
Abstract
Phosphate-shelled brachiopods differ in filter-feeding lifestyle, with Lingula anatina an active infaunal burrower, and Discinisca tenuis a shallow marine epibenthic animal. The shells of these animals are built of organophosphatic constituents, the organic fibres/sheets reinforced with calcium phosphate to provide a sophisticated ultrastructural robustness. This investigation examined the nature of the organic fibres in order to improve understanding of how living organisms produce hierarchically structured biomaterials. Unlike powdered samples commonly used in previous studies, organic fibres were isolated for the first time and the shell fractions were purified, in order to study the content and nature of the biopolymer fibres. Biochemical methods including Calcofluor staining revealed a chitin matrix. Ultrastructural analysis, thermal gravimetric analysis, and spectroscopic analyses show that the core polysaccharide framework is composed of layers of β-chitin sheets and/or fibrils that are coated with a fibrous organic matrix. There is more chitin matrix in the L. anatina shells (26.6 wt.%) compared to the D. tenuis shells (12.9 wt.%). Taken together, the data show that the chitin matrix contributes to increased skeletal strength, making L. anatina highly adapted for life as an active burrower. In comparison, D. tenuis contains less chitin and lives as attached epibenthos in a shallow marine environment.
Graphical abstract

First spectroscopic evidence of β-chitin sheets in recent organophosphatic brachiopods
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In nature, living organisms are highly organized and are capable of producing three-dimensional hierarchically structured materials such as teeth, bones, and seashells. These biomaterials are inorganic–organic nanocomposites with exceptional structures and functions. Inorganic components comprise biominerals, that is, calcium carbonates and phosphates or silica [1,2,3], whereas organic macromolecules constitute a combination of soluble- and insoluble-organic components, primarily peptide-, protein- and polysaccharide-based layers which provide a scaffolding for the inorganic phase [4,5,6]. Water insoluble macromolecules are most frequently built from monomers-based either on sugars or peptides. Sugar monomers and biomolecules based on peptides are present throughout the animal kingdom [5, 7, 8]. The former are chemically modified, most commonly by amination, and are referred to as chitin, whereas the product of subsequent deacetylation of chitin is known as chitosan [8, 9]. As for the biomolecules based on peptides, these can either be functional or structural proteins comprised of a variety of fibrous, tubular, and triple helices in the case of collagen [5, 10].
Chitin is semicrystalline in nature and has three polymorphic forms: α-, β-, and γ-chitin [11,12,13,14]. Polysaccharide α-chitin is the most common, in which the chains of α-chitin are arranged in an anti-parallel manner with strong intermolecular hydrogen bonding [12]. β-Chitin chains are parallel to each other, with relatively weak intermolecular hydrogen bonding [11]. Chains of γ-chitin are a mixture of α- and β-chitin, with two parallel chains in one direction and a third one in the opposite direction [13, 14]. Typically, chitin is the structural polymer to be found in three-dimensional architectures of fungi, yeasts, diatoms, sponges, corals, annelids, and mostly in arthropods, including insects, arachnids, and crustaceans [10, 13, 15, 16]. In the layered cuticle of arthropod exoskeletons, chitin is cross-linked with substantial amounts of proteins to form variably oriented yet organized fibres [17, 18]. In the matrix sheets of calcareous bivalve shells, the core polysaccharide framework is composed of interlamellar structure of β-chitin, and is frequently reinforced and/or composited with extracellular matrix [4, 19, 20], other biomolecules include glycosaminoglycans [21,22,23], and/or inorganic components [2, 4]. These composite materials are high-strength yet lightweight, and provide functional flexibility, support ultrastructures, mechanical stability and compressive strength [2, 24, 25]. Living organisms use a combination of organic matrices and inorganic constituents, in various ratios, to produce extraordinary arrays of diverse shell morphologies which eventually facilitate adaptation to a diverse suite of habitats [26, 27].
Brachiopods are a diverse group of marine bivalved, sessile, filter-feeding lophophorate animals currently classified into three subphyla: Linguliformea, Craniiformea and Rhynchonelliformea [28]. Craniiformea and Rhynchonelliformea utilize calcium carbonate and ~ 2% of an organic matrix to hierarchically assemble ultrastructures of the calcitic shells [29]. In contrast, linguliform brachiopods such as L. anatina and D. tenuis use organic biopolymers reinforced with calcium phosphate (Ca-phosphate) to produce the shells [1, 30]. Linguliform brachiopods are an important ancestral animal group, with an unexcelled fossil record that originated some 530 million years ago in the early Cambrian as part of the emergence of biomineralization across disparate animal clades [28, 31]. Linguliform brachiopods exhibit distinctive features including a ciliated lophophore, chitinous chaetae, as well as a fleshy pedicle for anchoring to (or within) the substrate [28, 32]. Typical D. tenuis uses a pedicle for attachment to hard substrates, so as to survive wave action in the steeper slopes of the shallow seafloor [26]. In contrast, L. anatina has an elongate pedicle to prop itself up with the shell against the sediment. The downward U-shaped burrowing is accomplished by cyclical complex motions of the valves against the sediment, with the pedicle trailing behind the shell [26, 27, 32,33,34]. Definitive evidence for an active U-shaped burrowing lingulid lifestyle is unknown from the Palaeozoic, where the obolids burrowed pedicle first [32,33,34]. Lingulides have occurred in a vertical burrowed life position since the early Palaeozoic while the first appearance of U-shaped burrows constructed by lingulids likely occurred during the Mesozoic [35, 36]. Although burrowing has been connected with naturally occurring demineralization [37] and maceration [35], the survival of Lingulidae over time has been attributed to the depth of burrows [26, 32,33,34]. For instance, the shell of L. anatina is more flexible and has remained well adapted to deep burrows as a refuge against bioerosion of the substrate and predation since the early Palaeozoic [26, 33,34,35].
