
Abstract
Heart failure (HF) is a growing epidemic with high morbidity and mortality at an international scale. The apelin–APJ receptor pathway has been implicated in HF, making it a promising therapeutic target. APJ has been shown to be activated by a novel endogenous peptide ligand known as Elabela (ELA, also called Toddler or Apela), with a critical role in cardiac development and function. Activation of the ELA–APJ receptor axis exerts a wide range of physiological effects, including depressor response, positive inotropic action, diuresis, anti-inflammatory, anti-fibrotic, and anti-remodeling, leading to its cardiovascular protection. The ELA–APJ axis is essential for diverse biological processes and has been shown to regulate fluid homeostasis, myocardial contractility, vasodilation, angiogenesis, cellular differentiation, apoptosis, oxidative stress, cardiorenal fibrosis, and dysfunction. The beneficial effects of the ELA–APJ receptor system are well-established by treating hypertension, myocardial infarction, and HF. Additionally, administration of ELA protects human embryonic stem cells against apoptosis and stress-induced cell death and promotes survival and self-renewal in an APJ-independent manner (X receptor) via the phosphatidylinositol 3-kinase/Akt pathway, which may provide a new therapeutic approach for HF. Thus, targeting the ELA–APJ axis has emerged as a pre-warning biomarker and a novel therapeutic approach against progression of HF. An increased understanding of cardiovascular actions of ELA will help to develop effective interventions. This article gives an overview of the characteristics of the ELA–apelin–APJ axis and summarizes the current knowledge on its cardioprotective roles, potential mechanisms, and prospective application for acute and chronic HF.
Similar content being viewed by others

Introduction
Internationally, heart failure (HF) is a leading cause of mortality and morbidity, especially in developed countries with an aging population [1, 2]. To address this tough challenge, new clinical therapies for HF need to be developed [3]. Apelin was firstly discovered in 1998 as the endogenous peptide capable of binding the APJ, which was originally described as an orphan G protein–coupled receptor (GPCR) [4, 5]. Clinical and experimental studies clearly support multiple cardioprotective effects of the apelin–APJ axis [6,7,8]. Recently, the apelin receptor has been shown to be activated by a novel endogenous peptide ligand known as Elabela (ELA, also called Toddler or Apela), with an important role in cardiovascular development and function [9, 10]. Unlike apelin, the expression of ELA is mainly enriched in embryonic stem cells (ESCs), pluripotent stem cells, the kidney, prostate, and vascular endothelium [9, 11,12,13]. Notably, ELA and apelin share the same receptor and exert similar biological effects. Therefore, a family consisting of ELA, apelin, and APJ named the apelinergic system plays cardiotonic, diuretic, depressor, and cardiorenal protective roles (Table 1) [14,15,16,17,18,19,20,21,22,23,24,25,26,27,28]. The underlying mechanisms of the ELA–APJ axis remain largely unclear despite their clinical importance. Furthermore, some studies have introduced different isoforms of ELA as agonists of APJ and future medications for HF [17, 18, 29]. This review discusses the biological characteristics of the ELA–APJ axis, its cardioprotective effects, and its potential mechanisms for treating HF.
Discovery of ELA
ELA is a hormonal peptide originally detected in ESCs in which it plays a role in embryonic development via secretion and combination with APJ [9, 10]. A previous study found that the transcript of the ELA/Apela/Toddler gene, AK092578, was likely to be a noncoding RNA in humans [30]. Given its high expression in undifferentiated ESCs and fast downregulation in differentiated ESCs, ELA functions as a secretory peptide with important effects in early stage of embryo. ELA has been demonstrated to have an essential role in heart genesis and proper endoderm differentiation in the zebra fish model via the combination of APJ receptor. The phenotype of ELA mutants in heart development resembles APJ mutants but cannot be duplicated by apelin mutants, implying that ELA is a new-found ligand of APJ [4, 14, 31]. Soon after, a short, conserved, and secreted peptide named Toddler was discovered during zebra fish embryogenesis with promotion of gastrulation movements [10]. These discoveries confirmed that the ELA–APJ receptor pathway participates in regulating circulation system and embryonic development [32]. The discovery of ELA, a novel APJ receptor agonist, gives rise to the intriguing prospect of adjusting the ELA–apelin–APJ receptor system in HF and other cardiovascular disorders.
Biological characteristics of ELA
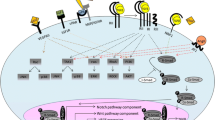
ELA gene contains three exons and is located on chromosome 4. ELA-54, as a full-length peptide, is detected in the Golgi apparatus, which is cleaved into ELA-32 as its mature secretory form (Fig. 1) [4, 9]. Mature ELA binds to the APJ receptor to become biologically active [4, 13, 14, 17, 18]. Positively charged amino acids in ELA peptides play a critical role in its receptor-binding affinity. ELA-11, without positively charged amino acids, displays a 100-fold decrease in affinity when compared with the longer ELA peptides with positively charged amino acids [13, 17]. Both ELA-32 and [Pyr1] apelin-13 possess the same affinity toward APJ in the β-arrestin assay. However, ML221, a nonpeptide small molecule antagonist, blocks the binding of ELA-32 and [Pyr1] apelin-13 with APJ to a similar extent as the positively amino acids. This provides additional evidence that these ligands may bind to the same or overlapping sites on the receptor [13]. Both ELA and apelin are detectable in human plasma at just subnanomolar levels, indicating that ELA and apelin may work as locally released autocrine/paracrine mediators, rather than as circulating hormones [13]. It was a well-established viewpoint that the levels of apelinergic system elements are diverse in different types of tissue/organ [11]. The levels of APJ, apelin, and ELA are higher in noncardiomyocyte cells than in cardiomyocytes in the hearts of C57/BL6 mice. However, levels of ELA should not be regarded as the equivalent of apelin. Both APJ and apelin expression levels were lower in fibroblasts than in endothelial cells (ECs), but the expression levels of ELA showed no remarkable difference between the two cell types [15]. These data help us to understand the complexity of the apelinergic system and potential differences between apelin and ELA from a unique perspective.

