
Abstract
Olea europaea subsp. cuspidata (Wall. & G. Don) Cif. is one of the six subspecies important for domestication of olive described as having valuable breeding traits. It is distributed from South Africa to the Middle East and the Mediterranean region to China mainly at mid to high altitudes with adequate precipitation. The currently rather fragmented Olea europaea subsp. cuspidata populations likely represent outposts of a formerly larger and more connected area and are hence vulnerable to additional stresses such as erratic weather extremes and anthropogenic influences. This may result in further fragmentation of stands, shifts in demography, increased clonal growth, lower genetic diversity, and higher differentiation between olive stands. In this study, we present data on Olea europaea subsp. cuspidata growing in the Hajar Mountains, Sultanate of Oman, a large area subjected to many anthropogenic disturbances. We used dendrological traits and genetic parameters to infer its stand and population genetic structure, levels of genetic diversity, and differentiation. To accomplish this, 366 individual trees in total were inventoried and 188 of them genotyped using 12 microsatellite markers. Dendrological measures indicated disturbances mainly due to human activities. Height and crown area of individuals averaged 3.7 m (ranging from 0.2 to 12 m) and 14 m (ranging from 1 to 18 m), respectively. The stand conditional index was mainly 50%. The identified genetic variation within the sampled areas was relatively moderate. The expected and observed heterozygosity equalled 0.55–0.58 and 0.62–0.64, respectively. All individual genotypes were unique indicating the absence of clones. Though significant genetic differentiation was found, there was no evidence of population substructure suggesting effective pollen and long-distance seed dispersal in the past. However, changes in gene flow patterns may be reflected in the next (sibling) generation. Further studies are necessary to detect any changes in the future and to develop sustainable conservation strategies for this species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plant communities in tropical and subtropical regions are commonly exposed to extreme weather and climatic conditions including high-temperature fluctuations, variable precipitation patterns, and intense radiation. Along with recent climate changes, anthropogenic activities, such as overexploitation, habitat destruction and fragmentation, and introduction of new species, have accelerated ecosystem alterations (Goudie and Viles 2013) resulting in often massive ecological disturbances and fragile biological conditions with unpredictable consequences. Becklin et al. (2016) reported that alteration of precipitation regimes and temperature may disturb the regular patterns of plant morphology, physiology, and life history, which can lead to low plant viability and germination success of seeds (Anderson 2016). Furthermore, disturbances may cause shifts in demography which in turn cause alterations in genetic settings (genetic diversity and differentiation, genetic bottlenecks, gene flow patterns) worldwide (Kassa et al. 2017; MacLaren 2016; Tardieu 2013; Wesche et al. 2005). Responses to changing and harsher environmental conditions can result in a shift in the distribution of mountain plant communities (Grabherr et al. 1994; Lenoir et al. 2008) and a switch from sexual to asexual reproduction (Braatne et al. 1996), respectively, among others.
Such adverse conditions and their effects are also assumed for the open forest community of high altitudinal regions in the Hajar Mountains (the highest area of the Sultanate of Oman with a maximum altitude of 2976 m a.s.l.) dominated by Olea europaea subsp. cuspidata (Wall. & G.Don) Cif. and Juniperus seravschanica Kom. (Zeravschan or Persian juniper). Studies by Al-Kalbani and Price (2015; data range 1979–2012) and Al Sarmi and Washington (2011; data range 1980–2008) indicated that in one decade, mean winter and annual temperatures have risen by 0.85 °C and 0.27 °C, respectively, while the annual precipitation dropped by 67 mm and 9 mm, respectively. Therefore, in addition to recent and accelerated anthropogenic disturbances such as urban encroachments (housing area expansion, road constructions) and touristic activities (campfires), this mountain range is considered highly vulnerable (Al Charaabi and Al-Yahyai 2013; Al-Kalbani et al. 2014; Patzelt 2015a, b; Victor 2008). Consequently, these changes are likely to affect the stands of Olea europaea subsp. cuspidata, nonetheless clear-cut causes are not yet understood.
Olea europaea subsp. cuspidata is one of six sub-species distributed in Asia, Africa, the Mediterranean region, and Macaronesia with less oil yield and smaller fruits than the economically important and widely cultivated ancient cultigen O. europaea subsp. europaea L. (European olive) in the Mediterranean Basin (Díaz et al. 2006; Rallo et al. 2000). Olea europaea subsp. cuspidata is the most widespread subspecies distributed naturally from the southern to north-eastern part of Africa and from the Arabian Peninsula to China and is restricted to regions with adequate precipitation (± 600 mm) including higher elevation areas. It was introduced into Australia, Hawaii, New Zealand, and Norfolk Island (Green 2002). This diploid (2n = 46) and monoecious tree species is mostly outcrossing and wind pollinated, though insect pollination has been also observed (Canale and Loni 2010; Kassa et al. 2017). Seeds are typically dispersed by small mammals and birds (Abiyu et al. 2015; Cuneo and Leishman 2006). So far, clonal reproduction for this subspecies has not yet been proven, though related subspecies demonstrate clonal growth (Baali-Cherif and Besnard 2005). The tree plays an important role as forage and shade tree for agro-pastoral activities, and fruits are eaten by local people (Ghazanfar 2018).
This study was set up to test the hypotheses that (1) remoteness from human activity positively affects stand structure and viability of O. europaea subsp. cuspidata, (2) clonal growth of the species may exist in drier (sloped) conditions promoted by lack of water that suppresses successful germination of seeds, and (3) genetic structure shows bottleneck effects and signs of inbreeding.
Knowledge about this subspecies is insufficient, and the overall goal of this study therefore was to evaluate the vulnerability of the subspecies based on genetic data that can be potentially used to develop conservation strategies for this wild valuable genetic resource.