Lingulid shells are used as paleoenvironmental proxies due to their complete fossil record and their distribution in modern oceans [28, 38]. Despite their palaeontological importance, the growth mechanism of brachiopod shells including linguliforms, is not clearly understood, and regarding the organic biopolymers, detailed structural and molecular analyses are topical in order to understand the mechanisms and formations of lingulid shells. Although biochemical studies and spectroscopic analyses on powdered brachiopod samples have identified proteins associated with chitin from linguliform shells [39,40,41], the composite fibres have not been characterized, so little is known about the nature of the organic sheets. Chemical fixation and scanning electron microscopy (SEM) were used to propose that the organic fibrils in linguliform shells are composed of chitin, a dense mat of fibrils which are probably collagenous, and membranous laminae within the shell layers21, 42. However, these previous studies were based on a more or less complete mixture of organic biopolymers and Ca-phosphate mineral. It is necessary to establish an easy and efficient method to reveal the composition of the organic sheet layers in brachiopod shells. The approach of this study is to use field emission gun-SEM (FEG-SEM), light and fluorescence microscopy, and comparison of the imaged region with natural composite biominerals analysed by microRaman spectroscopy and Fourier transform infrared (FTIR) spectroscopy. The goal of this study is to identify and characterize the major organic frameworks in lingulid brachiopod shells. This investigation facilitates a clearer understanding of morphological structures of fossilized, recent phosphate-shelled brachiopods and the biosynthesis of organic–inorganic composites, and the design of new biomaterials.
Materials and methods
Materials
Three phosphatic brachiopod shells of Lingula anatina Lamarck, 1801 and Discinisca tenuis Sowerby, 1847 (SI Fig. 1) were used for this study, including both recently alive and fresh specimens (live). Recently alive shells of D. tenuis and L. anatina were collected from Walvis Bay, Namibia, and from Moreton Bay, Queensland, Australia, respectively. The fresh (live) specimens of L. anatina used in this study were collected in the Bay of Guangxi, China, and were preserved in 10% formalin. While long exposure of biological samples to formalin does not alter the organic composition, it does diminish the fracture toughness due to etching/removal of calcium phosphate. Shells of brachiopods are laminated and are covered with a non-mineralized organic layer, the periostracum, which ranges from 1 to 30 µm thick [43, 44].
Sample preparation and thermal gravimetric analysis
The three shell samples were mechanically cleaned with a scalpel, and then washed with Milli-Q water to remove organic and inorganic contaminants. Samples were soaked in hydrogen peroxide (35%; Chem-Supply, UN2014) for about 2 h to remove surface organic detritus. Varied chemical procedures, extraction temperatures, and different times have been described to achieve optimal demineralization and deproteinization of chitin matrix [45, 46]. For this study, each shell sample was demineralized with 5% acetic acid at ambient temperature for about 48 h. Subsequently, shell samples were washed several times with Milli-Q water, followed by deproteinization and/or depigmentation with 5% sodium hydroxide at ambient temperature for ~ 48 h. During these procedures the samples were monitored intermittently using Fourier transform infrared (FTIR) spectroscopy. After 24 h in sodium hydroxide, several layers were peeled off the shells using tweezers: these are termed the organic fibres. The remaining shell samples and the organic fibres were collected from the solution, washed in Milli-Q water until a pH of about 6.8 was reached, and then rinsed in cold acetone twice. The shell samples and organic fibres were then dried at room temperature.
Shells were broken into a few mm-sized pieces and heated using a thermal gravimetric analyzer (TGA) model 2050 (TA Instruments, USA) equipped with a differential thermal gravimetric (DTG) analyzer. About 10 mg of sample was measured at a heating rate of 10 °C/min over a temperature interval from 25 to 900 °C. The moisture content, organic biopolymer content, and residual ash content were determined from mass losses. The analyses were recorded twice for each sample.
α-Chitin extracted from shrimps (Sigma-Aldrich) and squid pen β-chitin were used as standards for this study. A squid pen was prepared by degrading any possible inorganic matrix with 5% hydrochloric acid at room temperature for 24 h. After washing with Milli-Q water, the squid pen sample was treated with 5% sodium hydroxide at 65 °C for 6.5 h, and was washed with Milli-Q water until a pH of about 6.8 was reached.
Calcofluor white staining and chitinase digestion test
For biochemical staining, isolated organic fibres from the brachiopod shells were rinsed with Milli-Q water, and then 25 µL of a solution of 10 g sodium hydroxide in 90 mL Milli-Q water and 10 mL glycerine were added to the samples. After 40 s, a 0.1% Calcofluor White M2R (Sigma-Aldrich; Fluorescent Brightener F-3543) solution was added and the samples were kept in the dark for ~ 30 min. Samples were rinsed twice with Milli-Q water and dried at room temperature. Images of the stained samples were captured using 40 × objective on a Zeiss fluorescence microscope system (Zeiss Axioskop 40 FL system) equipped with the Axiocam 506 mono high-resolution camera and Zeiss ZEN2 lite software. The images were acquired below the saturation level of the camera, and all images shown are representative of results.
For the chitinase digestion process, sodium hydroxide-etched samples and organic fibres were rinsed with phosphate buffer (pH 6.0), then treated with chitinase (EC 3.2.1.14, No. C-6137, Sigma-Aldrich) from Streptomyces griseus in a phosphate buffer (pH 6.0; 1 mg/mL). Control samples were treated with phosphate buffer without chitinase. The treated and control samples were incubated at 25 °C for ~ 20 h and later rinsed with Milli-Q water. Samples were allowed to dry at room temperature, before imaging without prior coating using a Phenol XL Desktop back-scattered detector scanning electron microscope (SEM).
MicroRaman and Fourier transform infrared spectroscopy
Raman spectra were recorded at room temperature using a Horiba Jobin Yvon LabRAM HR Evolution spectrometer equipped with an Olympus BX41 microscope and an automated x–y stage. A red wavelength helium–neon laser (633 nm) was used and scattered light was dispersed by a grating with 600 grooves/mm (750 nm spacing). Rayleigh radiation was blocked using an edge filter. Spectra of the organic fibres were recorded in the 400–1800 cm−1 range with an acquisition time of 60 s, 10 accumulations, and a delay time of 3 s. Twenty spectra were measured for all samples. The Raman spectrometer was calibrated before and after measurement using the 520.46–520.60 cm−1 peak of a silicon wafer. Data acquisition and spectra treatments were carried out with the commercially available program LabSpec v6 (HORIBA Jobin Yvon GmbH).
