholding the image to a binary black and white image to display the grain boundary structure, and finally software measurement of the spatial quantities of the grains. Data of equivalent diameter of grains were processed in a statistical software package (Origin, OriginLab, Massachusetts, USA) to determine the grain length distribution and average grain length. Shapiro–Wilk test was performed to validate the normality of the grain length distribution. All grain sizes were analyzed by one-way analysis of variance (ANOVA) and the post hoc Tukey's test, with significance set at p < 0.05. The grain lengths given by each of the three methods are reported hereafter as the grain sizes and a grain size correction factor to give true grain size was not applied.
Results and discussion
Physical properties
The relative bulk densities of the sintered samples as measured by hydrostatic weighing are 99.62% (1400 °C), 99.95% (1500 °C), and 99.95% (1600 °C), and the amounts of open porosity and closed porosity in all samples were close to zero. These density and porosity data indicate that the samples are all sintered virtually to full density which agrees with microstructural images in which there no pores are visible. Also, the TEM imaging of samples shown in Fig. 2 revealed only occasional pores, these typically being of submicron size and located at the grain boundaries. Collectively, these data indicate that all sintered samples are within experimental error of them being completely densified (Fig. 3).

TEM images of 3Y-TZP grains after sintering, vacuum annealing, and air post-annealing

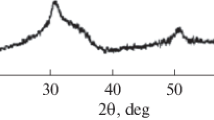
Representative diffraction pattern of polished surfaces of samples [this sample sintered at 1400 °C (t = tetragonal)]
The quantitative XRD data for the sintered samples was based on full-surface scanning of triplicate samples for each sintering, annealing and postannealing sequence, giving a total of nine samples (Fig. 3). This conventional method involved using the intensities of the (111) and (11\({\overline{\text{1}}}\)) monoclinic peaks and the (101) tetragonal peak [32, 33] revealed the presence of ~ 1 vol% monoclinic phase at 1600 °C only (Fig. 4). No cubic phase was observed in any samples, as evidenced by the absence of the (004) cubic peak situated between the (004) and (400) tetragonal peaks at 72°–76° 2θ. The tetragonality of the starting powder was determined using the same method and this was determined to be 23.82 wt% (Fig. 5). However, all sintered samples exhibited a small shoulder to the peak at 30° 2θ, which is indicative of the presence of a rhombohedral polymorph [30, 34]. These two trace polymorphs appear to have been eliminated upon annealing and post-annealing. The low levels of these secondary polymorphs are consistent with the view that the samples are effectively 3Y-TZP. Hence, subsequent discussion about grain growth is considered in terms of a single-phase tetragonal zirconia; in particular, contribution of the tet → mono phase transformation to measured grain size is negligible such that the second part of Eq. (7) can be ignored, and so, Eqs. (1) and (3) are sufficient for the subsequent analysis of grain growth in this work.

X-ray diffraction patterns of samples sintered at 1400 °C, 1500 °C, and 1600 °C

Diffraction pattern of starting powder (m = monoclinic, t = tetragonal)
Comparison of microstructure of sintered samples
Figure 6 shows representative SEM images of the sintered-only samples as a function of sintering temperature and holding time. (Only surface microstructures of sintered samples are shown here to indicate the gross differences in appearance and grain size; corresponding bulk microstructures were similar but with only slightly larger grain size.) It can be seen that the microstructures are virtually fully dense with no obvious porosity which is consistent with the materials having relative bulk densities very close to 100%. The microstructures are typical of equiaxed, single-phase grain structures (such as TZP materials) in that there is a distribution in grain sizes and planar, convex, and concave grain boundaries also are apparent. In this type of microstructure, normal grain growth (as distinct to abnormal or exaggerated grain growth) occurs when there is sufficient thermal energy available to provide the activation energy required for atomic diffusion (of the diffusion mechanism(s) involved). Thus, temperature is a key parameter in the consideration of grain growth and appreciable grain growth only occurs if the temperature is sufficiently high [27]. The driving force for diffusion and grain growth can be described in terms of the pressure difference between convex and concave boundaries surfaces in that curved grain boundaries (arcs) migrate to their centers of curvature, and so, effectively larger grains (convex/scalloped) grow at the expense of smaller grains (concave/spherical) [35, 36].

Representative SEM (FEI Nova NanoSEM 450) images of surfaces of the sintered samples with 1, 2, and 4 h holding time
The determination of grain size by SEM examination was based on the assessment of the samples used for phase quantification by XRD. Each of the nine samples was examined in four different locations on the top and cross section each. In addition, three more samples were subjected to a separate sequence of sintering and post-annealing. Consequently, a total of 81 SEM images were obtained. The grain sizes at both the surface and bulk of the initial sintered samples as measured by intercept method, planimetric method, and grain size statistics by image analysis are summarized in Table 2. The data presented are for an initial sintering time of 2 h (this time being typical of that used in the preparation of dental ceramics) and give an initial comparison between the three grain size measurement methods and also between surface and bulk grain sizes. The data show the following main trends:
-
•There is a progressive increase in grain size as sintering temperature increases (as expected, discussed further later).