Materials and methods
Study sites and plant sampling
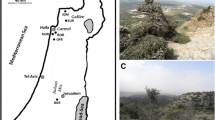
The field study was conducted in Al Jabal Al Akhdar (hereafter named as Akhdar) and Jabal Shams (herein named Shams) area in the Hajar Mountains range of 2110–2507 m a.s.l. (Fig. 1) in October 2016 and approved by the Ministry of Environmental and Climate Affairs (MECA, permit number 15/2016) of Sultanate of Oman. Three plateaus and their adjacent slopes (~ smallest sampling units with distances of 0.5–1.0 km between them) were selected per each of the two mountains ((3 plateaus + 3 slopes) × 2 mountains = 12 sites in total). The field design furthermore included six nested locations (3 locations (plateaus + slopes) × 2 mountains = 6 locations) (Table 1). In addition, the two mountains were analysed as two separate populations. All slope sites selected were north-exposed to reduce bias and to avoid exposure effects on plant growth and stand density. Per site, one reference point was assigned randomly. Around this predefined reference point, a group of the closest 30–32 individuals was recorded (Fig. 2a, b). Finally, 366 individual trees were sampled. For each individual, spatial coordinates were recorded with a hand-held GPS receiver (Vista HCx eTrex, accuracy ± 2 m, Garmin® Ltd., Southampton, Ireland). The following individual dendrological parameters were recorded: height and crown diameter (both in m), number of alive and dead trunks, diameter at breast height (DBH), and diameter at base (both in cm) for individuals smaller than 1.3 m. Tree condition index was used as proxy for tree vitality and estimated based on foliage cover, partitioned into four classes (100%, 75%, 50% and 25%, Fisher and Gardner 1995), knowing that this parameter is rather subjective. In addition, the presence of human disturbance (pruning and cutting) was recorded. Five healthy leaves from each individual were collected and air dried. Out of the 366 individuals, a subset of 188 individuals (15–16 individuals nearest to the reference point of each of the 12 sites) were chosen for further genetic analyses.

Maps of the study region and its sampling sites (white dots) in the Hajar Mountains range, Sultanate of Oman

Examples of a plateau (a) and slope (b) site displayed as top and intersectional views each of Olea europaea subsp. cuspidata trees in the Hajar Mountains range, Sultanate of Oman
DNA extraction and microsatellite marker analysis
Total genomic DNA was extracted from 188 individuals using one air-dried leaf per tree and the DNeasy™ 96 Plant Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturers’ protocol.
We selected the most suitable and reproducible SSR markers from the published data for our study. Eleven nSSRs [ssrOeUA-DCA1, ssrOeUA-DCA3, ssrOeUA-DCA4, ssrOeUA-DCA5, ssrOeUA-DCA7, ssrOeUA-DCA9, ssrOeUA-DCA18 (Sefc et al. 2000), PA(ATT)2 (Saumitou-Laprade et al. 2000), EMO2 (De la Rosa et al. 2002), GAPU71 (Carriero et al. 2002), and UDO99-011 (Cipriani et al. 2002)] and 15 EST-SSRs [OeUP-02, OeUP-06, OeUP-09, OeUP-10, OeUP-11, OeUP-16, OeUP-18, OeUP-23, OeUP-29, OeUP-33, OeUP-38, OeUP-41, OeUP-42, OeUP-45, and OeUP-46 (Arbeiter et al. 2017)] were originally selected for testing in O. europaea subsp. cuspidata. All these markers were tested for amplification and polymorphism in four samples per each of all four different geographic sites. Four markers (ssrOeUA-DCA1, GAPU71, OeUP-06, and EMO2) producing multi-locus patterns and weak amplification products were additionally tested in 16 samples from the first two sites. Among the 26 markers tested, 17 generated single locus polymorphic products, five were monomorphic (ssrOeUA-DCA5, OeUP-09, OeUP-11, OeUP-18, and OeUP-29), and the remaining four produced multi-locus patterns (OeUP-23, OeUP-38, and OeUP-46) or no amplification (ssrOeUA-DCA7).
Finally, 12 polymorphic microsatellite markers consisting of six nSSRs and six EST-SSRs were used for genetic analysis considering their consistent amplification and reliable unambiguous genotyping results (Table 2). The samples were amplified using three PCR amplification multiplexes: (1) PCRI-UDO99-011, ssrOeUA-DCA9, and OeUP-16 labelled by HEX together with ssrOeUA-DCA4 labelled by 6-FAM; (2) PCRII-ssrOeUA-DCA18 and GAPU71 labelled by HEX together with OeUP-02 and OeUP-10 labelled by 6-FAM; 3) PCRIII-OeUP-41 labelled by HEX together with ssrOeUA-DCA3, OeUP-42, and OeUP-45 labelled by 6-FAM. The following touchdown PCR program was used on a Biometra TProfessional Thermocycler (Analytik Jena, Germany): an initial denaturation for 15 min at 95 °C, 12 touchdown cycles each with 1 min at 94 °C, 1 min at 60 °C (decreased by 1 °C per cycle), and 1 min at 72 °C followed by 24 regular cycles each with 1 min at 94 °C, 1 min at 48 °C, and 1 min at 72 °C followed by a 20 min final extension at 72 °C. For cost-efficient PCRs, a tailed primer approach was used (Kubisiak et al. 2013; Schuelke 2000). The PCR was carried out in a 14 µl PCR reaction mix containing 1.5 µl MgCl2 (25 mM), 1 µl dNTPs (2.5 mM each dNTP), 0.2 µl Taq DNA Polymerase (5 U/µl), 1.5 µl 10X reaction buffer, 0.2 µl (5 picomol/µl) forward primer with a M13-specific sequence (5′-CACGACGTTGTAAACGAC-3′) added to the 5′-end of the primer (Kubisiak et al. 2013; Schuelke 2000) and 0.5 µl (5 picomol/µl) of the PIG-tailed (5′-GTTTCTT-3′, Brownstein et al. 1996) reverse primer (Sigma-Aldrich, St. Louis, MO, USA) (Table 2), 1.0 µl of (5 picomol/µl) dye labelled (6-FAM/HEX) M13 primer, 7.1 µl H2O, and 1.0 µl DNA (~ 0.6 ng/µl). An automatic capillary sequencer ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) was used to separate the PCR products. GeneMapper v. 4.0 (Applied Biosystems, Foster City, CA, USA) was used to record the alleles after careful visual inspection.