FTIR measurements were conducted as described previously [1], with 64 accumulations and a resolution of 2 cm−1. Background spectra were measured at the start of each analysis. Spectra treatments were performed using the software Origin LabPro 2017. A linear baseline was subtracted for the spectra and for the amide I region, over the range of 1580–1720 cm−1. Peak fitting of the spectra was performed to reveal the different components based on Gaussian functions.
Scanning electron microscope imaging
Both organic fibres and shell samples were mounted on aluminium SEM sample holders and gold coated for imaging, prior to analysis with a JEOL JSM-7100F field emission gun SEM at an electron energy of 10 kV and a 10 mm walking distance. For the chitinase digestion test, samples were evaluated without coating with a Phenol XL Desktop back-scattered detector SEM at an electron energy of 15 kV and ~ 5 mm walking distance.
Results and discussion
Results
Vibrational spectroscopy—microRaman and FTIR spectroscopy
Raman spectra of organic fibres from the shell samples (Fig. 1) show absorption bands that are typical of chitin (N-acetyl-D-glucosamine). The band assignments are listed in Table 1, where they are compared with the spectra of standard α-chitin and squid pen β-chitin, and literature data [1, 20, 47,48,49,50,51,52]. The spectra of the organic fibres reveal the presence of C=O (carbonyl) functional groups based on the amide I stretching vibration at ~ 1654 cm−1, and correspond primarily to the β-form of chitin. The deconvolutional fit of the amide I band in the 1580–1720 cm−1 range (SI Fig. 2) has some ubiquitous features with that of standard α-chitin, but the vibrational bands have different area percentages (SI Table 1). The rocking CH2 vibrations at 1376 cm−1 and the amide III bands in the 1202–1327 cm−1 range are predominantly assigned to organic biopolymers (Fig. 1). The CH deformation of the β-glycosidic bond band at 897 cm−1 for D. tenuis and at ~ 902 cm−1 for L. anatina is present at 895 cm−1 in standard α-chitin and at 902 cm−1 in β-chitin. Bands at 1003 cm−1 in L. anatina and 1005 cm−1 in D. tenuis are not typically present in standard α-chitin and β-chitin, and are attributed to the aromatic C–C and/or C–O stretch of fibrous insoluble protein [1, 53]. A similar β-chitin peak was reported from diatom species Thalassiosira rotula [47] and Riftia pachyptila tubeworm [52]. The vibrational bands at ~ 856 cm−1 and ~ 820 cm−1 are assigned to a combination of C–C and C–O stretching, and appear in L. anatina only. Ca-phosphate vibrational bands at 964 cm−1, 608 cm−1 and 581 cm−1 are only present in the D. tenuis spectrum (Fig. 1; SI Fig. 3). Identification of Ca-phosphate components was most likely due to the fact that both Ca-phosphate and the framework of organic biopolymers form complexes to produce shell ultrastructure.

Raman spectra (acquired using a 633-nm laser) of chitin fibre from brachiopod shells: Recent Lingula anatina (RL), fresh Lingula anatina (FL), recent Discinisca tenuis (DT), and pure β-chitin (βC) and α-chitin (αC) as a reference. The baselines of the spectra are corrected. The amide I single band at ~ 1654 cm−1 is due to chitin fibre, and is characteristic of β-chitin. This is dissimilar from α-chitin that possesses double bands at 1617 and 1654 cm−1. The amide I band of D. tenuis is broad compared to the L. anatina shells. See SI Fig. 2 for band deconvolution. D. tenuis also contains prominent phosphate (vPO43−) vibrational modes at 964 cm−1, 608 cm−1 and 581 cm−1. See Table 1 for band assignments
FTIR spectroscopy was used to probe and/or identify the molecular and functional properties of the organic fibres from the shell samples, and for comparison with the α-chitin standard and squid pen β-chitin (Fig. 2). The amide I of the insoluble matrix of L. anatina shows a broad band at 1630–1635 cm−1, similar to β-chitin. However, the spectrum of D. tenuis organic matrix reveals a broad band with a partial splitting pattern in two bands at 1652 cm−1 and 1634 cm−1, while standard α-chitin has two bands at 1656 cm−1 and 1621 cm−1. The band at 1550 cm−1 is assigned to amide II, while bands in the 1203–1308 cm−1 range are attributed to amide III. The stretching C-O bands in the 1153–950 cm−1 range (Fig. 2) are indicative of chitin. The fingerprint region of the organic fibres of the phosphate-shelled brachiopods are doublet bands at 702–708 cm−1 and 691–692 cm−1, suggesting crystalline chitin. The stretching band of the OH group for shell samples is centred at ~ 3424 cm−1, with a shoulder near 3532 cm−1. Other spectra bands for amide A (3282–3272 cm−1), amide B (3106–3095 cm−1), and the C-H stretching regions in the 2952–2851 cm−1 range are all characteristics of crystalline chitin and/or a protein motif. The bands and functional groups are assigned in detail in Table 1.

FTIR spectra of chitin fibre from brachiopod shells: Recent Lingula anatina (RL), fresh Lingula anatina (FL), recent Discinisca tenuis (DT), and pure β-chitin (βC) and α-chitin (αC) as a reference. a Shows a larger wavenumber range (4000–600 cm−1) than the expanded range (1800–600 cm−1) in (b)
Biochemical analyses of chitin matrix
Organic fibres from the shell samples were stained with the blue fluorescent stain Calcofluor White, also known as Fluorescent Brightener M2R (Fig. 3; SI Fig. 4). Calcofluor White forms hydrogen bonds with β-(1–3)- and β-(1–4)-linked polysaccharides [20], enabling identification and visualization of the chitin matrix [15].