-
•Grain size is consistently smaller for surface compared with bulk (but not necessarily statistically significant in all cases) for each sintering temperature and grain size measurement method. This is attributed to the bulk of each sample receiving effectively a longer heat treatment time owing to the retention of heat in the bulk relative to the surface as the sample quickly cools from the sintering temperature. Although the samples were of 2 mm thickness, whereas typical dental restorations are ≤1 mm thickness [37], this result is significant because it highlights the need to minimize thermal gradients in order to ensure uniform microstructural development in zirconia dental restorations as well as the avoidance of damage induced by thermal shock.
-
•Grain size increases in order of the grain size measurement method from planimetric method to intercept method to grain size statistics by image analysis for both surface and bulk at each sintering temperature. This suggests that although there appears to be a systematic difference in grain size values between each of the grain size measurement methods, they each shown same relative changes in grain size between surface and bulk as a function of the sintering temperature. Hence, each measurement technique shows the same meaningful trend in grain size, and so, only one method is needed to consider the effect of heat treatment on the grain size in the rest of this work—the intercept method is used because it tends to be the method most commonly used in the literature for ceramics.
Grain size measurements of sintered, annealed, and post-annealed samples
Table 3 reports the grain sizes of the samples (measured by intercept method) after the initial sintering (at 1400 °C, 1500 °C, and 1600 °C) for various times (1, 2, and 4 h) as well as the subsequent heat treatment on each temperature–time combination of annealing (in vacuum at 750 °C for 1 min.) and then post-annealing (in air at 750 °C for 1 min.). Figure 7, 8 and 9 show the effects of temperature (sintering), time (hold time), and heat treatment (sintering → annealing → post-annealing) on grain growth. Table 3 gives the data variations in terms of averages and standard deviations, the data in Figs. 7 and 8 give them in terms of the actual ranges, while Fig. 9 shows histograms of the particle size distributions.

Experimental 3Y-TZP average grain sizes (line-intercept method) and grain size ranges for sintered, annealed, and post-annealed samples (abscissa is the initial sintered hold time, green = surface, red = cross section)

Experimental 3Y-TZP average grain sizes (intercept method) and size ranges for sintered, annealed, and post-annealed samples

SEM images and grain size distributions (from image analysis to grain size statistic method) of cross sections of 3Y-TZP after sintering for 2 h (at 1400 °C, 1500 °C, or 1600 °C), then annealing for 1 min at 750 °C under vacuum, and then post-annealing for 1 min at 750 °C in air
These data can be considered firstly in terms of the effect of heat treatment conditions (temperature and time) on the grain size of the initial sintered samples at the sample surface and bulk. Consistent with typical behavior expected from theory and reported in the literature, grain growth occurs with increasing sintering temperature and time typical of the generally observed sigmoidal kinetics of the process [38]. The grain size at the surface is consistently smaller than that in the bulk in almost all samples. The grain size being slightly higher in the bulk is attributed to the cooling rate of the furnace being slower than the heating rate such that on cooling, the interior of the samples would remain at higher temperature compared with the surface and hence effectively received more heat work. However, in no sample is the grain size statistically different (at the p = 0.05 level) between the surface and the bulk, and so, this effect, if real, is not significant.
Figure 7 shows that the grain size increases with increasing sintering temperature and increasing time as expected from fundamental energetics considerations and the changes in grain size are typical for 3Y-TZP [23, 39] for the sintering temperatures used (1400–1600 °C). In comparison, only a small, if any, increase in grain size occurred for the subsequent heat treatment steps of annealing and then post-annealing progression (both at 750 °C). This indicates that the sintering temperature provides the major effect on grain growth, whereas subsequent heating at the much lower temperature of 750 °C does not contribute significantly. This is to be expected since the amount of diffusion occurring in the latter would be does not very much owing to much reduced thermal energy resulting is substantially slower diffusion [40].
The data in Table 3 are now considered data in terms of the effect of subsequent stepwise annealing (750 °C for 1 min. in vacuum) and post-annealing (750 °C for 1 min in air) steps. Considering the effect of annealing first, it gave statistically significant (at p > 0.05 level) increases in grain size (irrespective of whether the grain size was measured in the bulk or at the surface) for many of the starting sintered materials with this tending to occur for lower sintering temperatures and shorter times, these being the materials which would have had smaller grain sizes initially and hence greater propensity for further grain growth. Critically, this demonstrates that despite the fact that the thermal energy input for the annealing condition (750 °C for 1 min) is very modest compared to those for the initial sintering conditions of 1400–1600 °C for 1, 2, or 4 h, it nonetheless gave a statistically significant increase in grain size. In contrast, the post-annealing step (750 °C for 1 min. in air) did not give statistically significant (at p > 0.05 level) increases in grain size except for the sample sintered at the lowest temperature for the shortest time (1400 °C/1 h). Also, a control set consisting of samples sintered at each of the sintering temperatures (1400 °C, 1500 °C, 1600 °C) for 2 h subjected directly to the post-annealing step did not give any statistically significant (at p > 0.05 level) changes in grain size (data not shown here). Collectively, the data demonstrate that the grain size is given primarily by the particular sintering temperature and time with a small, but statistically significant, subsequent increase in grain size then given by the annealing step but little or no further increase with the post-annealing step.