Data analysis
GenAlEx v6.5 (Peakall and Smouse 2012) was used to calculate percentage of polymorphic loci (PPL), the mean number of all (Na), effective (Ne) and private alleles (NPA), and observed (Ho) and expected (He) heterozygosity (Nei 1973), including the standard error of the mean each. For comparative analysis, the samples were grouped based either on their location (six groups, each including plateau and adjacent slope), plateau versus slope site (two groups), or Akhdar vs. Shams (two groups). Genetic parameters were calculated for all geographic groupings. To check for generation-dependent effects on genetic parameters, age class (assuming increasing DBH as proxy for increasing tree age) quartiles (≤ 7.2 cm, ≤ 10.7 cm, ≤ 16.1 cm and > 16.1 cm; n = 160) were created from trees higher than 1.3 m. The GenAlEx software was also used to check for the probability of identity (probability that two unrelated individuals will have the same genotype) and to detect identical multi-locus genotypes (match function). The MICRO-CHECKER software (van Oosterhout et al. 2004) was used to test for the presence of null alleles and to estimate their frequency. To check for usability of markers, Polymorphic Information Content (PIC) was calculated as PIC = 1 − ∑ (Pi)2, where Pi is the proportion of samples carrying the ith allele of a particular locus. GenAlEx was applied to check for deviations from Hardy–Weinberg equilibrium (HWE). The Genepop 4.3 software (Rousset 2008) was used to calculate FIS values (Weir and Cockerham 1984) for all markers. Significance values were corrected for multiple testing using the false discovery rate (FDR) method in the p.adjust function with R 3.4.3 software (R Core Team 2017). The BOTTLENECK v. 1.2.02 software (Piry et al. 1999) was used to test whether observed heterozygosity and allele frequency distribution were affected by bottleneck events in the recent past. Non-excess of heterozygosity assumes L-shaped allele frequency distributions, which are expected under mutation-drift equilibrium and lack of bottlenecks. The program was run for the two-phased model (TPM) of mutation choosing a 0.7 proportion of the stepwise mutation model (SMM) in the TPM and a 0.3 variance of the geometric distribution for TPM with 10,000 iterations. The Wilcoxon rank test was applied to test for significance. AMOVA, fixation indices (FST, FIS and FIT), the standardized F′ST (Meirmans 2006), Jost’s estimate of differentiation Dest (Jost 2008), pairwise genetic differentiation with 999 permutations, and Nei’s genetic distance between groups were calculated using GenAlEx.
A Principal Coordinate Analysis (PCoA) based on Nei´s unbiased genetic distance (Nei 1978) was also performed with GenAlEx.
The STRUCTURE v. 2.3.4 software (Pritchard et al. 2010) was used to examine the entire sample for population structure by inferring the number of potential subpopulations or genetic clusters (K) in a sample based on the Bayesian model-based clustering method and considering correlated allele frequencies for an admixture model. The admixture model potentially improves the clustering of closely related species or populations (Lind and Gailing 2013). The program was set to 10 independent runs for each value of potential clusters (K). Each K from 1 to 10 included a burn in period of 50,000 MCMC followed by 106 iterations. One of the 12 SSR markers, OeUP-16, likely contained null alleles at high frequency based on the MICRO-CHECKER analysis (see results below) and was hence excluded from STRUCTURE analysis since the presence of null alleles might adversely affect the efficiency of assigning individuals correctly in STRUCTURE (Carlsson 2008).
The STRUCTURE HARVESTER 0.6.94 software (Earl and VonHoldt 2012) was used to extract the most likely number of clusters (K) based on the rate of change in the log probability of data between successive K values as suggested by Evanno et al. (2005). We used the CLUMPAK software (Kopelman et al. 2015) to generate the summary and the graphical presentation of STRUCTURE results.
To check for large-scale spatial genetic patterns, isolation-by-distance (Mantel test with 999 permutations) was assessed with GenAlEx. Fine-scale spatial genetic structure (FSGS) was analysed for each site by spatial autocorrelation analysis using SPAGeDi v. 1.5 (Hardy and Vekemans 2002). To quantify FSGS, the Sp statistic (Vekemans and Hardy 2004) was calculated as Sp=− bF/(1 − F1), where bF is the regression slope of F(d) over the natural logarithm of distance, and F1 is the average kinship coefficient referred to as a co-ancestry estimate (Loiselle et al. 1995) based on all loci between pairs of individuals of the first distance class. The kinship coefficients were plotted against 10 m distance classes. The distance classes were defined so that 50% of all trees were represented at least once in the interval, and the coefficient of variation (CV) of the number of times when each individual was represented was ≤ 1. Upper and lower 95% confidence intervals of kinship coefficients were determined through 10,000 permutations of individuals among distance classes.
Results
Dendrological parameters
The height and crown width of individuals averaged 3.7 m (ranged from 0.2 to 12.0 m) and 14 m (ranged from 1 to 18 m) (Table 3), respectively. The number of alive and dead trunks varied only marginally from three to four (Table 3). DBH size classes showed a bell-shaped distribution, which was slightly right-skewed due to a few very old trees (Fig. 3). Trees smaller than 1.3 m showed an L-shaped distribution and were heavily browsed, resulting in a dwarfed habitus. DBH and diameter at base varied strongly among sites and locations, although less between mountains (data not shown). The plateau sites exhibited larger DBH values then their adjacent slopes, a trend that was not observed for diameter at base. Only one individual had fruits. The condition index averaged in total 50%, while trees from plateaus of Shams showed the lowest vitalities. In terms of cuttings (branches and stems), human influence was pronounced on Akhdar, while almost absent on Shams (Table 3).