Light microscope a, b and fluorescence microscope (a′, b′) images of the selected organic fibres after calcofluor white staining. (a, a′) Recent Lingula anatina shell; b, b′ Discinisca tenuis shell. See SI Fig. 4 for the unstained isolated organic fibres. Blue fluorescent images were recorded through an exposure time of 60 ms
When the shell samples were treated with Streptomyces griseus chitinase, no clear evidence of chitinase activity was apparent under a light microscope. However, SEM imaging showed that the chitin-bearing fibres of the lingulid shells were damaged and disorganized (Fig. 4). Unlike the control samples (SI Fig. 5), chitinase targeted the chitin in the shells, leading to degradation and perforation. There was no distinctive difference between the isolated organic fibres and the NaOH-treated samples (not shown).

Back-scattered scanning electron microscope images of the sodium hydroxide-treated brachiopod shells digested with chitinase Streptomyces griseus in phosphate buffer. (a, a′) Recent Lingula anatina shell; b, b′) Discinisca tenuis shell. Images were obtained after 20 h of incubation in chitinase solution. See SI Fig. 5 for control test
Morphology of the organic fibre extracts
Surface imaging of the organic fibres shows ultrastructural features of biopolymers, although the fibre morphology varies from shell to shell (Fig. 5). Organic fibres in L. anatina contain pores between the fibres (Fig. 5a), and closer imaging of the radiating units of the ridges reveals organic granules (Fig. 5a’). The organic fibre extract from D. tenuis shows lamellar units (Fig. 5b) that are comparable with the rheomorphic folding discussed by previous authors [21]. This suggests that the polymeric structure of the polysaccharide is preserved after acid (demineralization) and sodium hydroxide (deproteination) treatments. A polymeric structure as rheomorphic folding is observed in D. tenuis (Fig. 5b’), unlike the fibrous structure found in the organic fibre extract of L. anatina.

Field emission gun scanning electron microscope images of β-chitin organic fibre from brachiopod shells. a Representative fibre surface of Lingula anatina showing pores and radiating units of the ridges. a′ The ridge is expanded to show nanoparticle granules. b, b′ External surface of Discinisca tenuis organic fibre showing a lamellar ultrastructure
Structural imaging of brachiopod shells after demineralization and deproteination
In the shells of L. anatina, there are apatite-rich lamellae and organophosphatic-rich lamellae (Fig. 6a, b). The thickness of the apatite-rich layers is up to 840 nm, whereas the thickness of the organophosphatic-rich lamellae is up to 1.5 µm (Fig. 6a). In the organophosphatic lamellae, the organic sheet laminations are parallel to the Ca-phosphate sub-lamellae (Fig. 6b), but do not penetrate into the apatite-rich lamellae which are approximately perpendicular (Fig. 6b). Within the apatite-rich laminae of L. anatina, there are nanoscale fibrils that are mineralized with Ca-phosphate nanoparticles (~ 50–70 nm) to form a fibrous biocomposite (Fig. 6c). FEG-SEM imaging of L. anatina reveals composite organic sheets with Ca-phosphate granules (SI Fig. 6a). The organic sheets or lamellae coated with nanoparticle granules (in SI Fig. 6a) represent several layers of organic sheets that were peeled off the shells for the characterization of the chitin matrix (see experimental). Some biomineralized nanofibrils are arranged approximately perpendicular to the micro-laminations (SI Fig. 6a).

Field emission gun scanning electron microscope images of sodium hydroxide-treated Lingula anatina a–c and Discinisca tenuis d–f shells. a Vertical fracture surface of a valve of a recent L. anatina shell showing apatite-rich (Ap-rich) and organophosphatic-rich (OrgPhos-rich) laminae. b Organic sheets/fibres (blue arrows) in the organophosphatic-rich layer are highlighted. These sheets are arranged roughly perpendicular to the apatite-rich layer, but do not penetrate into it. c Biomineralized ruptured fibrils (green arrows) on the fracture surface of an apatite-rich layer. The nanoparticle granule sizes are in the ~ 50–70 nm range. d Vertical fracture surface of a valve of D. tenuis shell depicting canals. e Nanoparticle granules in the ~ 50–100 nm range and organic fibrils (white arrows). f Vertical fracture section of a valve of D. tenuis shell showing nano-rods (dashed arrows), mosaics (mc), and spherical micro-apatite in the 50–60 nm range (arrowheads). Asterisk in a denotes the inner surface of the L. anatina shell. Arrowhead in d points towards the inner surface of the D. tenuis shell
After demineralization and deproteination, the shell of D. tenuis reveals only one major and continuous compact layer that is penetrated by canals with diameters of ~ 90–120 nm (Fig. 6d). This conspicuous sets of canals consists of various aggregates of organic-coated granules in the 50–100 nm range (Fig. 6e; SI Fig. 6b). As shown in Fig. 6f, the shell of D. tenuis is composed of ~ 50–60 nm spherical Ca-phosphate nanoparticles, with the aggregates of organic-coated granules naturally compacted to form mosaics (see Fig. 6f), similar to a previous study[21]. Rod-shaped crystallites (Fig. 6f) may perhaps be self-organized chitin fibrils, which are coated with proteins before mineralization. The apatitic components entwine with the organic fibrils (Fig. 6e, white arrows), resulting in a stratiform succession of microscale laminae [42, 44].
Thermal gravimetric and differential thermal gravimetric analyses
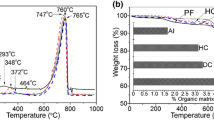
TGA and DTG analyses of brachiopod shell samples, standard α-chitin, and standard β-chitin were carried out to detect degradation features in the organic matrix. TGA weight loss and multistage decompositional steps occur for the brachiopod shells, α-chitin, and β-chitin (Fig. 7). The first weight loss of 5.1 wt% to 5.6 wt% for both L. anatina and D. tenuis occurs over the 30–200 °C range, and is attributed to removal of water. The second weight loss of 26.6 wt% for L. anatina and 12.9 wt% for D. tenuis (Fig. 7a) is recorded in the 200–650 °C range and is interpreted to be due to the combustion of a complex mixture of shell macromolecules, including chitin fibre and a protein moiety [54, 55], that is occluded in the apatitic shells. The maximum DTG value for the commercially obtained α-chitin is recorded at 357 °C, whereas the peak shift occurs at 335 °C for L. anatina and 347 °C for D. tenuis, comparable with the squid pen β-chitin standard at 341 °C (Fig. 7b). These maximum DTG values for the shells are in the 250–350 °C range for thermal stability of β-chitin [54, 55]. Lower temperature peaks are considered to be a measure of lower thermal stability [54]. However, the DTG thermogram of L. anatina is similar to α-chitin, and shows a broad peak at around 600 °C (Fig. 7b), which is attributed to the thermal degradation of a pyranose ring and the decomposition of residual carbon [54]. DTG thermograms of the shells reveal apatitic peaks at 840 °C for L. anatina and 740 °C for D. tenuis. The residual mass after heating to 900 °C is 65.9 wt% for L. anatina and 79.6 wt% for D. tenuis. This suggests the presence of inorganic constituents that were not extracted during the acid-treatment stage.