Figure 9 shows grain size distribution of each microstructure (based on its grain intercept size), and each distribution appears to be an approximately symmetrical, normal intercept size distribution which suggests that normal grain growth has occurred in the TZP samples without any non-uniform grain growth. Figure 10 shows normal probability plot of grain length distribution indicating, at the 0.05 level, the data were significantly drawn from a normally distributed population. With increasing sintering temperature, there is some minor narrowing of the distribution curves, and the subsequent annealing and post-annealing possibly also gave slight change in the shape of the curves, but these changes are considered to be minor and within experimental error. Collectively, the grain size distribution curves indicate grain growth behaviors typical of uniform (isotropic) growth of equiaxed grains, specifically: As the average size of the grains increases, the shape of the grain size distribution (given by Eq. (10)) remains largely unchanged and effectively the microstructure just scales to a larger size. For ceramics in general, non-uniform grain growth can occur as a result of localized impurities, inhomogeneities, or discontinuous or exaggerated grain growth giving a change in the shape of the grain size distribution and, where this is pronounced, a result in a bimodal grain size distribution. Also, for Y-TZP ceramics in particular, a bimodal grain size distribution can occur as a result of phase partitioning in which there is localized segregation of the composition to give small tetragonal grains (lowered Y content) and large cubic grains (increased Y content) [41]. The consistent unimodal shape of the grain size distribution curves suggests that non-uniform grain growth has not occurred to any measurable extent, and so, it is concluded that all grain growth given by the heat treatment steps involved normal diffusion-controlled grain growth processes.

Log-normal probability plot of the grain size distribution with significant level of 0.05
In considering the additional grain growth given by the annealing and post-annealing steps, it is helpful to report these as relative changes with respect to the initial sintered grain size for each sintering temperature–hold time combination. Table 4 shows these changes in grain size expressed as a percentage of the grain size of the material in the initial as-sintered condition calculated using Eqs. (11) and (12)
where Sint = after sintering, Ann = after annealing, Post = post-annealing.
Table 5 simplifies the data in Table 3 by reporting the grain size averaged for the top surface and bulk. As mentioned, the exposed surface values not only the enhanced radiant heating on the top surfaces but also from the thermal insulation from the dense sintered Al2O3 sample support on the underside. Since the samples were of 2 mm thickness and typical dental restorations are ≤ 1 mm thickness [37], the effect of the reduced thermal gradient across the latter was simulated by using the average of the surface and cross-sectional grain sizes for the former.
Consideration of grain growth of sintered samples
First it is necessary to consider grain growth for the initial sintered 3Y-TZP samples (using the average of surface and bulk grain sizes for each sample). The calculated linear regression coefficients, which are measures of the fit to linearity for grain size vs time as per Eq. (1), suggest the most likely grain growth mechanism. As shown in Table 6, the highest regression value was for a grain growth exponent of n = 2. From consideration of the grain growth exponents in Table 1, this suggests that grain growth during the initial sintering of the 3Y-TZP is controlled one or more of the following mechanisms: (1) pore control facilitated by vapor diffusion; (2) boundary control in a pure system; or (3) boundary control by a high-solubility solute in a doped system. The relevancy of each of these to the grain growth behavior in the 3Y-TZP is now considered.
The values for \(G^{n} - G_{0}^{n}\) in Eq. (1) were determined using n = 2 in order to determine the partial pre-exponential constants, the results of which are given in Table 7. The fitting of these data against t–t0, where t0 = 1 h (3600 s), is shown in Fig. 11.

Determination of partial pre-exponential constants (K = slope) from Eq. (1) for 3Y-TZP sintered at 1400 °C–1600 °C for soak times of 1, 2, and 4 h (n = 2)
For 1600 °C samples, it is apparent that grain growth stopped due to either a loss in driving force and/or a change in mechanism. For the latter, TEM-EDS data reported by others [42] suggest that this sample is likely to have exhibited extensive segregation of yttrium to the grain boundaries, which effectively pinned the grain boundaries, thereby effectively preventing further grain growth. This occurred only for 1600 °C since the kinetics are faster, hence the only temperature for which this situation was reached within the heat treatment times used in this study. For this reason, only the first two times for 1600 °C are used in the kinetics calculation.