Distribution of the diameter at breast height (DBH) and diameter at base measured on 366 Olea europaea subsp. cuspidata trees in the Hajar Mountains range, Sultanate of Oman
Genetic richness, diversity, and differentiation
In total, 147 different microsatellite alleles were found for all 12 microsatellite markers. Probability of observing identical genotypes by chance was extremely low (4.3−13), and no identical multi-locus genotypes were found in the entire sample. The mean number of alleles per locus was 12.3 with the lowest number of alleles (Na = 2) at both EST-SSR loci OeUP-42 and -45 and the highest number of alleles (Na = 31) at locus ssrOeUA-DCA9 (Table 4). The effective number of alleles per locus averaged 5.6. The observed heterozygosity (Ho) ranged from 0.03 (OeUP-45) to 0.89 (ssrOeUA-DCA18) with a mean value of 0.57. The average expected heterozygosity was higher than the observed one (He = 0.64 vs. Ho = 0.57). Fixation index (FIS) and genetic differentiation (FST) for the 12 markers averaged 0.09 (ranging from − 0.41 to 0.15) and 0.02 (ranging from 0.02 to 0.04), respectively (Table 4). The standardized differentiation measures F’ST and Jost´s Dest averaged 0.03 (ranging from 0.003 to 0.18) and 0.02 (ranging from 0.0001 to 0.17), respectively (Table 4). PIC ranged between 0.28 and 0.82 (Table 4).
The HWE tests per locus and in the six locations showed overall low indication of homozygote excess (about 25% of the markers demonstrated homozygote excess, mostly statistically insignificant and likely due to null allele presence) (Table 5). The OeUP-16 marker indicated a pronounced excess of homozygotes with significant deviation from HWE in all six locations, except for Akhdar 2 (Table 5). The ssrOeUA-DCA4, ssrOeUA-DCA9, GAPU71, UDO99-011, and OeUP-41 markers were only occasionally indicating deviations from HWE and null allele excess. Deviations from HWE and excess of homozygotes were observed for the same markers also for the mountain and site groups. FIS ranged from − 0.064 to 0.396.
Microsatellite loci were polymorphic for any grouping (Table 6). The average number of alleles (Na) per grouping was nearly twice as high as the number of effective alleles (Ne; Table 6). The number of private alleles (NPA) ranged between 3 and 12 (location), 36–51 (mountain), and 24–41 (site) (Table 6). Expected (He) and observed (Ho) heterozygosities showed nearly identical values ranging between 0.61 and 0.65 and 0.61 and 0.66, respectively (Table 6). Genetic diversity parameters declined with increasing age, while number of private alleles did not follow this trend (Table 6).
An L-shaped distribution of allele frequencies was observed for any grouping, indicating the absence of pronounced bottleneck effects (data no shown). Pairwise Nei´s genetic distance ranged from 0.03 to 0.09, being highest for Shams 3 against all others (Table 7). The F-index values were low, but differed significantly from zero for any grouping (Table 8). While FIS and FIT between groupings differed only marginally, the FST value between the two mountains (Akhdar and Shams) and among the six locations was about two and four times higher, respectively, as compared to plateau and slope sites (Table 8). However, the FST values were very small in general.
Population structure
The PCoA did not reveal any clear substructure among the sampled individuals (Fig. 4) demonstrated by the low discriminatory power of the first two coordinates that explained 12.9% of the total genetic variation observed. Together with the third coordinate they still explained only 17.8% of the total variation. These data were in agreement with the STRUCTURE analysis results, indicating little or no genetic structure. The most likely number of clusters (K) was eight, although less likely it could also equal four or six (Fig. 5). However, most individuals expressed some degree of balanced share of cluster assignments irrespective of the actual number of clusters and grouping. Therefore, neither PCoA and STRUCTURE nor FST values revealed any obvious population substructure or strong differentiation among O. europaea subsp. cuspidata trees.

Principal Coordinates Analysis (PCoA) based on genetic distances between 188 Olea europaea subsp. cuspidata individual trees from four sample sites in the Hajar Mountains range, Sultanate of Oman

The STRUCTURE analysis for all 188 Olea europaea subsp. cuspidata individuals in the Hajar Mountains range, Sultanate of Oman. DeltaK (mean (|L″(K)|)/s.d.(L(K)) and mean log-likelihood Ln(K) ± s.d. (dashed line) inferred by Evanno method and visualized by STRUCTURE HARVESTER (upper plot). The three lower plots present distribution of admixture (Q-values) for 12 studied sites based on the most plausible number of clusters (K) equalled four, six and eight clusters with vertical multi-coloured bars representing likelihood of individual tree genotypes to belong to one of the clusters
Spatial genetic structure
Large-scale spatial analyses based on a Mantel test revealed a low but significant correlation between genetic and geographic distances (r = 0.139, p = 0.04). Fine-scale spatial analyses revealed a positive significant spatial autocorrelation at the Akhdar 3 slope and Shams 2 plateau sites in the first (0–10 m) and second (10–20 m) distance intervals, respectively (Fig. 6). A negative significant autocorrelation was observed for the Shams 2 plateau site at the third (20–30 m), for the Akhdar 3 slope site at the seventh (70–80 m), and for the Shams 3 slope site at the second (10–20 m) distance intervals. Overall, the observed patterns demonstrated no or weak autocorrelation.

Fine-scale spatial genetic structure autocorrelograms of the average kinship coefficient (F(d), black solid line) according to Loiselle et al. (1995) with 95% confidence interval (grey area) along spatial distance (m) for each site of 188 Olea europaea subsp. cuspidata trees in the Hajar Mountains. Circles indicate significant autocorrelation (see geographic data on the sites in Table 1)
Discussion
Dendrology
The obtained dendrological data indicated clearly location and site-specific responses. Signs of human activities such as pruning of branches and logging of trees on Shams seemed to be less visible, which can be historically explained: until recently, only few and small settlements exist on Shams. This could be attributed to a scarce water availability and rather steep terrain (higher water run-off) in comparison to the neighbouring Jabal Al Akhdar. The generally sinking groundwater levels in the region (Al-Kalbani and Price 2015) and the overall more severe water approachability on Jabal Shams might thus also affect the existence of O. europaea subsp. cuspidata, visible in pronounced die-backs on Shams. Additionally, livestock browsing was apparently higher on Shams considering the number of dwarf shrubs (diameter at base) as proxy for high livestock intensities. This observation might be related to the slightly different livelihoods on both peaks; Shams serves predominantly as a rangeland area for the piedmont villages, while mountain villages on Akhdar traditionally practice terrace gardening with nowadays increasing off-farm activities such as tourism and administrative jobs.