a Thermal gravimetric analysis data and b differential thermal gravimetric analysis data of shell biominerals (LA: Lingula anatina, DT: Discinisca tenuis), and pure β-chitin (βC) and pure α-chitin (αC) as a reference. The bar chart in a represents the calculated total polysaccharide chitin contents in the 200–650 °C range. L. anatina and D. tenuis comprise 26.6 wt.% and 12.9 wt.% of the total polysaccharide matrix, respectively
Discussion
The presented data provide a much better characterization of organic fibres in two organophosphatic brachiopod shells that have well established lineages in the fossil record, compared to previous studies [41]. After purification, the thermal degradation results confirm the presence of a chitin matrix in the shells of two representative taxa belonging to separate superfamilies of lingulid organophosphatic brachiopods, L. anatina (Linguloidea) and D. tenuis (Discinoidea). The L. anatina shells are composed of twofold more abundant chitin matrix compared to the D. tenuis shells. Chitin is rarely found in a pure state in nature, and is most commonly linked covalently with proteins [13, 56]. Pure chitin can be difficult to isolate, for example in mollusc shells [4, 19] and brachiopod shells [41]. Nevertheless, modern chitin staining techniques, such as the use of the fluorochrome Calcofluor White which binds to D-glucopyranose polysaccharides enable identification of chitin [15, 22], see Fig. 3. Calcofluor White is widely used for the rapid identification of chitin in chitin-containing organisms, i.e. fungi [15, 57], chytrid parasites on phytoplanktonic eukaryotes and prokaryotes [58], and in the shells of molluscs [20, 22, 59].
In this study, chitinase perforated and/or degraded the studied samples, thus confirming the presence of a chitin matrix in the shells of brachiopods. In the shell of L. anatina, the chitin layer was not readily degraded by chitinase [39]. Chitin matrix is covalently linked with other biomolecules, or may be masked by other organic matrices such as glycosaminoglycans and extracellular matrix [1, 39, 42]. Also, when chitin forms complexes with other biomolecules such as melanin, bromotyrosine, and/or inorganic components such as calcium carbonate, chitinase has little or no effect on such chitin-containing biomaterials using light and fluorescent microscope [15, 60]. Still, after chitinase activity, the SEM images (Fig. 4) are comparable to the SEM images of fossilized brachiopod shell structures from the lower Cambrian [61]. The basic effect of chitinase in this study extends our understanding concerning the evolution of biomineral formation in phosphatic small shelly fossils [61, 62].
Chitin can be readily distinguished using vibrational spectroscopy, such as by Raman and FTIR spectroscopy, due to its distinctive bands [1, 47,48,49,50,51]. The analyses presented here using vibrational spectroscopy reveal the biochemical composition of brachiopod shells, with chitin fibres entwined with minor shell proteins, supporting previous work [41]. The spectral bands of the organic fibres compare closely with the chitinous system containing amino-polysaccharide chitin (Figs. 1 and 2). The FTIR and Raman spectra bands associated with the amide I vibrational modes reveal broad and complex bands that are centred at high frequencies and are attributed to β-chitin. The Raman band is centred at ~ 1655 cm−1 for brachiopod shells, and is commonly assigned to stretching of the C=O group hydrogen bonded to the N–H group of the neighbouring intra-sheet chain [49]. In the FTIR spectra, the amide I position is at 1652 cm−1 for D. tenuis. The band shifts by ~ 19 ± 3 cm−1 towards a lower frequency for L. anatina (Table 1). The FTIR spectra 1634 cm−1 band for D. tenuis and a 1646 cm−1 band for L. anatina are present as a shoulder, and are likely due to defects in the crystalline structure of the chitin fibre, or possibly a shell-associated protein. The deconvolutional fit of the amide I band for the FTIR spectra in the 1580–1720 cm−1 range also reveals a band at ~ 1622 cm−1, compared with the α-chitin band at 1620 cm−1 (SI Fig. 7). The occurrence of this band may show a specific hydrogen bond of carbonyl (C=O) with the hydroxymethyl group of the next chitin residue of the same chain, or may be due to the presence of the enol form of the amide moiety [49, 63, 64]. A ~ 1003 cm−1 vibrational ring peak is visible in the Raman spectra of the brachiopod shells, and is commonly observed in the published spectra of chitin [47, 52] and collagen [53, 65]. Recent analyses provide comprehensive data on D. tenuis and L. anatina shells, with identification of a collagen matrix as the primary protein motif within the inorganic matrix of phosphate-shelled brachiopods [1].
Total organic macromolecules in the shells of D. tenuis is 24.6 wt.% and for L. anatina is 40.6 wt.% [1]. After demineralization and deproteination, the amount of organic macromolecules reduced to 12.9 wt.% for D. tenuis and 26.6 wt.% for L. anatina. The extraction methods and calculation from TGA reveal the presence of phosphatic soluble organic macromolecules in L. anatina (14.0 wt.%) and D. tenuis (11.7 wt.%). Based on the previous work of Lévêque et al. [66], there may be soluble organic matrices in Ca-phosphate shells that greatly contribute and/or influence the nucleation and growth of brachiopod shells. The inorganic matrix provides the structural strength, stiffness and rigidity for many invertebrate shells, including organophosphatic brachiopod shells [67]. However, polysaccharide chitin offers increased strength and flexibility [67, 68].