Using Eqs. (3–6), the values for the logarithms of the three K slopes from Eq. (1) were fitted against 1/T in order to determine the \(D_{0}^{\prime }\) in Eq. (2). This plot is shown in Fig. 12. The activation energy Q was calculated from Eq. (4) using the slope and the gas constant R. The calculated activation energy is 598.66 kJ/mol which is in close approximate to published value of activation energy for the grain growth of 3YTZP [43,44,45]. However, all published value considered the solute drag (n = 3) as diffusion mechanism and it was used to calculate activation energy which is in contrary to boundary control solute drag (high-solubility solute) (n = 2) mechanism of the present study.

Determination of activation energy for grain growth of sintered 3Y-TZP at 1400 °C–1600 °C (n = 2)
For relatively pure, in single-phase materials such as doped zirconia, the amount of grain growth that takes place during the initial and intermediate stages of solid-state sintering is relatively small owing to the pinning effect of continuous porosity located along the grain boundaries. The final stage of sintering is characterized by pore closure in which the continuous pores close to form discrete pores located on the grain boundaries. These discrete pores have a greatly diminished ability to impede grain boundary movement such that the final stage of sintering is also characterized by substantial grain growth. 3Y-TZP is considered to be a doped system (Y dopant in ZrO2 host lattice), and as such, the mechanism of boundary control in a pure system (i.e., n = 2 in Table 1) does not apply to it. However, the mechanism of boundary control by a high-solubility solute in a doped system (also n = 2 in Table 1) is applicable and agrees with the value of n = 2 obtained from the kinetics data. This agrees with a solute drag mechanism proposed by Lee and Chen [46] based on a space charge model in which segregation of aliovalent solute cations to the grain boundaries establishes a positive charge at the grain boundary with an adjoining space charge region, consisting of an excess of oxygen vacancies and a deficit of cation vacancies, extending into the bulk. In this present work, the aliovalent solute cations are the yttrium cations (\({\text{Y}}_{{{\text{Zr}}}}^{\prime }\)), and the short-range attraction between them and the oxygen vacancies (\(V_{{\text{O}}}^{ \cdot \cdot }\)) at the grain boundaries results in a drag force for cation diffusion which lowers the grain boundary mobility.
Slightly different to the present work, although their 3Y-TZP grain size data do not allow clear differentiation between grain growth mechanisms, Sagel-Ransijn et al. [39] considered that grain growths exponents were likely to be either values of 2 or 3 corresponding to “normal grain growth” or “solid solution drag control,” respectively. These presumably correspond to the mechanisms in Table 1 of “boundary control—pure system” and “boundary control—solute drag (low-solubility solute),” respectively, although the latter mechanism does not make sense since yttrium has a relatively high solubility in zirconia. They also suggested that grain growth in the 3Y-TZP was possibly inhibited by yttrium segregation to the grain boundaries and tetragonal/cubic phase partitioning, although the role of lattice diffusion (n = 2) was not discounted.
Consideration of grain growth of annealed and post-annealed samples
In general, although diffusion can occur at significantly lower temperatures than the practical sintering temperature, it is usually considered that this diffusion usually involves the lower activation energy diffusion mechanisms involving evaporation–condensation and/or surface diffusion and occurs for materials with extremely high surface areas (notably the powders and initial powder compacts) and leads to particle coarsening and the formation of interparticle sinter bonds, both of these prior to any actual significant densification or the extensive grain growth associated that occurs during the final stage of solid-state sintering. The annealing process in this work, however, is very different to this in that the ceramics were already sintered to near-theoretical density and had already undergone significant grain growth such that their microstructures were “thermodynamically mature,” and in particular, any further grain growth would be expected to require significant thermal treatment—that is, heat treatment involving temperatures and times greater than or at least comparable to those used in the initial sintering.
The data of relative changes in grain size (Table 4) contradict the above description of expected behavior in that there is a significant increase in grain size in the range of ~ 10–20% for almost of the sintered starting materials when heat-treated in the first annealing step (750 °C) under vacuum. This result is surprising because, although the 3Y-TZP shows a possibility for further grain growth as evidenced by the samples sintered at lower temperatures and/or shorter times undergoing grain growth at higher temperatures and/or longer times, hence demonstrating that there is a thermodynamic driving force for grain growth, the following two considerations of kinetics would suggest otherwise: (1) the annealing temperature 750 °C is significantly lower than that of the initial sintering temperatures used to produce the ceramics such that it would have been expected that the amount of thermal energy available would be insufficient the activation energy required for any further grain growth; and (2) even if there was sufficient thermal energy available, a mere 1 min for the annealing would be expected to be insufficient time for any significant further grain growth to occur. Post-annealing at 750 °C in air gave significant further increase in grain size only for the initial sintered material of the lowest temperature and hold time (i.e., 1400 °C and 1 h). This is possibly because this material, being the smallest starting grain size, would have the greatest propensity for any further grain growth. While there were some small increases in grain size of the other starting materials annealed in air, these were not statistically significant (and some were negative indicating that the grain sizes were within experimental error of the real average grain size).