The bell-shaped distribution of size class > 1.30 m clearly indicates an ongoing over-aging process of the entire population, which was different from O. europaea subsp. cuspidata population structures observed in Ethiopia (Bekele 2000) with typical L-shaped age class distributions indicative for relatively high proportions of young trees. In our study, trees with heights smaller than < 1.30 m indicate high recruitment rates (L-shape distribution). The older aged individuals with heights smaller than < 1.30 m were also present, but have been kept short due to browsing.
Given the different ecological conditions on both mountains, hypothesis 1 cannot be satisfactorily answered, as human influence seems higher on Akhdar, while livestock significantly affects stands on Shams.
Genetic richness and diversity
Although clonal propagation would be potentially possible and the sampling design should allow us to detect it, we did not find any evidence of vegetative propagation. Similarly, Kassa et al. (2017) did not find clonal colonies (genets) of O. europaea subsp. cuspidata in Ethiopia. However, Baali-Cherif and Besnard (2005) identified genets of O. europaea subsp. laperrinei in Algeria based on nine nSSRs, of which ssrOeUA-DCA3 and -DCA 9 have been also used in our study. The rate of clonal reproduction in Algeria was relatively high − 16% of genets were represented by two or more ramets and the area covered was more than 80 m2 in one of the locations. The absence of genets in our sample indicates an exclusive sexual reproduction mode in this population, probably also for the entire subspecies. This observation would merit further studies in additional stands of the Hajar Mountains as well as other populations, for instance in Dohfar (S-Oman) and neighbouring countries. Therefore, the clonal propagation proposed in hypothesis 2 must be rejected.
Genetic diversity was rather moderate (Ho = 0.55–0.58, He = 0.62–0.64), although slightly lower as compared to the same species in an Ethiopian forest studied by Kassa et al. (2017) using nSSR markers (Ho = 0.66–0.72, He = 0.74–0.77). Four of those SSR markers have also been used in our study, namely GAPU71 (Ho = 0.90 vs. 0.74 in our study, He = 0.92 vs. 0.80 in Kassa et al. 2017), ssrOeUA-DCA3 (Ho = 0.52 vs. 0.82, He = 0.40 vs. 0.86), ssrOeUA-DCA18 (Ho = 0.93 vs. 0.89, He = 0.86 vs. 0.88), and UDO099-011 (Ho = 0.93 vs. 0.81, He = 0.91 vs. 0.86). Erre et al. (2010) and Baali-Cherif and Besnard (2005) found also similar heterozygosity levels in Olea europaea subsp. laperrinei (He = 0.41–0.89 and 0.45–0.87, respectively). The lower heterozygosity level in our study can be explained by the inclusion of the EST-SSR markers leading to overall lower mean values. The EST-SSR markers alone generated much lower average heterozygosity levels (Ho = 0.33, ranging from 0.03 to 0.49 and He = 0.40, ranging from 0.03 to 0.83) than the nSSR markers (Ho = 0.81, ranging from 0.74 to 0.89 and He = 0.87, ranging from 0.80 to 0.94). The EST-SSR based values are also lower than values obtained by Arbeiter et al. (2017) (Ho = 0.68, ranging from 0.29 to 0.96 and He = 0.64, ranging from 0.51 to 0.79) and (Kassa et al. 2017) (Ho = 0.76, ranging from 0.40 to 0.92 and He = 0.79, ranging from 0.52 to 0.93) for the same subset of EST-SSR markers suggesting possibly a lower adaptive variation for the populations under study. Nevertheless, our data on genetic diversity are in agreement with the SSR data found in other published studies of related subspecies. The observation of decreasing diversity with increasing size classes, however, was unexpected and has been to our knowledge only rarely observed as for instance for Quercus rubra in southern New England (USA) by Gerwein and Kesseli (2006). Authors suggested some recovery of alleles when forest cover increases, which might be true also for our study indicating a formerly stronger fragmented population with reduced gene flow subjected to loss of genetic diversity. Alternatively, selected genotypes with augmented growth could have resulted in low genetic diversity with increasing size. Our results, however, do not provide evidence for selected genotypes with comparable genetic composition. Moreover, since we have used DBH as a proxy for age, but have not determined tree age by annual ring counts, these observations remain vague and, if they are not accidental, could have been caused by other factors.
Genetic differentiation and population structure
Genetic differentiation among geographic groups was very low in general, but significant in all cases. The low differentiation among the six sampling locations (FST = 0.012, F′ST = 0.036) and even lower differentiation between sites (FST = 0.004, F′ST = 0.009) and mountains (FST = 0.007, F′ST = 0.020) suggest effective gene flow (at least in the recent past) and a still effectively large population size, supported by an absent or at least a weak fine-scale genetic structure. As not all markers demonstrated significant excess of homozygotes, the fixation index (FIS) cannot serve as a proxy for inbreeding.
The low genetic differentiation measures found for the samples can be explained by long-distance dispersal. Extensive wind pollination and long distance seed dispersal by mammals and birds may ensure comparatively high genetic diversity even in isolated mountain populations of O. europaea subsp. cuspidata as likewise suggested for related O. europaea subspecies (Baali-Cherif and Besnard 2005; Besnard et al. 2007, 2009, 2013; Breton et al. 2006; Kassa et al. 2017). Potential mammal vectors for O. europaea subsp. cuspidata seeds (fruits) dispersal in the Hajar Mountains are livestock (goats and sheep) and feral donkeys nowadays. Livestock species very likely contributed to the dispersal activity in the last decades and still do. However, other taxa such as birds of the genera Blackcaps (Sylvia sp.) and Thrushes (Turdus sp.) are known to be likely effective seed dispersal agents for O. europaea (Rey et al. 1997). Although a reduced gene flow can be expected due to a rough terrain, absence of sufficient flowering trees, and the fragmentation of stands (Abiyu 2012; Kassa et al. 2017), we did not find any environmental or demographic effect on subpopulation structure. Even a ~ 20 km distance between the mountain peaks seems to have no effect on the substructure. Perhaps, it is due to the connection via a mountain crest that hardly goes beyond 1800 m. Wadis (deep valley occasionally filled with water) between mountains are narrow and are unlikely to be major barriers for anemochory and zoochory. This finding was furthermore substantiated by the low correlation between genetic and geographic distances. However, this correlation was significant demonstrating that gene flow is or was relatively limited and that isolation by distance might have played (and certainly still does) an important role in differentiation of olive populations.