L. anatina is clearly laminated with an alternating chitinous system and biomineralized chitin-phosphate layers. In contrast, lamination of the mineralization framework is less pronounced in the sessile epibenthic D. tenuis [67, 68]. Biomineralized laminae provide strength to the shells of L. anatina, and allow bending flexibility [68], a factor that may be important in the complex shell-first burrowing of living lingulids [27, 32,33,34]. However, flexibility is less important for the shallow marine epibenthic phosphate-shelled brachiopods [67]. Further, L. anatina shells contain an organic primary layer that protects the secondary layer from incipient cracks that can occur during the movement of the animal through the sediment [42, 43]. The flexible secondary layer of L. anatina consists of stacks of alternating mineralized and non-mineralized β-chitin sheets [42], see Fig. 6. Chitin chains lie parallel with the c-axes of the apatite components, and chitin fibrils are usually coated or covered with spherical and/or elongated shapes of Ca-phosphate particles [1, 39, 42]. The organized structure of the L. anatina shell helps divert fractures, and thus provides high flexibility while the animal burrows itself into and through the sediment [67, 68]. The analyses reported here reveal the architectural structures and polysaccharide matrix of two important phosphate-shelled brachiopods and demonstrate further a practical bioanalytical approach for analysing lophotrochozoan shells.
Conclusions
-
1.
Based on FEG-SEM, vibrational spectroscopic analyses, and biochemical studies, phosphate-shelled brachiopods (Lingula anatina and Discinisca tenuis) contain chitin fibre composites with a fibrous protein concentration, which create suitable microenvironments for inorganic nanoparticle aggregation.
-
2.
The polysaccharide-apatitic matrix composition of the shells of L. anatina is distinctly different from that of D. tenuis. For L. anatina, the polysaccharide fibre component is internally reinforced by a nanoscale Ca-phosphate component and is distinctively laminated. D. tenuis used a different shell architecture, because it contains a continuous compact layer with canals and a stratiform succession of microscale laminae.
-
3.
This study highlights the differences in the shell ultrastructure of L. anatina and D. tenuis. L. anatina has a distinctive two-layer structure with apatite-rich lamellae and organophosphatic-rich lamellae. D. tenuis has a major and continuous compact layer, with organic fibrils entwined or intercalated with a Ca-phosphate mineral. However, based on the extraction methods and calculation from TGA, the amount of soluble shell macromolecules is similar, 14.0 wt.% for L. anatina and 11.7 wt.% for D. tenuis.
-
4
It is proposed that the high polysaccharide chitin matrix content of L. anatina may contribute to its complex burrowing mechanism, in which the valves function as flexible digging tools, combined with the injection of water in a shell-first burrowing action that produces U-shaped burrows. The shell of L. anatina is composed of chitin sheets or lamellae, forming a laminar shell design that produces a flexible shell. Taken together, a flexible shell gives an advantage for burrowing and prompt movements in the burrow [26, 27, 32,33,34,35].
References
Agbaje OB, George SC, Zhang Z, Brock GA, Holmer LE (2020) Characterization of organophosphatic brachiopod shells: spectroscopic assessment of collagen matrix and biomineral components. RSC Adv 10(63):38456–38467
Barthelat F, Yin Z, Buehler MJ (2016) Structure and mechanics of interfaces in biological materials. Nat Rev Mater 1(4):1–16
Wysokowski M, Jesionowski T, Ehrlich H (2018) Biosilica as a source for inspiration in biological materials science. Am Mineral 103(5):665–691
Agbaje OBA, Shir IB, Zax DB, Schmidt A, Jacob DE (2018) Biomacromolecules within bivalve shells: Is chitin abundant? Acta Biomater 80:176–187
Neville AC (1993) Biology of fibrous composites: development beyond the cell membrane. Cambridge University Press, New York
Tsurkan D, Wysokowski M, Petrenko I, Voronkina A, Khrunyk Y, Fursov A, Ehrlich H (2020) Modern scaffolding strategies based on naturally pre-fabricated 3D biomaterials of poriferan origin. Appl Phys A 126(5):1–9
Gupta NS (2010) Chitin: formation and diagenesis. Springer Science & Business Media, Berlin, Germany
Kim SK (2010) Chitin, chitosan, oligosaccharides and their derivatives: biological activities and applications. CRC Press, Taylor and Francis Group, Florida
Pillai CK, Paul W, Sharma CP (2009) Chitin and chitosan polymers: chemistry, solubility and fiber formation. Prog Polym Sci 34(7):641–678
Ehrlich H (2010) Chitin and collagen as universal and alternative templates in biomineralization. Int Geol Rev 52(7–8):661–699
Dweltz N (1961) The structure of β-chitin. BBA 51(2):283–294
Minke R, Blackwell J (1978) The structure of α-chitin. J Mol Biol 120(2):167–181
Rudall K (1963) The chitin/protein complexes of insect cuticles. Adv Insect Phys 1:257–313
Jang MK, Kong BG, Jeong YI, Lee CH, Nah JW (2004) Physicochemical characterization of α-chitin, β-chitin, and γ-chitin separated from natural resources. J Polym Sci A Polym Chem 42(14):3423–3432
Tsurkan MV, Voronkina A, Khrunyk Y, Wysokowski M, Petrenko I, Ehrlich H (2020) Progress in chitin analytics. Carbohydr Polym 252:117204. https://doi.org/10.1016/j.carbpol.2020.117204
Rudall K, Kenchington W (1973) The chitin system. Biol 48(4):597–633