From the above observations, it is apparent that the vacuum condition of the annealing at 750 °C facilitated significant additional grain growth in the “thermodynamically mature” 3Y-TZP ceramics and this occurred at a fraction of the initial sintering temperatures (0.61, 0.58, and 0.55 of the absolute sintering temperatures of 1674 K, 1774 K, and 1874 K, respectively) and for a very small fraction of the initial sintering times (0.017, 0.008, and 0.004 of the sintering times of 1, 2, and 4 h, respectively). The significant effect of the vacuum condition on grain growth is considered in terms of the effect on the defect structure of the 3Y-TZP and, in turn, the effect of this on the diffusion process(es) involved in the grain growth (both of these with respect to the normal 3Y-TZP processed in air).
Grain growth is influenced by the rate of diffusion involved in the grain growth mechanism(s) operating and this is strongly influenced by the overall defect chemistry of the 3Y-TZP. Considering simple zirconia (ZrO2) in air as a reference, it consists of Schottky defects of cation vacancies (zirconia) and anion (oxygen) vacancies. Under atmospheric conditions (normal temperature, pressure, and oxygen partial pressure), the intrinsic defect chemistry of pure zirconia (ZrO2) is characteristically that of an anion-deficient oxide (MO1-x, or specifically ZrO2-x here) with the non-stoichiometry accommodated by the formation of oxygen vacancies and electrons as written in Kröger–Vink notation for the two basic charge compensation mechanisms:
Electronic:
As a consequence of this non-stoichiometry, the concentration of oxygen vacancies exceeds that of the zirconia vacancies. Equation (13) shows that, when heating in air is involved and the conventional ionic charge compensation is assumed [16, 39, 47], the equilibrium would shifted to the left owing to the oxygen concentration gradient imposed by the \(\frac{1}{2}{\text{O}}_{2 } \left( g \right)\). (i.e., equivalent to air) such that the oxygen vacancy concentration in the ZrO2 is set by the partial pressure of oxygen (PO2 = 0.2 in air).
The addition of Y2O3 into solid solution in the host ZrO2 lattice and assumed substitutional solid solubility [48] introduces one oxygen vacancy for each YO1.5 added in accordance with the YO1.5 stoichiometry compared with ZrO2, and the thermodynamic need for these extrinsic atomic defects to be compatible with the intrinsic defect structure gives the following defect equations:
Electronic:
where
\({\text{Y}}_{{\text{Y}}}^{x}\) = Yttrium ion on yttrium lattice site, Nil net charge.
\({\text{O}}_{{\text{O}}}^{x}\) = Oxygen ion on oxygen lattice site, Nil net charge.
\({\text{Y}}_{{{\text{Zr}}}}^{\prime }\) = Yttrium ion on zirconium lattice site, − 1 net charge.
\({\text{Zr}}_{{{\text{Zr}}}}^{\prime }\) = Zr reduction (Zr4+ → Zr3+), − 1 net charge.
\({\text{Zr}}_{{\text{S}}}^{x}\) = Zirconium ion migrated to surface and neutralized, Nil net charge.
\(V_{{\text{O}}}^{ \cdot \cdot }\) = Oxygen vacancy, + 2 net charge.
\(h^{ \cdot }\) = Hole, + 1 net charge.
According to Eq. (13), annealing of the as-sintered 3Y-TZP ceramics under vacuum conditions (Po2 ≈ 0.426 kPa for 1 min at 750 °C) would thermodynamically drive the equation to the right resulting an increase in the concentration of oxygen vacancies, hence giving more vacancies for the diffusion of oxygen during grain growth. However, in general, diffusion in a metal oxide involves codiffusion of cations and anions and is rate-limited by the diffusion rate of the slowest species. For zirconia in particular, it is generally accepted that the diffusion coefficients for grain boundary diffusion and possibly lattice diffusion of the zirconium and dopant cations are many orders of magnitude (~ 4–7) smaller than those of the oxygen anions [47] and that the diffusion kinetics involved in sintering and grain growth are rate-controlled by the diffusion rates of the cationic species [47]. It is known that two-stage sintering followed by annealing in air can suppress grain growth by grain boundary pinning [49, 50]. The present work reveals that similar heat treatment also resulted in the suppression of grain growth. However, in contrast, two-stage sintering followed by annealing under vacuum at only 750 °C for 1 min resulted in grain growth. As indicated, this could be attributed to the significant role of oxygen vacancy formation, associated enhancement of diffusion, and resultant grain growth. Nevertheless F, in the present work even if the annealing under vacuum gave an increase in oxygen vacancy concentration, this by itself may not be sufficient to explain the apparent enhanced diffusion that would be required for the significant additional grain growth (since oxygen anions are not the rate-controlling species). However, it is likely that an increase in oxygen vacancy concentration gives reduced constraint of the overall host ZrO2 crystal lattice such that there is less localized lattice (ionic) bonding and lattice strain to overcome during diffusion [51] not only for the oxygen anions but also the Zr and Y cations, thus effectively making it easier for codiffusion to occur (effectively lowering the activation energy for diffusion, thus enabling more diffusion to occur at the low temperature of 750 °C). Furthermore, this effect may have specific effect on the solute drag grain growth mechanism in that movement of the solute cloud associated with the grain boundary would be easier/faster, hence facilitating grain growth at lower temperatures/shorter times.