The regulatory roles and underlying mechanisms of ELA-APJ axis in heart failure. (a) ELA-54 is the full-length peptide via transcription and translation. ELA-54 is subsequently cleaved into the mature secretary peptide ELA-32 in Golgi apparatus, which exerts its biologicl effects by autocrine or paracrine. (b) The combination of ELA and APJ receptor activates intracellular signaling pathways, which partialy atributes to the recruitment of β-arrestin and internalization of the ELA-APJ complex. In addition, ELA binds an unkown receptor (X receptor), instead of APJ receptor, and plays a key role in the cell growth, survival, and self-renewal of hESCs or the OCCC cell lines by activating the PI3K/Akt phosphorylation signaling pathway or by suppressing the p53 signaling pathway. More importantly, the ELA-APJ receptor axis has been shown to regulate cellular differentiation, apoptosis, inflammation, oxidative stress, fibrosis, remodeling, vasodilation, cardiac contractility, cellular survival, and stem cell renewal in heart failure through the MEK/ERK, GATA4/Tbx5, AMPK/mTOR, and TGFβ/MMP9 signaling pathways, respectively. ELA, Elabela; X receptor, unknown receptor; hESCs, human embryonic stem cells; ECs, endothelial cells; HUVECs, human umbilical vein endothelial cells; CMs, cardiomyocytes; CFs, cardiofibroblasts; OCCC, ovarian clear cell carcinoma cell line; ER, endoplasmic reticulum; ERK1/2, extracellular signal-regulated kinase 1/2; MEK1/2, mitogen-activated protein kinase/external-signal regulated kinase; mTORC1, mammalian target of rapamycin complex 1; ACE, angiotensin converting enzyme; Ang II, angiotensin II; AT1, angiotensin II type 1 receptor; NF-κB, nuclear factor κ-B; TGF-β, transforming growth factor-β; MMP9, matrix metallopeptidase 9; TNF-α, tumor necrosis factor-α
The changes of ELA and apelin levels in patients with HF
Because of the importantly biological effect of the apelinergic system, clinical trials have been conducted to reveal the differences and changes between patients and healthy controls. Children with HF secondary to congenital heart disease had lower levels of serum apelin than health controls. Further analysis indicated that the level of serum apelin had a negative correlation with prognosis of patients with HF [33]. Optimized drug therapy, including treatment with spironolactone, led to increased serum apelin-12 levels among HF patients with reduced left ventricular (LV) ejection fraction (LVEF) [34]. Apelin level was significantly lower in dialyzed patients with cardiovascular diseases and had a negative association with cardiac function [35]. Low levels of plasma apelin were associated with more severe LV systolic and diastolic dysfunction in hypertensive patients [36]. Although some researches revealed no correlation between apelin levels of peripheral venous blood and the severity and prognosis of HF, the myocardial apelin production has been shown to be reduced in humans with LV systolic dysfunction [37]. A possible explanation for these conflicting conclusions could be the existence of diverse isomers. Whether the samples were extracted also influenced the accuracy of serum ELA levels [38]. To date, there is still no clinical trial to explore the exact serum levels of ELA between patients with HF and healthy population. Further efforts should be made to investigate whether the serum level or the expression of ELA in the cardiovascular system are different between patients with and without HF.
The ELA–APJ axis and cardiac contractility
As a key signaling pathway regulating cardiovascular hemodynamics, apelin has been proven to participate in hemodynamic regulation since it was first discovered [39, 40]. Apelin-13, the most potent isoform of apelin, participates in the development of HF [19, 41, 42]. Furthermore, apelin-13 optimizes the cardiac workload and reduces vascular resistance to improve the cardiac index [43]. ELA, as a second endogenous ligand of APJ, has a similar inotropic effect. The acute infusion of ELA-32 or apelin-13 increased the heart rate and cardiac output without changing the stroke volume but showed lower systemic vascular resistance in a rat model of septic shock [16]. This phenomenon was significantly blunted but not completely eliminated by an APJ antagonist. The improvement in LV function was observed after ELA perfusion and may thus be beneficial to the mediation of cardiac contractility according to the Frank–Starling law [16]. A significant increase in LV developed pressure (LVDP), an indirect parameter reflecting the contractility, was observed in the isolated heart after administration of ELA and its analogs [17]. LV fractional shortening (LVFS) is another important parameter representing the contractility of the heart [44]. Both apelin-13 and ELA increased LVFS and the heart rate in vivo [17]. Recently, it was discovered that ELA-32 had a dose-dependent increase in both LVEF and right ventricular ejection fractions in rats [13]. Administration of ELA-32 promotes cardiac output and the maximal rate of rise of LVDP in the monocrotaline (MCT)-induced pulmonary arterial hypertension (PAH) rats [13]. Additionally, ELA may mediate vasodilatory effects, contributing to the declining perfusion pressure in a dose-dependent manner. Treatment with ELA alleviates myocardial ischemia and modulates metabolism, ultimately contributing to the improvement of myocardial contractility [15].
The ELA–APJ axis and humoral regulation
Fluid overload plays a key role in the development of HF and the participation of apelin in the regulation of fluid homeostasis has been well established [17, 20, 21]. ELA expression is detected in renal collecting tubules, suggesting that it has potential biological effects on fluid homeostasis [22]. It has been reported that both ELA and apelin increased urine flow rates in rats, and ELA injection induced approximately 5-fold higher maximal increases in the plateau responses of urine flow rates than apelin. Regarding fluid homeostasis, both apelin-13 and ELA increase daily urinary output, which is accompanied by decreased urine osmolality and electrolyte excretion [17, 22]. The inhibition of sodium and chloride fractional excretion and the increase in diuretic may be partly responsible for the fluid homeostasis regulation of ELA. Collectively, these results suggest that compared with apelin, ELA exerts a more potent biological effect on the kidney, indicating that ELA may represent a promising diuretic agent.