Fabritius H, Sachs C, Raabe D, Nikolov S, Friák M, Neugebauer J (2011) Chitin in the exoskeletons of arthropoda: from ancient design to novel materials science, In Chitin, Springer 35–60
Vincent JF (2002) Arthropod cuticle: a natural composite shell system. Compos Part A Appl Sci Manuf 33(10):1311–1315
Osuna-Mascaró AJ, Cruz-Bustos T, Marin F, Checa AG (2015) Ultrastructure of the interlamellar membranes of the nacre of the bivalve Pteria hirundo, determined by immunolabelling. PLoS ONE 10(4):e0122934. https://doi.org/10.1371/journal.pone.0122934
Agbaje OB, Dominguez JG, Jacob DE (2021) Organic biopolymers of venus clams: Collagen-related matrix in the bivalve shells with crossed-lamellar ultrastructure. Biochem Biophys Rep 26:100939. https://doi.org/10.1016/j.bbrep.2021.100939
Williams A, Cusack M, Buckman JO (1998) Chemico–structural phylogeny of the discinoid brachiopod shell. Philos Trans R Soc B 353(1378):2005–2038
Agbaje OB, Thomas DE, Dominguez JG, Mclnerney BV, Kosnik MA, Jacob DE (2019) Biomacromolecules in bivalve shells with crossed lamellar architecture. J Mater Sci 54(6):4952–4969. https://doi.org/10.1007/s10853-018-3165-8
Oudot M, Shir IB, Schmidt A, Plasseraud L, Broussard C, Neige P, Marin F (2020) A nature’s curiosity: the Argonaut “Shell” and its organic content. Crystals, MDPI 10(9):839. https://doi.org/10.3390/cryst10090839
Meyers MA, Chen PY, Lin AYM, Seki Y (2008) Biological materials: structure and mechanical properties. Prog Mater Sci 53(1):1–206
Naleway SE, Porter MM, McKittrick J, Meyers MA (2015) Structural design elements in biological materials: application to bioinspiration. Adv Mater 27(37):5455–5476
Emig CC (1997) Ecology of inarticulated brachiopods, Treatise on invertebrate paleontology, part H, brachiopoda (revised) 1: 473–495
Thayer CW, Steele-Petrovic HM (1975) Burrowing of the lingulid brachiopod Glottidia pyramidata: its ecologic and paleoecologic significance. Lethaia 8(3):209–221
Harper DA, Popov LE, Holmer LE (2017) Brachiopods: origin and early history. Palaeontology 60(5):609–631
Williams A (2000) Treatise on invertebrate paleontology: Part H, Brachiopoda, Linguliformea, Craniiformea, and Rhynchonelliformea (part), The Geological Society of America Inc. and The University of Kansas, Lawrence Vol. 3
Williams A, Brunton H, Carlson S, Alvarez F, Baker P, Bassett M, Boucot A, Carter J, Cocks L, Cohen B (2007) Treatise on Invertebrate Palaeontology (Part H, Brachiopoda Revised). Volume 6: Supplement, Boulder, CO
Veizer J, Ala D, Azmy K, Bruckschen P, Buhl D, Bruhn F, Carden GA, Diener A, Ebneth S, Godderis Y (1999) 87Sr/86Sr, δ13C and δ18O evolution of Phanerozoic seawater. Chem Geol 161(1–3):59–88
Zhang Z, Shu D, Han J, Liu J (2005) Morpho-anatomical differences of the Early Cambrian Chengjiang and Recent lingulids and their implications. Acta Zool 86(4):277–288
Savazzi E (1986) Burrowing sculptures and life habits in Paleozoic lingulacean brachiopods. Paleobiology 12:46–63
Savazzi E (1991) Burrowing in the inarticulate brachiopod Lingula anatina. Palaeogeogr Palaeoclimatol Palaeoecol 85(1–2):101–106
Kowalewski M (1996) Taphonomy of a living fossil; the lingulide brachiopod Glottidia palmeri Dall from Baja California. Mexico Palaios 11(3):244–265
Zonneveld JP, Beatty TW, Pemberton SG (2007) Lingulide brachiopods and the trace fossil Lingulichnus from the Triassic of western Canada: implications for faunal recovery after the end-Permian mass extinction. Palaios 22(1):74–97
Ehrlich H, Koutsoukos PG, Demadis KD, Pokrovsky OS (2009) Principles of demineralization: modern strategies for the isolation of organic frameworks: Part II. Decalcification Micron 40(2):169–193
Murdock DJ (2020) The ‘biomineralization toolkit’ and the origin of animal skeletons. Biol 95(5):1372–1392
Iwata K (1981) Ultrastructure and mineralization of the shell of Lingula unguis Linne, (Inarticualte Brachiopod), Journal of the Faculty of Science, Hokkaido University. Series 4, Geology and mineralogy 20(1): 35–65
Jope M (1979) The protein of brachiopod shell—VI. C-terminal end groups and sodium dodecylsulphate-polyacrylamide gel electrophoresis: molecular constitution and structure of the protein. Comp Biochem Physiol Part B Biochem Mol Biol 63(1):163–173
Neary MT, Reid DG, Mason MJ, Friščić T, Duer MJ, Cusack M (2011) Contrasts between organic participation in apatite biomineralization in brachiopod shell and vertebrate bone identified by nuclear magnetic resonance spectroscopy. J R Soc Interface 8(55):282–288
Williams A, Cusack M, MacKay S (1994) Collagenous chitinophosphatic shell of the brachiopod Lingula. Philos Trans R Soc B 346(1316):223–266
Merkel C, Griesshaber E, Kelm K, Neuser R, Jordan G, Logan A, Mader W, Schmahl WW (2007) Micromechanical properties and structural characterization of modern inarticulated brachiopod shells. J Geophys Res Biogeosci 112:G02008
Williams A, Cusack M (1999) Evolution of a rhythmic lamination in the organophosphatic shells of brachiopods. J Struct Biol 126(3):227–240
Percot A, Viton C, Domard A (2003) Characterization of shrimp shell deproteinization. Biomacromol 4(5):1380–1385
Younes I, Rinaudo M (2015) Chitin and chitosan preparation from marine sources. Structure, properties and applications. Mar Drugs 13(3):1133–1174
Brunner E, Ehrlich H, Schupp P, Hedrich R, Hunoldt S, Kammer M, Machill S, Paasch S, Bazhenov V, Kurek D (2009) Chitin-based scaffolds are an integral part of the skeleton of the marine demosponge Ianthella basta. J Struct Biol 168(3):539–547