In comparison to other population genetic studies of olive subspecies that mostly employed random sampling approaches, an effect of our sampling design on the obtained results should also be considered. Although no closely related or clonal trees have been found, neighbouring trees were slightly more related than more distant trees based on the spatial autocorrelation analysis. Finally, the STRUCTURE analysis did find an ambiguous number of clusters, which can be interpreted as a lack of a strong population substructure. A large individual genetic admixture and rather arbitrary assignment of individuals to different clusters mean no obvious substructure except for the Shams 3 slope site that behaved slightly different. However, it is known that population structure algorithms in BAPS, GENELAND and STRUCTURE computer programs tend to overestimate K, especially when isolation-by-distance is strong (Frantz et al. 2009; Guillot et al. 2005; Pritchard et al. 2000). Therefore, the low, but nonetheless significant isolation-by-distance found in our study can partly explain the rather large number of clusters inferred in our sample by STRUCTURE analysis, although those numbers could be rather illusive. Nevertheless, simulations by Guillot and Santos (2009) found that such interferences are stronger particularly in populations with small dispersal distances and spatially irregular patterns (clumped sampling as ours). Moreover, Pritchard et al. (2010) even stated that they permit allele frequencies to be correlated across populations in STRUCTURE to allow accurate assignments of individuals in very closely related individuals, though possibly at the cost of making it likely to overestimate the number of clusters. Hence, we should not overemphasize the number of clusters, especially when population structure and overall differentiation measures are low. However, effects of more recent fragmentation events could be reflected in the next offspring generation suggesting the need for additional studies, particular with respect to a very low recruitment and die-backs that seems to be chronic for the population in the Hajar Mountains. Finally, hypothesis 3 must be rejected as no evidence on genetic bottlenecks and signatures of inbreeding were found.
Conclusions
We could not identify the driving forces of low regeneration and die-back of trees, but suggest cumulative abiotic and biotic effects as the most plausible cause for demographic change. Remoteness alone appears to be not an explanatory factor for the viability of stands. Therefore, we failed to answer hypothesis 1. The absence of identical multi-locus genotypes led us conclude that clonal growth is non-existent at least for the studied individuals and reject hypothesis 2. The studied populations contained relatively high levels of genetic variation and low differentiation based on both nSSR markers that are likely selectively neutral and EST-SSR markers that could be under selection. Overall, bottleneck effects and inbreeding expected under the prevalent conditions have not been found prompting us to reject also hypothesis 3. However, comparatively high levels of genetic variation do not preclude that this population could become vulnerable, especially considering their fragmentation, the lack of regeneration, over-aging, and ongoing climate variations. The unexpectedly higher diversity found in younger size classes merits further studies of sibling generations to better address the questions regarding sustainability of this population, and whether they need special protection.
Data availability
Data can be shared upon request.
References
Abiyu A (2012) The role of seed dispersal, exclosures, nurse shrubs and trees around churches and farms for restoration of ecosystem diversity and productivity in the Ethiopian Highlands. BOKU-University of Natural Resources and Applied Life Sciences, Wien
Abiyu A, Teketay D, Glatzel G, Gratzer G (2015) Tree seed dispersal by African civets in the Afromontane Highlands: too long a latrine to be effective for tree population dynamics. Afr J Ecol 53:588–591. https://doi.org/10.1111/aje.12198
Al Charaabi Y, Al-Yahyai S (2013) Projection of future changes in rainfall and temperature patterns in Oman. J Earth Sci Clim Change 4:154–161. https://doi.org/10.4172/2157-7617.1000154
Al Sarmi S, Washington R (2011) Recent observed climate change over the Arabian Peninsula. J Geophys Res. https://doi.org/10.1029/2010JD015459
Al-Kalbani MS, Price MF (2015) Sustainable aflaj water management in Al Jabal Al Akhdar, Sultanate of Oman. Water Resour Manag VIII 1:27–38
Al-Kalbani MS, Price MF, Abahussain A, Ahmed M, O’Higgins T (2014) Vulnerability assessment of environmental and climate change impacts on water resources in Al Jabal Al Akhdar, Sultanate of Oman. Water 6:3118–3135. https://doi.org/10.3390/w6103118
Anderson JT (2016) Plant fitness in a rapidly changing world. New Phytol 210:81–87. https://doi.org/10.1111/nph.13693
Arbeiter AB, Hladnik M, Jakše J, Bandelj D (2017) Identification and validation of novel EST-SSR markers in olives. Sci Agric 74:215–225. https://doi.org/10.1590/1678-992x-2016-0111
Baali-Cherif D, Besnard G (2005) High genetic diversity and clonal growth in relict populations of Olea europaea subsp. laperrinei (Oleaceae) from Hoggar, Algeria. Ann Bot 96:823–830. https://doi.org/10.1093/aob/mci232
Becklin KM, Anderson JT, Gerhart LM, Wadgymar SM, Wessinger CA, Ward JK (2016) Examining plant physiological responses to climate change through an evolutionary lens. Plant Physiol 172:635–649. https://doi.org/10.1104/pp.16.00793
Bekele T (2000) Plant population dynamics of Dodonaea angustifolia and Olea europaea ssp. cuspidata in dry Afromontane forests of Ethiopia. Ph.D. Thesis