The ELA–APJ axis and blood pressure regulation
Hypertension is an independent risk factor for HF and a major public health problem among the aging population, contributing to worldwide morbidity and mortality [1, 2, 4]. A growing number of factors are responsible for hypertension, such as the renin–angiotensin system (RAS), the apelinergic system, genetics, obesity, dyslipidemia, sedentary lifestyle, and diabetes [1, 4, 7, 14, 45,46,47]. Apelin is a vasodilator and exerts hypotensive actions. The intravenous injection of apelin in rats transiently reduces systolic and diastolic blood pressure (BP) levels [46, 47]. Similar to apelin, ELA infusion also downregulates BP levels in mice. ELA has also been shown to stimulate the angiogenesis of human umbilical vascular ECs (HUVECs) and to induce the relaxation of aortas through the activation of APJ receptor in mice [12, 14]. These results indicate that ELA also exerts a hypotensive effect in vivo and improves cardiac function, which is why there is now a preliminary clinical study focused on circulating ELA levels in patients with essential hypertension. Preliminary results show that compared with healthy subjects, circulating levels of ELA are significantly reduced in patients with hypertension [48]. Consistent with this finding, serum levels of ELA are significantly decreased in preeclamptic women [49, 50]. These findings support the theory that ELA is an important regulator of BP levels and may bring benefits to HF. PAH is a disease of the small pulmonary arteries characterized by vascular proliferation and remodeling. In consequence, the progressive increase in pulmonary vascular resistance induces right-sided HF [51]. Patients with PAH have lower plasma apelin levels than controls [52]. Some researchers attribute PAH to the loss of apelin [53]. Similarly, ELA expression is also reduced in patients with PAH and rodent models of PAH (Table 1) [13]. ELA attenuated monocrotaline (MCT)-induced PAH by alleviating right ventricular hypertrophy and lessening fully muscularized vessels (Fig. 1) [13]. In addition, ELA possesses vasodilatory property, which effectively improves myocardial blood supply to mitigate the impairment due to myocardial ischemia (Fig. 1) [13, 15]. Therefore, ELA reduces BP levels not only in the systemic circulation but also in the pulmonary circulation, both of which are likely to be beneficial to patients with HF.
The ELA–APJ axis and cardiac remodeling
Myocardial fibrosis and inflammation are major factors participating in adverse cardiac remodeling [23, 54, 55]. Sepsis is associated with a remarkable elevation in inflammation and easily induces HF [56]. Treatment with ELA has been demonstrated to lower myocardial inflammation, limit myocardial injury, and significantly improve cardiorenal function with optimal preload, afterload, and inotropic effects in a sepsis animal model (Table 1) [16]. In turn, myocardial inflammation plays a critical role in the development of cardiac fibrosis, cardiac remodeling, and HF [24, 55, 57, 58]. Ischemic cardiac myopathy manifests as HF and is accompanied by chronic active inflammation. Adverse cardiac remodeling is the primary characteristic of myocardial fibrosis and dysfunction. In a rat model of myocardial infarction (MI), pretreatment with ELA significantly reduced the severity of cardiac fibrosis and improved hemodynamic parameters obtained from the invasive catheter examination, such as LV systolic pressure, LV end-diastolic pressure, and maximal positive and negative first derivative of LV pressure (Table 1) [25]. Administration of ELA further limited the area of cardiac fibrosis in a murine MI model with downregulated levels of profibrotic genes, including transforming growth factor-β (TGFβ), latent TGFβ-binding protein 2, periostin, and collagen 8a [14], suggesting that ELA blunts the process of myocardial fibrosis and the loss of cardiomyocytes secondary to MI, thereby preserving cardiac function.
HF is associated with pathological hypertrophy and maladaptive ventricular remodeling which is an adaptive response for pressure overload to increase the contractility of cardiomyocytes [26, 27, 59, 60]. The ELA–APJ axis has been exhibited to suppress cardiac hypertrophy and remodeling, as measured by heart weight to body weight ratio and LV wall thickness in a mice model of transverse aortic constriction (TAC), by suppressing the levels of pro-hypertrophic factors atrial natriuretic factor and β-myosin heavy chain (Table 1) [14].
Angiogenesis is another aspect of cardiac remodeling in response to stress, but ELA prevents cardiac remodeling by regulating neovascularization [9]. A special research focus on the coronary vasculature revealed that the ELA–APJ signaling is required for sinus venosus–derived progenitors, which revascularizes the heart by creating new vasculature from the outside in [26]. ELA stimulated the formation of tubular structures and increased the number of branch points in a concentration-dependent manner, indicating an angiogenic effect [12]. Such an angiogenic effect may improve cardiac function and cardiac remodeling after MI. This was supported by the fact that ELA-32 attenuated right ventricular hypertrophy in MCT-induced PAH rats. The adverse remodeling of the vasculature was significantly lessened by ELA-32 including fully muscularized vessels and arteriolar wall thickness (Table 1) [13].
Moreover, ELA is also considered to be a factor affecting cell growth and cell cycle progression, meaning that it may affect cardiac fibroblasts (CFs) and ECs in impaired hearts (Fig. 1) [25, 27, 59]. This in vitro work demonstrated that apoptosis after MI was suppressed by administration of ELA in the experimental group compared with controls [25]. Stem cell exhaustion has been recognized as a common factor in age-related diseases such as HF. Recently, Yi et al. revealed that addition of human recombinant ELA peptide to the ovarian clear cell carcinoma (OCCC) cell lines promoted cell growth and migration by suppressing the p53 signaling pathway while gentic ELA knockdown augmented p53 pathway in OCCC cell lines using CRISPR/Cas9 via an APJ receptor-independent manner (Fig. 1) [59]. Intriguingly, ELA binds an unkown receptor (X receptor), instead of APJ receptor, and promotes human ESCs (hESCs) self-renewal and differentiation via the activation of the phosphatidylinositol 3-kinase (PI3K)/Akt phosphorylation and TGFβ signaling [27], suggesting that ELA functions as an endogenous hormonal peptide and signals through X receptor (Fig. 1). However, what is still unclear is whether ELA also mediates cardiovascular and renal protective effects through the X receptor in adult animals and humans. These results revealed that ELA plays a crucial role on stem cells, providing a new approach for HF treatment.