Other than the above defect chemistry, another consideration is the possible effect on the role of liquid phases in densification and grain growth. It is well known that the densification of zirconia to theoretical density requires the presence of an exceedingly thin (~ 1–10 nm) but finite liquid layer at the grain boundaries which, although the ceramic sinters fundamentally by a solid-state sintering process, the presence of the liquid gives a much lower activation energy for diffusion along the grain boundaries, hence enabling densification to occur at typical processing temperatures [47, 52]. Although there are no published data demonstrating this explicitly for zirconia, since grain growth involves similar diffusion mechanism as densification (i.e., grain boundary diffusion) it stands to reason that the liquid phase may also be implicit grain growth and, in particular, that an increase in the amount of this phase may promote grain growth (as is typical for liquid phase sintering). The reducing conditions of the annealing step may give possible reduction of Zr4+ to Zr3+ (this correlating to a slight darkening of color in the zirconia) which could increase the inherent chemical reactivity of the zirconia and in particular propensity to form a liquid eutectic phase (at the grain boundaries) and at lower temperatures [53]. However, this is discounted in the current work because high-resolution transmission electron microscopy (HRTEM) used to examine the grain structure and grain boundary morphology (data not shown here) revealed that the glassy grain boundaries present in the sintered samples were noticeably diminished in thickness (and in many places not discernable other than at the grain triple points) in the annealed samples.
Lastly, further heating in air at the same temperature (750 °C) during the post-annealing step did not produce any further significant increase in grain size for the majority of samples. This is attributed to the oxygen vacancy concentration returning to whatever high levels (i.e., reduction in \(V_{{\text{O}}}^{ \cdot \cdot }\) concentration) such that the lattice relaxation was no longer present and hence the diffusion rate returning to that of “normal” in-air conditions which, since was at 750 °C rather than whichever sintering temperature, did not give significant diffusion, and hence no further grain growth.
Conclusions
The present work aims to clarify the experimental method to examine grain growth in 3Y-TZP. Since commonly used techniques for preparing microstructural samples of 3Y-TZP for grain imaging risk grain growth and/or tet → mono phase transformation (both of which may alter the grain size), the procedure of pre-polishing bisque-fired samples represents an important development in the microstructural characterization of these ceramics that, combined with precise SEM imaging, it allows the direct imaging and accurate size measurement of unaltered grains. These measurements are recommended to be done according to ASTM E112-96 (2004), Standard Test Methods for Determining Average Grain Size [54].
The experimental work focused on progressive thermal treatments used commonly by dentists and dental technicians for the fabrication of 3Y-TZP dental restorations. Densification (≥ 99.62% was done by sintering at 1400–1600 °C for 1–4 h and this was followed by typical layering or glazing conditions of heating at 750 °C for 1 min under mechanical vacuum (annealing) and in air (post-annealing). The trends for grain growth as a function of temperature and time were approximately logarithmic and linear, respectively. Grain growth was by grain boundary control rate-limited by solute drag of high-solubility solute. Significantly, it was found that subsequent vacuum annealing produced a small but significant increase in grain size of most samples (as much as 25%; Table 4), this tending to be more predominant for those samples sintered initially at low temperatures and/or shortest times since these samples being of smaller initial grain size, hence having more opportunity for grain growth. This finding is significant because it demonstrates for the first time that heat treatment in vacuum at temperatures (e.g., as done in layering) well below the initial sintering temperature steps can significantly increase grain size in sintered 3Y-TZP—this has important consequence for control of grain size and, in particular, where grains should be retained below the critical size of ~ 300 nm for aging resistance. 3Y-TZP sintered at 1400 °C is not usually used in dental practice owing to it having a high grain boundary density and hence low translucency which is esthetically undesirable, but the vacuum annealing may overcome this to some extent. More generally, this work demonstrates that low-temperature vacuum annealing can produce a small but significant increase in grain size and the associated decrease in grain boundary density may attenuate light scattering resulting in increasing translucency of the 3Y-TZP ultimately giving a noticeable improvement in visual esthetic of a dental restoration.
Change history
23 February 2023
This article was revised to add the missing Open Access funding note.