Besnard G, Christin PA, Baali-Cherif D, Bouguedoura N, Anthelme F (2007) Spatial genetic structure in the Laperrine’s olive (Olea europaea subsp. laperrinei), a long-living tree from the central Saharan mountains. Heredity 99:649–657. https://doi.org/10.1038/sj.hdy.6801051
Besnard G, Baali-Cherif D, Bettinelli-Riccardi S, Parietti D, Bouguedoura N (2009) Pollen-mediated gene flow in a highly fragmented landscape: consequences for defining a conservation strategy of the relict Laperrine’s olive. CR Biol 332:662–672. https://doi.org/10.1016/j.crvi.2009.02.003
Besnard G, El Bakkali A, Haouane H, Baali-Cherif D, Moukhli A, Khadari B (2013) Population genetics of Mediterranean and Saharan olives: geographic patterns of differentiation and evidence for early generations of admixture. Ann Bot 112:1293–1302. https://doi.org/10.1093/aob/mct196
Braatne J, Rood S, Heilman P (1996) Life history, ecology, and conservation of riparian cottonwoods in North America. In: Stettler R, Bradshaw T, Heilman P, Hinckley T (eds) Biology of Populus and its implications for management and conservation. Canadian Science Publishing NRC Research Press, Ottawa, pp 57–85
Breton C, Tersac M, Bervillé A (2006) Genetic diversity and gene flow between the wild olive (oleaster, Olea europaea L.) and the olive: several Plio-Pleistocene refuge zones in the Mediterranean basin suggested by simple sequence repeats analysis. J Biogeogr 33:1916–1928. https://doi.org/10.1111/j.1365-2699.2006.01544.x
Brownstein MJ, Carpten JD, Smith JR (1996) Modulation of non-templated nucleotide addition by Taq DNA polymerase: primer modifications that facilitate genotyping. Biotechniques 20:1004–1010. https://doi.org/10.2144/96206st01
Canale A, Loni A (2010) Insects visiting olive flowers (Olea europaea L.) in a Tuscan olive grove. J Zool 92:95–98
Carlsson J (2008) Effects of microsatellite null alleles on assignment testing. J Hered 99:616–623. https://doi.org/10.1093/jhered/esn048
Carriero F, Fontanazza G, Cellini F, Giorio G (2002) Identification of simple sequence repeats (SSRs) in olive (Olea europaea L.). Theor Appl Genet 104:301–307. https://doi.org/10.1007/s001220100691
Cipriani G, Marrazzo MT, Marconi R, Cimato A, Testolin R (2002) Microsatellite markers isolated in olive (Olea europaea L.) are suitable for individual fingerprinting and reveal polymorphism within ancient cultivars. Theor Appl Genet 104:223–228. https://doi.org/10.1007/s001220100685
Cuneo P, Leishman MR (2006) African Olive (Olea europaea subsp. cuspidata) as an environmental weed in eastern Australia: a review. Cunninghamia 9:545–577
De la Rosa R, James CM, Tobutt KR (2002) Isolation and characterization of polymorphic microsatellites in olive (Olea europaea L.) and their transferability to other genera in the Oleaceae. Mol Ecol Notes 2:265–267. https://doi.org/10.1046/j.1471-8286.2002.00217.x
Díaz A, de la Rosa R, Martín A, Rallo P (2006) Development, characterization and inheritance of new microsatellites in olive (Olea europaea L.) and evaluation of their usefulness in cultivar identification and genetic relationship studies. Tree Genet Genomes 2:165–175. https://doi.org/10.1007/s11295-006-0041-5
Earl DA, VonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Erre P, Chessa I, Muñoz-Diez C, Belaj A, Rallo L, Trujillo I (2010) Genetic diversity and relationships between wild and cultivated olives (Olea europaea L.) in Sardinia as assessed by SSR markers. Genet Resour Crop Evol 57:41–54. https://doi.org/10.1007/s10722-009-9449-8
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Fisher M, Gardner AS (1995) The status and ecology of a Juniperus excelsa subsp polycarpos woodland in the northern mountains of Oman. Vegitatio 119:33–51. https://doi.org/10.1007/bf00047369
Frantz AC, Cellina S, Krier A, Schley L, Burke T (2009) Using spatial Bayesian methods to determine the genetic structure of a continuously distributed population: clusters or isolation by distance? J Appl Ecol 46:493–505. https://doi.org/10.1111/j.1365-2664.2008.01606.x
Gerwein JB, Kesseli RV (2006) Genetic diversity and population structure of Quercus rubra (Fagaceae) in old-growth and secondary forests in southern New England. Rhodora 108:1–18. https://doi.org/10.3119/05-9.1
Ghazanfar SA (2018) Edible wild plants: a case study from Oman. In: Ozturk M, Hakeem KR, Ashraf M, Ahmad MSA (eds) Global perspectives on underutilized crops. Springer International Publishing, Cham, pp 207–216
Goudie AS, Viles HA (2013) The earth transformed: an introduction to human impacts on the environment. Wiley, Hoboken
Grabherr G, Gottfried M, Pauli H (1994) Climate effects on mountain plants. Nature 369:448. https://doi.org/10.1038/369448a0
Green PS (2002) A revision of Olea L. (Oleaceae). Kew Bull 57:91–140. https://doi.org/10.2307/4110824
Guillot G, Santos F (2009) A computer program to simulate multilocus genotype data with spatially autocorrelated allele frequencies. Mol Ecol Resour 9:1112–1120. https://doi.org/10.1111/j.1755-0998.2008.02496.x
Guillot G, Estoup A, Mortier F, Cosson JF (2005) A spatial statistical model for landscape genetics. Genetics 170:1261–1280. https://doi.org/10.1534/genetics.104.033803
Hardy OJ, Vekemans X (2002) SPAGeDI: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620. https://doi.org/10.1046/j.1471-8286.2002.00305.x
Jost LOU (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026. https://doi.org/10.1111/j.1365-294X.2008.03887.x
Kassa A, Konrad H, Geburek T (2017) Landscape genetic structure of Olea europaea subsp. cuspidata in Ethiopian Highland forest fragments. Conserv Genet 18:1463–1474. https://doi.org/10.1007/s10592-017-0993-z
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) CLUMPAK: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191. https://doi.org/10.1111/1755-0998.12387