The ELA–APJ axis and cardiorenal dysfunction
Cardiorenal syndrome is a pathophysiologic disorder of the heart and kidney whereby acute or chronic dysfunction in one organ affects the other. Evidence reveals a close relationship between acute kidney injury (AKI) and HF, with the treatment for AKI contributing to anti-HF therapy [61]. Because of the high expression of ELA in the kidney, ELA may exert protective effects against AKI. The mRNA level of ELA was greatly decreased in mice with cardiorenal damage. The mature peptide (ELA-32) or the 11-residue furin-cleaved fragment (ELA-11) was administered to hypoxia–reperfusion (H/R)-injured or adriamycin-treated renal tubular cells in vitro. Both ELA-32 and ELA-11 significantly inhibited the H/R-induced DNA damage response and suppressed the DNA damage response, fibrosis, inflammation, and apoptosis. All of these contributed to the improvement of the renal tubular lesions and renal dysfunction [18]. These data indicated that ELA has positive effects on the prevention of AKI. ELA preserved the normal glomerular architecture and alleviated renal fibrosis in rodents fed a high-salt diet [62]. Three months of sustained ELA gene therapy augmented levels of ELA and preserved the glomerular structure, attenuated renal fibrosis, and suppressed the expression of fibrosis-associated genes in the kidneys of Dahl salt-sensitive rats with a high-salt diet [62, 63]. There has been new evidence supporting renal protection derived from ELA. In addition to the suppression of inflammation and fibrosis, ELA also protected podocytes from injury [63]. Such direct evidences support that ELA plays an active role in kidney protection and anti-HF therapy, especially in cardiorenal syndrome. In an observational clinical study, 80 patients with type 2 diabetes were divided into different arms according to their baseline urinary albumin/creatinine ratio (ACR). ELA has been shown to be a significantly negative correlation with the ACR, and the serum ELA levels might be a novel clinical predictor of diabetic nephropathy [64]. This clinical evidence indirectly verified the protective effects of ELA on renal function in the diabetes population. Importantly, ELA maintains the cardiovascular integrity of both the mother and the fetus during pregnancy by normalizing hypertension and decreasing proteinuria [65]. Taken together, the ELA–APJ axis appears to protect the kidneys from a wide degree of insults, thereby contributing to the maintenance of cardiac function in the context of HF.
Underlying mechanisms of the ELA–APJ axis
ELA exerts its cardioprotective effects in an APJ receptor–dependent manner in most studies, although other alternate receptors may exist [4, 14, 27, 50, 59]. Ho et al. demonstrated that ELA-KO, not apelin-KO, pregnant mice displayed preeclampsia-like symptoms and that infusion of ELA could alleviate these symptoms [65]. ELA-KO mice placentas were not rescued by apelin but by ELA infusion, again suggesting differences of ELA and apelin in signaling pathways. The preeclampsia alleviation is likely achieved through APJ receptor signaling in ECs. However, they also do not rule out a possible contribution from additional unidentified ELA receptors [4, 65]. X receptor, a poorly understood alternate receptor, is firstly reported in human embryos to sustain survival and self-renewal of hESCs (Fig. 1) [27]. Many studies have drawn the above conclusion via ligand-binding assays or by comparing wild-type and APJ-KO animal models [14, 17]. The hyperactive RAS plays a crucial role in the progression of HF. Angiotensin II (Ang II), an important element of the RAS, is generated by angiotensin-converting enzyme (ACE) and degraded by ACE2 [66], which is also a co-receptor for the severe respiratory syndrome coronavirus 2 (SARS-CoV2) entry for the novel coronavirus 2019 pneumonia and regulates the fundamental cellular biology of cardiomyocytes, CFs, and coronary ECs in animal models and patients with HF by blocking the RAS [67,68,69]. Unsurprisingly, pharmacological antagonism of the RAS using ACE inhibitors (ACEI) or Ang II type 1 receptor (AT1) blockers (ARB) is a cornerstone of current medical therapy for patients with HF. ACE2, by virtue of its action on Ang I and Ang II, has been successfully shown to be an endogenous ACEI or ARB at the cellular level [67]. Myocardial ACE2 protein levels decrease in pressure-overload HF, suggesting an inverse relationship between its levels and disease progression. Loss of ACE2 resulted in worsened pathological remodeling, contributing to systolic dysfunction and ventricular dilation [68]. Apelin, another vasoactive peptide substrate of ACE2, upregulates ACE2 levels in the heart to disrupt the balance between ACE and ACE2, while ELA reduces the ratio of ACE/ACE2 in a different manner from apelin to affect the RAS activation [68,69,70,71]. Apelin upregulates protein expression of ACE2, while ELA downregulates ACE expression in the heart [14, 71]. Apelin has been shown to increase ACE2 promoter activity in vitro and upregulate ACE2 expression in failing hearts in vivo. In TAC mice, ELA negatively regulates the expression of Brahma-related gene-1 and forkhead box M1, which upregulates ACE expression in the heart [13, 70]. The apelin–APJ axis activated ACE2 promoter activity, while ELA dose-dependently decreased ACE promoter activity via APJ. Therefore, ELA neutralizes the adverse effects of the RAS to provide cardiac protection. ELA has been exhibited to combine with APJ receptor and activate Gi signaling, which exerts diverse effects, including diuresis and vasodilatation [4, 22]. Activated Gi signaling may contribute to a decrease in cAMP production, the stimulation of extracellular-regulated protein kinases (ERKs), and an increase in intracellular calcium mobilization [12, 15]. Correspondingly, the suppression of ERK1/2 phosphorylation activation markedly decreased the positive inotropy effect induced by ELA [15], indicating cardiovascular protective effects of the ELA–APJ axis via the regulation of ERK1/2 phosphorylation signaling. The apelin–APJ axis is a potent inotrope/vasodilator via modulation of ACE2 and ERK1/2 phosphorylation signaling [71]. ELA can activate Gi protein signaling and β-arrestin-dependent pathways, which mediate GPCR internalization to trigger the downstream signaling pathway [13]. Apelin improves cardiac dysfunction after myocardial I/R injury by inhibiting myocardial apoptosis and oxidative stress, along with upregulation of endothelial nitric oxide synthase (eNOS) levels and of PI3K/Akt and activation of ERK1/2 phosphorylation signaling [72]. Administration of ELA protects hESCs against apoptosis and stress-induced cell death and promotes survival and self-renewal in an APJ-independent manner via the PI3K/Akt pathway and thus may also play a crucial role in regulating the apoptosis of cardiomyocytes under stress (Fig. 1) [27]. In addition, treatment with ELA suppresses the levels of TGF-β and matrix metallopeptidase 9 (MMP9) in a murine I/R injury model, which is recognized as a key effector molecule that participates in adverse myocardial hypertrophy, remodeling and interstitial fibrosis (Table 1 and Fig. 1) [18, 73, 74]. Notably, ELA exerts an anti-fibrosis effect in the heart and kidney by preventing the TGF-β signaling (Table 1) [18]. The underlying mechanisms responsible for cardiorenal protective roles of ELA may be involved in activation of the PI3K/Akt/mTOR signaling pathway (Fig. 1) [63]. Such novel findings may support the viewpoint that ELA promotes cell proliferation to tackle a series of unwanted pathological changes when HF occurs. It is well established that the effect of vasodilation induced by apelin depends on the functional endothelium. The hypotensive effect of apelin is mediated by endothelium-derived nitric oxide in both mice and humans [47, 75]. The combination of apelin and the APJ receptor induces the phosphorylation of eNOS via PI3K/Akt activation [76]. ELA relaxes mouse aortic blood vessels in a dose-dependent manner through a different way from apelin. This vasorelaxation did not disappear when pretreated with eNOS inhibitor L-NAME, suggesting that nitric oxide is not required for ELA-mediated vascular relaxation [12]. The hypotensive effect of ELA analogs was positively associated with the ability to elicit the internalization of APJ [17]. These observations have been reported in studies focusing on apelin, which demonstrate that the decline in mean arterial pressure induced by apelin is related to its ability to recruit β-arrestin and induce APJ internalization [77, 78]. Although the exact mechanism involved is still unknown, it is clear that ELA can enhance the cardiac-restricted transcription factors Tbx5 and GATA4 to promote the differentiation of hESCs into cardiomyocytes (Fig. 1) [28]. The primary signaling pathways are summarized to give us deeper understanding of ELA–APJ axis (Fig. 1). Due to the similar properties between apelin and ELA, whether the biological actions and underlying mechanisms of ELA are the same should be investigated in further studies.