Cárdenas G, Cabrera G, Taboada E, Miranda SP (2004) Chitin characterization by SEM, FTIR, XRD, and 13C cross polarization/mass angle spinning NMR. J Appl Polym Sci 93(4):1876–1885
Focher B, Naggi A, Torri G, Cosani A, Terbojevich M (1992) Structural differences between chitin polymorphs and their precipitates from solutions—evidence from CP-MAS 13C-NMR. FT-IR and FT-Raman Spectroscopy Carbohydr Polym 17(2):97–102
Galat A (1980) Study of the Raman scattering and infrared absorption spectra of branched polysaccharides. Acta Biochim Pol 27(2):135–142
Lavall RL, Assis OB, Campana-Filho SP (2007) β-Chitin from the pens of Loligo sp.: Extraction and characterization. Bioresour Technol 98(13):2465–2472
Ehrlich H, Maldonado M, Spindler KD, Eckert C, Hanke T, Born R, Goebel C, Simon P, Heinemann S, Worch H (2007) First evidence of chitin as a component of the skeletal fibers of marine sponges. Part I. Verongidae (Demospongia: Porifera). J Exp Zool B Mol Dev Evol 308(4):347–356
Wopenka B, Kent A, Pasteris JD, Yoon Y, Thomopoulos S (2008) The tendon-to-bone transition of the rotator cuff: a preliminary Raman spectroscopic study documenting the gradual mineralization across the insertion in rat tissue samples. Appl Spectrosc 62(12):1285–1294
Hassainia A, Satha H, Boufi S (2018) Chitin from Agaricus bisporus: Extraction and characterization. Int J Biol Macromol 117:1334–1342
Kaya M, Mujtaba M, Ehrlich H, Salaberria AM, Baran T, Amemiya CT, Galli R, Akyuz L, Sargin I, Labidi J (2017) On chemistry of γ-chitin. Carbohydr Polym 176:177–186
Kurita K (2006) Chitin and chitosan: functional biopolymers from marine crustaceans. Mar Biotechnol 8(3):203
Hoch H, Galvani C, Szarowski D, Turner J (2005) Two new fluorescent dyes applicable for visualization of fungal cell walls. Mycologia 97(3):580–588
Rasconi S, Jobard M, Jouve L, Sime-Ngando T (2009) Use of calcofluor white for detection, identification, and quantification of phytoplanktonic fungal parasites. Appl Environ Microbiol 75(8):2545–2553
Bezares J, Asaro RJ, Hawley M (2008) Macromolecular structure of the organic framework of nacre in Haliotis rufescens: implications for growth and mechanical behavior. J Struct Biol 163(1):61–75
Machałowski T, Wysokowski M, Tsurkan MV, Galli R, Schimpf C, Rafaja D, Brendler E, Viehweger C, Żółtowska-Aksamitowska S, Petrenko I (2019) Spider chitin: an ultrafast microwave-assisted method for chitin isolation from Caribena versicolor spider molt cuticle. Molecules 24(20):3736
Balthasar U (2004) Shell structure, ontogeny and affinities of the Lower Cambrian bivalved problematic fossil Mickwitzia muralensis Walcott, 1913. Lethaia 37(4):381–400
Devaere L, Holmer L, Clausen S, Vachard D (2014) Oldest mickwitziid brachiopod from the Terreneuvian of southern France. Acta Palaeontol Pol 60(3):755–768
Kumirska J, Czerwicka M, Kaczyński Z, Bychowska A, Brzozowski K, Thöming J, Stepnowski P (2010) Application of spectroscopic methods for structural analysis of chitin and chitosan. Mar Drugs 8(5):1567–1636
Rinaudo M (2006) Chitin and chitosan: properties and applications. Prog Polym Sci 31(7):603–632
Morris MD, Mandair GS (2011) Raman assessment of bone quality. Clin Orthop Relat Res 469(8):2160–2169
Lévêque I, Cusack M, Davis SA, Mann S (2004) Promotion of fluorapatite crystallization by soluble-matrix proteins from Lingula anatina shells. Angew Chem Int Ed 43(7):885–888
Merkel C, Deuschle J, Griesshaber E, Enders S, Steinhauser E, Hochleitner R, Brand U, Schmahl WW (2009) Mechanical properties of modern calcite-(Mergerlia truncata) and phosphate-shelled brachiopods (Discradisca stella and Lingula anatina) determined by nanoindentation. J Structur Biol 168(3):396–408
Schmahl WW, Griesshaber E, Merkel C, Kelm K, Deuschle J, Neuser RD, Göetz A, Sehrbrock A, Mader W (2008) Hierarchical fibre composite structure and micromechanical properties of phosphatic and calcitic brachiopod shell biomaterials–an overview. Mineral Mag 72(2):541–562
Acknowledgements
OBAA is grateful to Uppsala University for support through the VR Project number 2018-03390. The research for this paper was supported by the Swedish Research Council (VR Project no. 2018-03390 to LEH, GAB and SCG), by the National Natural Science Foundation of China (41720104002, 41621003 and 41890844 to NWU members), and by a Zhongjian Yang Scholarship to LEH from the Department of Geology, Northwest University, Xi’an. GAB’s research is also funded by a 1000 Talent Shaanxi Province Fellowship at Northwest University, Xi’an.
Funding
Open access funding provided by Uppsala University.
Author information
Authors and Affiliations
Contributions
OBAA designed the study, carried out the analyses, interpreted the data, and prepared the original manuscript. YL and ZZ provided fresh (live) specimens of Lingula anatina; LEH provided recently alive Discinisca tenuis shells, and GAB provided recently alive Lingula anatina shells. KCD carried out the fluorescence imaging. LEH, GAB and SCG coordinated the study. All authors reviewed the manuscript and gave final approval for publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Handling Editor: Stephen Eichhorn.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Agbaje, O.B.A., Brock, G.A., Zhang, Z. et al. Biomacromolecules in recent phosphate-shelled brachiopods: identification and characterization of chitin matrix. J Mater Sci 56, 19884–19898 (2021). https://doi.org/10.1007/s10853-021-06487-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-021-06487-9