Kubisiak TL, Nelson CD, Staton ME, Zhebentyayeva T, Smith C, Olukolu BA, Fang GC, Hebard FV, Anagnostakis S, Wheeler N, Sisco PH, Abbott AG, Sederoff RR (2013) A transcriptome-based genetic map of Chinese chestnut (Castanea mollissima) and identification of regions of segmental homology with peach (Prunus persica). Tree Genet Genomes 9:557–571. https://doi.org/10.1007/s11295-012-0579-3
Lenoir J, Gégout JC, Marquet PA, de Ruffray P, Brisse H (2008) A significant upward shift in plant species optimum elevation during the 20th century. Science 320:1768–1771. https://doi.org/10.1126/science.1156831
Lind JF, Gailing O (2013) Genetic structure of Quercus rubra L. and Quercus ellipsoidalis E. J. Hill populations at gene-based EST-SSR and nuclear SSR markers. Tree Genet Genomes 9:707–722. https://doi.org/10.1007/s11295-012-0586-4
Loiselle BA, Sork VL, Nason J, Graham C (1995) Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82:1420–1425. https://doi.org/10.2307/2445869
MacLaren CA (2016) Climate change drives decline of Juniperus seravschanica in Oman. J Arid Environ 128:91–100. https://doi.org/10.1016/j.jaridenv.2016.02.001
Meirmans PG (2006) Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evolution 60:2399–2402. https://doi.org/10.1111/j.0014-3820.2006.tb01874.x
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70:3321–3323. https://doi.org/10.1073/PNAS.70.12.3321
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Patzelt A (2015a) Photographic field guide to the plants of the Western Hajar mountains, Sultanate of Oman, with a complete checklist of vascular plant species. Sultan Qaboos University, Muscat
Patzelt A (2015b) Synopsis of the flora and vegetation of Oman, with special emphasis on patterns of plant endemism. Abhandlungen der Braunschweigischen Wissenschaftlichen Gesellschaft 282:317
Peakall R, Smouse PE (2012) GenALEx 6.5: genetic analysis in excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90:502–503. https://doi.org/10.1093/jhered/90.4.502
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pritchard JK, Wen X, Falush D (2010) Documentation for STRUCTURE software: version 234. Department of Human Genetics, University of Chicago, Chicago
Rallo P, Dorado G, Martín A (2000) Development of simple sequence repeats (SSRs) in olive tree (Olea europaea L.). Theor Appl Genet 101:984–989. https://doi.org/10.1007/s001220051571
Rey PJ, Gutiérrez JE, Alcántara J, Valera F (1997) Fruit size in wild olives: implications for avian seed dispersal. Funct Ecol 11:611–618. https://doi.org/10.1046/j.1365-2435.1997.00132.x
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Saumitou-Laprade P, Vassiliadis C, Epplen JT, Hardt C (2000) Isolation of microsatellite loci for paternity testing in Phillyrea angustifolia L. (Oleaceae). Mol Ecol 9:112–114. https://doi.org/10.1046/j.1365-294X.2000.00764-4.x
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234. https://doi.org/10.1038/72708
Sefc KM, Lopes MS, Mendonça D, Rodrigues Dos Santos M, Da Câmara Laimer, Machado M, Da Câmara MachadoA (2000) Identification of microsatellite loci in olive (Olea europaea) and their characterization in Italian and Iberian olive trees. Mol Ecol 9:1171–1173. https://doi.org/10.1046/j.1365-294X.2000.00954.x
Tardieu F (2013) Plant response to environmental conditions: assessing potential production, water demand, and negative effects of water deficit. Front Physiol 4:17. https://doi.org/10.3389/fphys.2013.00017
R Core Team (2017) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org
van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Vekemans X, Hardy OJ (2004) New insights from fine-scale spatial genetic structure analyses in plant populations. Mol Ecol 13:921–935. https://doi.org/10.1046/j.1365-294X.2004.02076.x
Victor R (2008) Are we losing Al Jabal Al Akhdar? An environmental evaluation of an arid mountain ecosystem in Oman. Int J Environ Stud 65:731–736. https://doi.org/10.1080/00207230802124499
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358. https://doi.org/10.2307/2408641
Wesche K, Ronnenberg K, Hensen I (2005) Lack of sexual reproduction within mountain steppe populations of the clonal shrub Juniperus sabina L. in semi-arid southern Mongolia. J Arid Environ 63:390–405. https://doi.org/10.1016/j.jaridenv.2005.03.014
Acknowledgements
We thank our colleagues Salim Al Rahbi, Omar Al Amri, Abdulrahman Al Hinai, and Zawan Al Qassabi from the Oman Botanic Garden for their assistance and care during field work. We are grateful to our colleague Alexandra Dolynska (Georg-August University of Göttingen), who assisted intensively in lab work. We thank Andreas Bürkert (Organic Plant Production and Agroecosystems Research in the Tropics and Subtropics, University of Kassel, Germany) for the generous financial and intellectual support.
Funding
Open Access funding enabled and organized by Projekt DEAL. There was no specific funding source.
Author information
Authors and Affiliations
Contributions
Conceptualization, AP, and MW; methodology, AP, GI, and MW; investigation, GI, NAH, MM, and MW; validation, GI, NAH, MM, and MW; formal analysis, GI, NAH, MM, and MW; data curation, GI, NAH, MM, and MW; writing—original draft preparation, NAH and MW; writing–review & editing, AP, GI, KVK, NAH, MM, MW, and OG; visualization, NAH, MM, and MW; resources: KVK, and OG; supervision, KVK, MW, and OG; project administration, AP, MW, and OG. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare there are no competing interests.
Ethics approval
Not applicable.
Informed consent
All authors agreed on the publication of the present work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Habib, N.A., Müller, M., Gailing, O. et al. Genetic diversity and differentiation of Olea europaea subsp. cuspidata (Wall. & G.Don) Cif. in the Hajar Mountains of Oman. Genet Resour Crop Evol 68, 865–883 (2021). https://doi.org/10.1007/s10722-020-01030-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-020-01030-2