Targeting the ELA–APJ pathway as novel therapies for HF
Given the multiple cardioprotective effects demonstrated in research studies, ELA and its diverse fragments/analogs could be the basis for novel pharmacologic therapies for HF (Table 1). Consequently, there is increasing research being devoted to creating these proteins. The Fmoc strategy is commonly used to synthesize peptides on solid phase. The Fmoc strategy, a special method of protein synthesis on solid phase, was employed to synthesize ELA and subsequently degraded ELA to identify the bioactive fragments [79]. ELA (19–32) which possesses a highly similar structure to apelin-13, binds to APJ, activates the Gαi1 and β-arrestin-2 signaling pathways to lower blood pressure levels, and exerts positive inotropic effects [17]. ELA-11 (ELA22-32) inhibited I/R injury–induced renal damage, including fibrosis, inflammation, and apoptosis, and preserved renal dysfunction in an AKI mouse model [18]. The different isoforms of ELA behave differently upon conformational and dynamic changes in an isoform-dependent manner [80]. Therefore, we need to make further efforts to identify more isoforms of ELA, and to identify which is the ideal agent for HF patients. Unfortunately, the short half-life of ELA hinders its clinical application [17]. Recently, a group of scientists added an Fc fragment to the N-terminus of ELA. This new synthetic peptide not only preserved the functional portion of ELA at the C-terminus but also greatly extended its half-life [25]. Cell therapy is a promising therapeutic approach to restore cardiac function. ELA was verified as a growth factor that promotes cardiomyocyte differentiation from transgenic hESCs to remedy the loss of cardiomyocytes in HF [28]. In the light of the limitation in translating lead compounds to clinical applications, investigators have identified and developed biased and nonbiased agonists toward APJ receptor and other GPCR signaling [32].
Conclusion
HF is a growing epidemic with high morbidity and mortality throughout the world. Acute and chronic HF are characterized by activation of several signaling pathways associated with pathological hypertrophy and maladaptive ventricular remodeling that in turn contributes to diminished systolic performance and diastolic function. The ELA–APJ axis has been shown to exert cardioprotective effects ranging from embryonic development to biological effects in adult kidney and cardiac tissues. The ELA–apelin–APJ axis has emerged as the dominant mechanism for the negative regulation of the Ang II-AT1 actions. Therefore, there are increasing efforts to harness this important biochemical and physiological property to develop novel therapies for HF targeting multiple cardioprotective mechanisms such as inotropic, vasodilatory, anti-inflammatory, antifibrotic, and diuretic effects (Table 1 and Fig. 1). To date, we do not fully understand the complicated system, including the various isoforms, relevant signaling pathways and adverse biological effects, especially in humans. These open questions and the short half-life of ELA are the main obstacles for the clinical application of the ELA–APJ axis. Every effort should be made to better understand the physiological effects of the ELA–APJ receptor signaling pathway since numerous clinical and experimental studies clearly support a physiological and pathophysiological role for the ELA–APJ axis in HF. Thus, targeting the ELA–APJ axis has emerged as a pre-warning biomarker and a novel therapeutic approach against progression of HF. An increasing understanding of cardiovascular actions of ELA will help develop effective interventions.