References
Kelly JR, Benetti P (2011) Ceramic materials in dentistry: historical evolution and current practice. Aust Dent J 56(1 Suppl):84–96
Meyenberg KH, Lüthy H, Schärer P (1995) Zirconia posts: a new all-ceramic concept for nonvital abutment teeth. J Esth Dent 7(2):73–80
Swain MV, Rose LRF (1986) Strength limitations of transformation-toughened zirconia alloys. J Am Ceram Soc 69(7):511–518
Chevalier J, Gremillard L, Deville S (2007) Low-temperature degradation of zirconia and implications for biomedical implants. Annu Rev Mater Res 37:1–32
Green DJ, Hannink RHJ, Swain MV (1989) Transformation toughening of ceramics. CRC Press, Boca Raton, FL
Lange FF (1982) Transformation toughening part 3 experimental observations in the ZrO2-Y2O3 system. J Mater Sci 17(1):240–246. https://doi.org/10.1007/BF00809059
Lu H-Y, Chen S-Y (1989) Low temperature ageing of t-ZrO2 polycrystals with 3 mol% Y2O3. J Am Ceram Soc 70(8):537–541
Rahaman MN (2003) Creamic processing and sintering, 2nd edn. CRC Press, Boca Raton
Cahn JW (1962) The impurity-drag effect in grain-boundary motion. Acta Metall 10(9):789–798
Brook RJ (1968) The impurity-drag effect and grain-growth kinetics. Scr Metall 2(7):375–378
Matsui K, Ohmichi N, Ohgai M, Yoshida H, Ikuhara Y (2006) Grain boundary segregation-induced phase transformation in yttria-stabilized tetragonal zirconia polycrystal. J Ceram Soc Jpn 114(1327):230–237
Matsui K, Yoshida H, Ikuhara Y (2018) microstructure-development mechanism during sintering in polycrystalline zirconia. Int Mater Rev 63(6):375–406
Viazzi C, Bonino JP, Ansart F, Barnabe A (2008) Structural study of metastable tetragonal YSZ powders produced via the sol–gel route. J Alloys Compd 452(2):377–383
Fabregas IO, Reinoso M, Otal E, Kim M (2016) Grain-size/(t″or c)-phase relationship in dense ZrO2 ceramics. J Eur Ceram Soc 36(8):2043–2049
Sakuma T, Yoshizawa Y (1992) The grain growth of zirconia during annealing in the cubic/tetragonal two-phase region. Mater Sci Forum 94:865–870
Kisi EH, Howard CJ (1998) Crystal structures of zirconia phases and their inter-relation. Key Eng Mater 153:1–36
Burke JE (1963) The effect of heat treatment on microstructure. In: Microstructure of ceramic materials. National Bureau of Standards Miscellaneous Publications, Pittsburgh
Minguela J, Slawik S, Mucklich F, Ginebra MP, Llanes L, Mas-Moruno C, Roa JJ (2020) Evolution of microstructure and residual stresses in gradually ground/polished 3Y-TZP. J Eur Ceram Soc 40(4):1582–1591
Frechette VD (1963) Experimental techniques for microstructure investigation in microstructure of ceramic materials. National Bureau of Standards Miscellaneous Publication, Pittsburgh
Flamant Q, Anglada M (2016) Hydrofluoric acid etching of dental zirconia Part 2: effect on flexural strength and ageing behavior. J Eur Ceram Soc 36(1):135–145
Nowicka A, El-Maghraby HF, Svancarkova A, Galuskova D, Reveron H, Gremillard L, Chevalier J, Galusek D (2020) Corrosionand low temperature degradationof 3Y-TZP dental ceramics under acidic conditions. J Eur Ceram Soc 40(15):6114–6122
Stevens R, Evans PA (1984) Transformation toughening by dispersed polycrystalline zirconia. Br Ceram Trans 83(1):28–31
Ruiz L, Readey MJ (1996) Effect of heat treatment on grain size, phase assemblage, and mechanical properties of 3mol% Y-TZP. J Am Ceram Soc 79(9):2331–2340
Taffner U, Carle V, Schafer U, Hoffmann MJ (2004) Preparation and microstructural analysis of high-performance ceramics. In: Voort GFV (ed) Metallography and microstructures, vol 9. ASM International, Almere
Scherrer P (1918) Bestimmung der grösse und der inneren struktur von kolloidteilchen mittels röntgenstrahlen. Nachr Ges Wiss Göttingen 26:98–100
Nath D, Singh F, Das R (2020) X-ray diffraction analysis by Williamson–Hall, Halder–Wagner and size-strain plot methods of CdSe nanoparticles—a comparative study. Mater Chem Phys 239(1):122021
Beck PA (1948) Effect of recrystallized grain size in grain growth. J Appl Phys 19(3):507–509
Pickering FB (1976) The basis of quantitative metallography. Monograph (Institute of Metallurgical Technicians). Metals & Metallurgy Trust for Institute of Metallurgical Technicians, London
Aljawfi RN, Alam MJ, Rahman F, Ahmad S, Shahee A, Kumar S (2018) Impact of annealing on the structural and optical properties of ZnO nanoparticles and tracing the formation of clusters via DFT calculation. Arab J Chem 13(1):2207–2218
Kitano Y, Mori Y, Ishitani A (1988) Rombohedral phase in Y2O3 partially stabilized ZrO2. J Am Ceram Soc 71(1):34–36
