
Abstract
Dynamic changes to the epigenome play a critical role in a variety of biology processes and complex traits. Many important candidate genes have been identified through our previous genome wide association study (GWAS) on milk production traits in dairy cattle. However, the underlying mechanism of candidate genes have not yet been clearly understood. In this study, we analyzed the methylation variation of the candidate genes, EEF1D and RPL8, which were identified to be strongly associated with milk production traits in dairy cattle in our previous studies, and its effect on protein and mRNA expression. We compared DNA methylation profiles and gene expression levels of EEF1D and RPL8 in five different tissues (heart, liver, mammary gland, ovary and muscle) of three cows. Both genes showed the highest expression level in mammary gland. For RPL8, there was no difference in the DNA methylation pattern in the five tissues, suggesting no effect of DNA methylation on gene expression. For EEF1D, the DNA methylation levels of its first CpG island differed in the five tissues and were negatively correlated with the gene expression levels. To further investigate the function of DNA methylation on the expression of EEF1D, we collected blood samples of three cows at early stage of lactation and in dry period and analyzed its expression and the methylation status of the first CpG island in blood. As a result, the mRNA expression of EEF1D in the dry period was higher than that at the early stage of lactation, while the DNA methylation level in the dry period was lower than that at the early stage of lactation. Our result suggests that the DNA methylation of EEF1D plays an important role in the spatial and temporal regulation of its expression and possibly have an effect on the milk production traits.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Milk production traits are the most important traits in dairy cattle. In the past decades, many studies have been carried out to reveal the genetic basis of milk production traits (Zhang et al. 1998; Farnir et al. 2002; Ashwell et al. 2004; Jiang et al. 2010; Mai et al. 2010) and a lot of candidate genes or QTL affecting milk yield and milk components have been reported (Viitala et al. 2006; Winter et al. 2002; Wang et al. 2013). Along with the development of cost-effective “omics” technology, many powerful tools are being used to identify functional genes (mutations) and their regulatory mechanisms recently, such as genome-wide association studies (GWAS) (Jiang et al. 2010; Mai et al. 2010; Meredith et al. 2012; Cole et al. 2011) and gene expression profiles (Wickramasinghe et al. 2012; Connor et al. 2013; Singh et al. 2013; Cui et al. 2014). Although these findings provide new insights into genetic basis of milk production traits, the underlying mechanism of potential candidate genes have not yet been clearly understood.
In our previous GWAS study in Chinese Holstein cattle, 105 significant SNPs associated with milk yield and composition traits were identified (Jiang et al. 2010). In the followed study, we used the target enrichment technology and next generation sequencing (NGS) to assess the candidate regions implicated by significant SNPs in our GWAS and then we conducted association analysis for 200 important variants revealed by NGS in a new dairy cattle population (Jiang et al. 2014). As a result, a total of 66 significant SNPs involved in 53 genes were identified. Of these, one SNP located in the promoter region of EEF1D showed strong association with milk yield, fat percentage and protein percentage with P values of 9.23E-06, 2.07E-15 and 1.26E-07, respectively (Jiang et al. 2014). Furthermore, one SNP in the promoter region of RPL8 was also identified to be associated significantly with milk yield, protein yield and fat percentage with P values of 2.62E-07, 6.63E-05 and 2.26E-15, respectively (Jiang et al. 2014). In addition, we investigated the mRNA expression of 20 significant candidate genes in different tissues in dairy cattle and most of them showed higher expression in mammary gland than in other tissues. In particular, both EEF1D and RPL8 had the highest expression in mammary gland among the 20 genes. Therefore, EEF1D and RPL8 were considered as two promising candidate genes for milk production traits in dairy cattle.
EEF1D encodes a subunit of the elongation factor-1 complex, which is responsible for the enzymatic delivery of aminoacyl tRNAs to the ribosome and functions as a guanine nucleotide exchange factor (Ogawa et al. 2004). RPL8 encodes the 60S ribosomal protein L8, which is a component of the ribosomes 60S subunit. This protein belongs to the L2P family of ribosomal proteins and is located in the cytoplasm. It has been reported that the mRNAs of components of the 60S subunit were highly abundant in bovine mammary and contributed to protein synthesis (Bionaz and Loor 2011). Both of these two genes are located on chromosome 14, which has been reported to harbor major QTLs for milk production traits, especially for fat percentage (Winter et al. 2002; Cole et al. 2011; Coppieters et al. 1998; Kaupe et al. 2007).