References
Alfaras I, Di Germanio C, Bernier M, Csiszar A, Ungvari Z, Lakatta EG, de Cabo R (2016) Pharmacological strategies to retard cardiovascular aging. Circ Res 118(10):1626–1642
Miller LW (2008) Heart failure: who we treat versus who we study. Cardiol Clin 26(1):113–125
Sacks CA, Jarcho JA, Curfman GD (2014) Paradigm shifts in heart-failure therapy — a timeline. New Engl J Med 371(11):989–991
Read C, Nyimanu D, Williams TL, Huggins DJ, Sulentic P, Macrae RGC, Yang P, Glen RC, Maguire JJ, Davenport AP (2019) International Union of Basic and Clinical Pharmacology. CVII. Structure and pharmacology of the apelin receptor with a recommendation that Elabela/Toddler is a second endogenous peptide ligand. Pharmacol Rev 71(4):467–502
Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX et al (1998) Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun 251(2):471–476
Ureche C, Tapoi L, Volovat S, Voroneanu L, Kanbay M, Covic A (2019) Cardioprotective apelin effects and the cardiac-renal axis: review of existing science and potential therapeutic applications of synthetic and native regulated apelin. J Hum Hypertens 33:429–435. https://doi.org/10.1038/s41371-019-0163-5
Folino A, Montarolo PG, Samaja M, Rastaldo R (2015) Effects of apelin on the cardiovascular system. Heart Fail Rev 20(4):505–518
Folino A, Accomasso L, Giachino C, Montarolo PG, Losano G, Pagliaro P, Rastaldo R (2018) Apelin-induced cardioprotection against ischaemia/reperfusion injury: roles of epidermal growth factor and Src. Acta Physiol 222(2):e12924
Chng SC, Ho L, Tian J, Reversade B (2013) ELABELA: a hormone essential for heart development signals via the apelin receptor. Dev Cell 27(6):672–680
Pauli A, Norris ML, Valen E, Chew GL, Gagnon JA, Zimmerman S, Mitchell A, Ma J, Dubrulle J, Reyon D, Tsai SQ, Joung JK, Saghatelian A, Schier AF (2014) Toddler: an embryonic signal that promotes cell movement via apelin receptors. Science 343(6172):1248636–1248636
Kawamata Y, Habata Y, Fukusumi S, Hosoya M, Fujii R, Hinuma S et al (2001) Molecular properties of apelin: tissue distribution and receptor binding. BBA-Mol Cell Res 1538(2–3):162–171
Wang Z, Yu D, Wang M, Wang Q, Kouznetsova J, Yang R et al (2015) Elabela-apelin receptor signaling pathway is functional in mammalian systems. Sci Rep. https://doi.org/10.1038/srep08170
Yang P, Read C, Kuc RE, Buonincontri G, Southwood M, Torella R et al (2017) Elabela/toddler is an endogenous agonist of the apelin APJ receptor in the adult cardiovascular system, and exogenous administration of the peptide compensates for the downregulation of its expression in pulmonary arterial hypertension. Circulation 135(12):1160–1173
Sato T, Sato C, Kadowaki A, Watanabe H, Ho L, Ishida J, Yamaguchi T, Kimura A, Fukamizu A, Penninger JM, Reversade B, Ito H, Imai Y, Kuba K (2017) ELABELA-APJ axis protects from pressure overload heart failure and angiotensin II-induced cardiac damage. Cardiovasc Res 113(7):760–769
Perjés Á, Kilpiö T, Ulvila J, Magga J, Alakoski T, Szabó Z et al (2016) Characterization of apela, a novel endogenous ligand of apelin receptor, in the adult heart. Basic Res Cardiol 111(1):2. https://doi.org/10.1007/s00395-015-0521-6
Coquerel D, Chagnon F, Sainsily X, Dumont L, Murza A, Côté J, Dumaine R, Sarret P, Marsault É, Salvail D, Auger-Messier M, Lesur O (2017) Elabela improves cardio-renal outcome in fatal experimental septic shock. Crit Care Med 45(11):e1139–e1148
Murza A, Sainsily X, Coquerel D, Côté J, Marx P, Besserer-Offroy É, Longpré JM, Lainé J, Reversade B, Salvail D, Leduc R, Dumaine R, Lesur O, Auger-Messier M, Sarret P, Marsault É (2016) Discovery and structure–activity relationship of a bioactive fragment of ELABELA that modulates vascular and cardiac functions. J Med Chem 59(7):2962–2972
Chen H, Wang L, Wang W, Cheng C, Zhang Y, Zhou Y, Wang C, Miao X, Wang J, Wang C, Li J, Zheng L, Huang K (2017) ELABELA and an ELABELA fragment protect against AKI. J Am Soc Nephrol 28(9):2694–2707
Goidescu CM, Anton FP, Leucuța DC, Mircea PA, Vida-Simiti LA (2016) Serum level of apelin-13 negatively correlated with NT-proBNP in heart failure patients. Rev Romana Med Lab 24(4):399–412
Miller WL (2016) Fluid volume overload and congestion in heart failure: time to reconsider pathophysiology and how volume is assessed. Circ Heart Fail 9(8):e002922
Cernaro V, Lacquaniti A, Lorenzano G, Loddo S, Romeo A, Donato V et al (2012) Apelin, plasmatic osmolality and hypotension in dialyzed patients. Blood Purif 33(4):317–323
Deng C, Chen H, Yang N, Feng Y, Hsueh AJW (2015) Apela regulates fluid homeostasis by binding to the APJ receptor to activate Gi signaling. J Biol Chem 290(30):18261–18268
Jia L, Li Y, Xiao C, Du J (2012) Angiotensin II induces inflammation leading to cardiac remodeling. Front Biosci 17(1):221–231
Ayoub KF, Pothineni NVK, Rutland J, Ding Z, Mehta JL (2017) Immunity, inflammation, and oxidative stress in heart failure: emerging molecular targets. Cardiovasc Drug Ther 31(5–6):593–608
Xi Y, Yu D, Yang R, Zhao Q, Wang J, Zhang H et al (2019) Recombinant Elabela-Fc fusion protein has extended plasma half-life and mitigates post-infarct heart dysfunction in rats. Int J Cardiol. https://doi.org/10.1016/j.ijcard.2019.04.089
Sharma B, Ho L, Ford GH, Chen HI, Goldstone AB, Woo YJ et al (2017) Alternative progenitor cells compensate to rebuild the coronary vasculature in Elabela- and APJ-deficient hearts. Dev Cell 42(6):655–666.e3
Ho L, Tan SYX, Wee S, Wu Y, Tan SJC, Ramakrishna NB, Chng SC, Nama S, Szczerbinska I, Chan YS, Avery S, Tsuneyoshi N, Ng HH, Gunaratne J, Dunn NR, Reversade B (2015) ELABELA is an endogenous growth factor that sustains hESC self-renewal via the PI3K/AKT pathway. Cell Stem Cell 17(4):435–447