AS 1774.5 (2014) Refractories and refractory materials—physical test methods. Method 5: determination of bulk density, apparent porosity and true porosity
Schmid HK (1987) Quantitative analysis of polymorphic mixes of zirconia by X-ray diffraction. J Am Ceram Soc 70(5):367–376
Evans PA, Stevens R, Binner JGP (1984) Quantitative X-ray diffraction analysis of polymorphic mixes of pure zirconia. Trans J Br Ceram Soc 83(2):39–43
Wertz M, Schmidt MB, Hoelzig H, Wagner M, Abel B, Kloess G, Hahnel S, Koenig A (2022) Rhombohedral phase formation in yttria-stabilized zirconia induced by dental technical tools and its impact on dental applications. Materials 15(13): 4471–4485
Brook RJ (1976) Controlled grain growth. In: Wang FFY (ed) Treatise on materials science and technology. Academic Press, New York, pp 331–364
Yue CH, Zhang L, Liao S, Gao H (2009) Kinetic analysis of the austenite grain growth in GCr15 steel. J Mater Eng Perform 19(1):112–115
Del Piva AMDO, Tribst JPM, Benalcazar Jalkh EB, Anami LC, Bonfante EA, Bottino MA (2021) Minimal tooth preparation for posterior monolithic ceramic crowns: effect on the mechanical behavior, reliability and translucency. Dent Mater 37(3):140–150
Kingery WD, Bowen HK, Uhlmann DR (1975) Introduction to ceramics. Wiley & Sons, Toronto
Sagel-Ransijn CD, Winnubst AJA, Burggraaf AJ, Verweij H (1997) Grain growth in ultrafine-grained Y-TZP ceramics. J Eur Ceram Soc 17(9):1133–1141
Ikuma Y, Tsubaki Y, Masaki T (1991) Oxygen diffusion in Y2O3-containing tetragonal zirconia polycrystals with different grain size. Yogyo Kyokaishi 99(1):101–103
Lange FF (1986) Transformation-toughened ZrO2: correlations between grain size control and composition in the system ZrO2–Y2O3. J Am Ceram Soc 69(3):240–242
Lin Y-J, Angelini P, Mecartney M (1990) Microstructural and chemical influences of silicate grain-boundary phases in yttria-stabilized zirconia. J Am Ceram Soc 73(9):2728–2735
Luo J (1998) Microstructure evolution and grain growth in the sintering of 3Y-TZP ceramics. J Mater Sci 33(22):5301–5309. https://doi.org/10.1023/A:1004481813393
Nieh TG, Wadsworth J (1989) Dynamic grain growth during superplastic deformation of yttria-stabilized tetragonal zirconia polycrystal. J Am Ceram Soc 72(8):1469–1472
Theunissen GSAM, Winnubst AJA, Burggraaf AJ (1993) Sintering kinetics and microstructure of nanoscale Y-TZP ceramics development. J Eur Ceram Soc 11(4):315–324
Lee IG, Chen IW (1988) Sintering ‘87. In: Somiya S, Shimada M, Yoshimura M, Watanabe R (eds) Sintering ‘87, vol 1. Elsevier Applied Science, London
Standard OC, Sorrell CC (1998) Densification of zirconia—conventional methods. Key Eng Mater 153–154:251–300
Stevens R (1986) Zirconia and zirconia ceramics, written for Magnesium Elektron, 2nd edn. Magnesium Elektron Ltd, Manchester
Wang CJ, Huang CY, Wu YC (2009) Two-step sintering of fine alumina–zirconia ceramics. Ceram Int 35(4):1467–1472
Lin FJT, De Jonghe LC, Rahaman MN (1997) Initial coarsening and microstructural evolution of fast-fired and MgO-doped Al2O3. J Am Ceram Soc 80(11):2891–2896
Nowotny J, Rekas M, Bak T (1998) Defect chemistry and defect-dependent properties of undoped and stabilised zirconia Bulk vs interface. Key Eng Mater 153–154:211–240
Mallinckrodt DV, Reynen P, Zografou C (1982) The effect of impurities on sintering and stabilization of Zr2(CaO). Interceram 31(2):126–129
Gionco C, Paganini MC, Giamello E, Burgess R, Di Valentin C, Pacchioni G (2013) Paramagnetic defects in polycrystalline zirconia: an EPR and DFT. Chem Mater 25(11):2243–2253
ASTM E112-96 (2004) Standard test method for determining average grain size. American Society for Testing and Materials, Philadelphia, pp 240–263
Acknowledgements
This work has been supported by the Australian Postgraduate Award (APA) and Australian Government Research Training Program (RTA). We are grateful for access to the characterization facilities provided by the Mark Wainwright Analytical Centre, UNSW Sydney.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author has no conflict of interests.
Additional information
Handling Editor: David Cann.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shahmiri, R., Standard, O.C., Hart, J.N. et al. Critical effects of thermal processing conditions on grain size and microstructure of dental Y-TZP during layering and glazing. J Mater Sci 58, 3854–3878 (2023). https://doi.org/10.1007/s10853-023-08227-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-023-08227-7