Wang Z, Huang J (2019) Apela promotes cardiomyocyte differentiation from transgenic human embryonic stem cell lines. Appl Biochem Biotechnol 189:396–410. https://doi.org/10.1007/s12010-019-03012-2
Shin K, Kenward C, Rainey JK (2017) Apelinergic system structure and function. Compr Physiol 8(1):407–450
Li L, Zhou Q, Li X, Chen L (2017) Elabela-APJ axis: a novel therapy target for cardiovascular diseases. Acta Biochim Biophys Sin 49(11):1042–1043
Charo DN, Ho M, Fajardo G, Kawana M, Kundu RK, Sheikh AY et al (2009) Endogenous regulation of cardiovascular function by apelin-APJ. Am J Physiol-Heart C 297(5):H1904–H1913
Zhong JC, Zhang ZZ, Wang W, McKinnie SMK, Vederas JC, Oudit GY (2017) Targeting the apelin pathway as a novel therapeutic approach for cardiovascular diseases. BBA-Mol Basis Dis 1863(8):1942–1950
El Amrousy D, El-Mahdy H (2018) Prognostic value of serum apelin level in children with heart failure secondary to congenital heart disease. Pediatr Cardiol 39(6):1188–1193
Topuz M, Cosgun M, Akkuş O, Bulut A, Sen O, Topuz AN, Caylı M (2016) Effect of spironolactone on plasma apelin-12 levels in patients with chronic systolic heart failure. Acta Cardiol Sin 32(6):690–697
Małyszko J, Małyszko JS, Koźminski P, Myśliwiec M (2006) Apelin and cardiac function in hemodialyzed patients: possible relations? Am J Nephrol 26(2):121–126
Przewlocka-Kosmala M, Kotwica T, Mysiak A, Kosmala W (2011) Reduced circulating apelin in essential hypertension and its association with cardiac dysfunction. J Hypertens 29(5):971–979
Chandrasekaran B, Kalra PR, Donovan J, Hooper J, Clague JR, McDonagh TA (2010) Myocardial apelin production is reduced in humans with left ventricular systolic dysfunction. J Card Fail 16(7):556–561
Georgiadou D, Boussata S, van Dijk M (2019) ELABELA measurements by commercial ELISA kits require sample extraction. Am J Physiol Endocrinol Metab 317(6):E1218–E1219
Yang P, Maguire JJ, Davenport AP (2015) Apelin, Elabela/toddler, and biased agonists as novel therapeutic agents in the cardiovascular system. Trends Pharmacol Sci 36(9):560–567
Pang H, Han B, Yu T, Zong Z (2014) Effect of apelin on the cardiac hemodynamics in hypertensive rats with heart failure. Int J Mol Med 34(3):756–764
Koguchi W, Kobayashi N, Takeshima H, Ishikawa M, Sugiyama F, Ishimitsu T (2012) Cardioprotective effect of apelin-13 on cardiac performance and remodeling in end-stage heart failure. Circ J 76(1):137–144
Kobayashi N, Takeshima H, Koguchi W, Ishikawa M, Sugiyama F, Mamada Y et al (2011) Cardioprotective effect of apelin-13 on cardiac performance and remodeling in Dahl rats with end-stage heart failure. J Card Fail 17(9):S171
Chagnon F, Coquerel D, Salvail D, Marsault E, Dumaine R, Auger-messier M et al (2017) Apelin compared with dobutamine exerts cardioprotection and extends survival in a rat model of endotoxin-induced myocardial dysfunction. Crit Care Med 45(4):e391–e398
de Simone G, Devereux RB, Roman MJ, Ganau A, Saba PS, Alderman MH, Laragh JH (1994) Assessment of left ventricular function by the mid-wall fractional shortening/end-systolic stress relation in human hypertension. J Am Coll Cardiol 23(6):1444–1451
Mehta PK, Griendling KK (2007) Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Phys 292(1):C82–C97
Lee DK, Cheng R, Nguyen T, Fan T, Kariyawasam AP, Liu Y et al (2001) Characterization of apelin, the ligand for the APJ receptor. J Neurochem 74(1):34–41
Ishida J, Hashimoto T, Hashimoto Y, Nishiwaki S, Iguchi T, Harada S, Sugaya T, Matsuzaki H, Yamamoto R, Shiota N, Okunishi H, Kihara M, Umemura S, Sugiyama F, Yagami K, Kasuya Y, Mochizuki N, Fukamizu A (2004) Regulatory roles for APJ, a seven-transmembrane receptor related to angiotensin-type 1 receptor in blood pressure in vivo. J Biol Chem 279(25):26274–26279
Li Y, Yang X, Ouyang S, He J, Yu B, Lin X et al (2019) Declined circulating Elabela levels in patients with essential hypertension and its association with impaired vascular function: a preliminary study. Clin Exp Hypertens 22:1–5
Deniz R, Baykus Y, Ustebay S, Ugur K, Yavuzkir Ş, Aydin S (2019) Evaluation of elabela, apelin and nitric oxide findings in maternal blood of normal pregnant women, pregnant women with pre-eclampsia, severe pre-eclampsia and umbilical arteries and venules of newborns. J Obstet Gynaecol 7:1–6
Zhou L, Sun H, Cheng R, Fan X, Lai S, Deng C (2019) ELABELA, as a potential diagnostic biomarker of preeclampsia, regulates abnormally shallow placentation via APJ. Am J Physiol Endocrinol Metab 316(5):E773–E781
Prins KW, Thenappan T (2016) World Health Organization Group I pulmonary hypertension. Cardiol Clin 34(3):363–374
Goetze JP, Rehfeld JF, Carlsen J, Videbaek R, Andersen CB, Boesgaard S, Friis-Hansen L (2006) Apelin: a new plasma marker of cardiopulmonary disease. Regul Pept 133(1–3):134–138
Chandra SM, Razavi H, Kim J, Agrawal R, Kundu RK, de Jesus PV et al (2011) Disruption of the apelin-APJ system worsens hypoxia-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol 31(4):814–820
Okuhara Y, Yokoe S, Iwasaku T, Eguchi A, Nishimura K, Li W, Oboshi M, Naito Y, Mano T, Asahi M, Okamura H, Masuyama T, Hirotani S (2017) Interleukin-18 gene deletion protects against sepsis-induced cardiac dysfunction by inhibiting PP2A activity. Int J Cardiol 243:396–403
Gruson D, Ahn SA, Rousseau MF (2011) Biomarkers of inflammation and cardiac remodeling: the quest of relevant companions for the risk stratification of heart failure patients is still ongoing. Biochem Med 1(3):254–263
Annane D, Bellissant E, Cavaillon JM (2005) Septic shock. Lancet 365(9453):63–78
Suthahar N, Meijers WC, Silljé HHW, de Boer RA (2017) From inflammation to fibrosis-molecular and cellular mechanisms of myocardial tissue remodelling and perspectives on differential treatment opportunities. Curr Heart Fail Rep 14(4):235–250
Passino C, Barison A, Vergaro G, Gabutti A, Borrelli C, Emdin M, Clerico A (2015) Markers of fibrosis, inflammation, and remodeling pathways in heart failure. Clin Chim Acta 443:29–38
Yi Y, Tsai SH, Cheng JC, Wang EY, Anglesio MS, Cochrane DR, Fuller M, Gibb EA, Wei W, Huntsman DG, Karsan A, Hoodless PA (2017) APELA promotes tumour growth and cell migration in ovarian cancer in a p53-dependent manner. Gynecol Oncol 147(3):663–671
Wang J, Wei L, Yang X, Zhong J (2019) Roles of growth differentiation factor 15 in atherosclerosis and coronary artery disease. J Am Heart Assoc 8(17):e012826
Lameire N, Van Biesen W, Vanholder R (2005) Acute renal failure. Lancet 365(9457):417–430
Schreiber CA, Holditch SJ, Generous A, Ikeda Y (2017) Sustained ELABELA gene therapy in high-salt diet-induced hypertensive rats. Curr Gene Ther 16(5):349–360
Zhang Y, Wang Y, Luo M, Xu F, Lu Y, Zhou X, Cui W, Miao L (2019) Elabela protects against podocyte injury in mice with streptozocin-induced diabetes by associating with the PI3K/Akt/mTOR pathway. Peptides 114:29–37
Zhang H, Gong D, Ni L, Shi L, Xu W, Shi M, Chen J, Ai Y, Zhang X (2018) Serum Elabela/toddler levels are associated with albuminuria in patients with type 2 diabetes. Cell Physiol Biochem 48(3):1347–1354
Ho L, van Dijk M, Chye STJ, Messerschmidt DM, Chng SC, Ong S, Yi LK, Boussata S, Goh GH, Afink GB, Lim CY, Dunn NR, Solter D, Knowles BB, Reversade B (2017) ELABELA deficiency promotes preeclampsia and cardiovascular malformations in mice. Science 357(6352):707–713
Koka V, Huang XR, Chung ACK, Wang W, Truong LD, Lan HY (2008) Angiotensin II up-regulates angiotensin I-converting enzyme (ACE), but down-regulates ACE2 via the AT1-ERK/p38 MAP kinase pathway. Am J Pathol 172(5):1174–1183
Patel VB, Zhong JC, Grant MB, Oudit GY (2016) Role of the ACE2/angiotensin 1–7 axis of the renin–angiotensin system in heart failure. Circ Res 118(8):1313–1326
Zhong J, Basu R, Guo D, Chow FL, Byrns S, Schuster M, Loibner H, Wang XH, Penninger JM, Kassiri Z, Oudit GY (2010) Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation 122(7):717–728
Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q (2020) Structural basis for the recognition of the SARS-CoV-2 by full-length human ACE2. Science. https://doi.org/10.1126/science.abb2762
Yang J, Feng X, Zhou Q, Cheng W, Shang C, Han P et al (2016) Pathological ACE2-to-ACE enzyme switch in the stressed heart is transcriptionally controlled by the endothelial Brg1–FoxM1 complex. Proc Natl Acad Sci USA 113(38):E5628–E5635
Zhang ZZ, Wang W, Jin HY, Chen X, Cheng YW, Xu YL, Song B, Penninger JM, Oudit GY, Zhong JC (2017) Pyr-apelin-13 is a negative regulator of angiotensin II-mediated adverse myocardial remodeling and dysfunction. Hypertension 70:1165–1175
Zeng XJ, Zhang LK, Wang HX, Lu LQ, Ma LQ, Tang CS (2009) Apelin protects heart against ischemia/reperfusion injury in rat. Peptides 30(6):1144–1152
Bunbupha S, Pakdeechote P, Maneesai P, Prachaney P, Boonprom P (2019) Carthamus Tinctorius L. extract attenuates cardiac remodeling in L-NAME-induced hypertensive rats by inhibiting the NADPH oxidase-mediated TGF-β1 and MMP-9 pathway. Ann Anat 222:120–128
Wu D, Huang D, Li LL, Ni P, Li XX, Wang B, Han YN, Shao XQ, Zhao D, Chu WF, Li BY (2019) TGF-β1-PML SUMOylation-peptidyl- prolyl cis–trans isomerase NIMA-interacting 1 (Pin1) form a positive feedback loop to regulate cardiac fibrosis. J Cell Physiol 234(5):6263–6273
Japp AG, Cruden NL, Barnes G, van Gemeren N, Mathews J, Adamson J, Johnston NR, Denvir MA, Megson IL, Flapan AD, Newby DE (2010) Acute cardiovascular effects of apelin in humans. Circulation 121(16):1818–1827
Zhong J, Yu X, Huang Y, Yung L, Lau C, Lin S (2007) Apelin modulates aortic vascular tone via endothelial nitric oxide synthase phosphorylation pathway in diabetic mice. Cardiovasc Res 74(3):388–395
Lee DK, Saldivia VR, Nguyen T, Cheng R, George SR, O’Dowd BF (2005) Modification of the terminal residue of apelin-13 antagonizes its hypotensive action. Endocrinology 146(1):231–236
Ceraudo E, Galanth C, Carpentier E, Banegas-Font I, Schonegge AM, Alvear-Perez R, Iturrioz X, Bouvier M, Llorens-Cortes C (2014) Biased signaling favoring Gi over β-arrestin promoted by an apelin fragment lacking the C-terminal phenylalanine. J Biol Chem 289(35):24599–24610
Behrendt R, White P, Offer J (2016) Advances in Fmoc solid-phase peptide synthesis. J Pept Sci 22(1):4–27
Huang SK, Shin K, Sarker M, Rainey JK (2017) Apela exhibits isoform- and headgroup-dependent modulation of micelle binding, peptide conformation and dynamics. BBA-Biomembranes 1859(5):767–778
Acknowledgments
We appreciate Dr. Lei Zhao for her efforts to help us submit this article.
Funding
This work was supported by the National Major Research Plan Training Program of China [91849111] and the National Natural Science Foundation of China [81770253; 81370362; 81670214], and the 1315 Personnel Training Plan and Talent project of Beijing Chaoyang Hospital Affiliated to Capital Medical University.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
The manuscript does not contain clinical studies or patient data.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ma, Z., Song, JJ., Martin, S. et al. The Elabela-APJ axis: a promising therapeutic target for heart failure. Heart Fail Rev 26, 1249–1258 (2021). https://doi.org/10.1007/s10741-020-09957-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-020-09957-5