Methylation of CpG islands in the promoter region of genes has been widely known to be involved in a variety of biology processes and complex diseases, such as embryo development (Smith et al. 2012; Smallwood et al. 2011) and tumorigenesis (Ronneberg et al. 2011; Dedeurwaerder et al. 2011). Recently, studies on DNA methylation profiling across the genome are increasing. Different genome-wide DNA methylation maps have been reported in many distinct tissues and organisms, such as human (Slieker et al. 2013; Davies et al. 2012; Day et al. 2013), chimpanzee (Pai et al. 2011) and rat (Hon et al. 2013). Many epigenetic studies revealed that the aberrant DNA methylation of CpG islands in the promoter regions results in inactivation of genes and plays an important role in tumor progression (Park et al. 2011; Cai et al. 2011). Differential CpG island methylation contributes to the gene expression by influencing transcription factors binding, altering genomic structure and regulating the microRNA expression levels (Jones and Liang 2009; Herman and Baylin 2003; Chellappan et al. 2010). However, few epigenetic modification studies have been reported for milk production traits in dairy cattle.
In this study, we analyzed the DNA methylation pattern of the CpG islands in the promoter regions of EEF1D and RPL8. Our results revealed that the DNA methylation level of one CpG island of EEF1D was significantly negatively correlated with the expression level of EEF1D in different tissues and different periods in dairy cattle. Our study provides more information on epigenetics in dairy cattle.
Materials and methods
Animals and tissue sample collection
Three lactating Chinese Holstein cows were selected from the Beijing Sanyuan Dairy Farm Center. All of them were fed in a standard environmental condition. Five tissue samples (heart, liver, mammary gland, ovary and muscle) from each individual were collected within 30 min after slaughter and stored at liquid nitrogen. The whole procedure for collection of tissue samples of all animals was carried out in strict accordance with the protocol approved by the Animal Welfare Committee of China Agricultural University (Permit number: DK996).
Western blotting analysis
Western blotting analysis was performed to detect the protein expression levels of EEF1D and RPL8 in different tissues. Total proteins were extracted from the five tissues samples (heart, liver, mammary gland, ovary and muscle) of three different individuals. A total of 60 µg of proteins were separated on 10% SDS–PAGE, and then transferred to PVDF membranes (BIO-RAD). After blocked with 5% skim milk for 1.5 h at room temperature, the membranes were incubated with the primary antibody at 4 °C over night (EEF1D: Abcam; RPL8: Santa Cruz Biotechnology; GAPDH: Santa Cruz Biotechnology), and then further incubated with corresponding HRP-conjugated secondary antibodies (Sigma) for 1 h at room temperature. The labeled bands were visualized by using the ECL kit (BIO-RAD). The Photoshop software was used to quantify the relative expression levels.
Methylation analysis
Genomic DNA was extracted from the five tissue samples of the three cows using the commercial kit (Tiangen Biotech, Beijing, China). The quantity and quality of DNA were measured using NanoDropTMND-2000c Spectrophotometer (Thermo Scientific, Inc.). The DNA was chemically modified by sodium bisulfate to convert all unmethylated cytosines to uracils while leaving methylcytosines unaltered using the EZ DNA Methylation-Gold Kit (Zymo Research, CA, USA). CpG islands in the promoter region were detected by using the CpG Plot web-tool (http://www.ebi.ac.uk/Tools/emboss/cpgplot/). For these regions, three pairs of primers were designed to carry out Methylation-Specific PCR (Table 1). After amplification, the PCR products were cloned into a pBLUE-T vector. The plasmid was then used to transform TOP competent cells. Ten colonies per sample were chosen randomly for sequencing.
Results
Analysis the promoter region of EEF1D
We first analyzed the expression of the EEF1D protein in the five tissues (heart, liver, mammary gland, ovary and muscle) of the three cows by using Western blotting. The results showed about two- to three-fold higher expression level in mammary gland than in other tissues (Fig. 1a), which is well in accordance with the mRNA expression of EEF1D in these tissues in our previous study (Jiang et al. 2014).

Relative protein expression of EEF1D (a) and RPL8 (b) in five tissues of three lactating cows detected by Western blotting. GAPDH was used as a control
Since no significant SNPs were found in the coding region of EEF1D in our previous association studies, we paid our attention to the promoter region. Two CpG islands (−2770 to −2444 bp and −2126 to −1808 bp upstream from ATG, respectively) were detected in the promoter region. The methylation status of the two CpG islands were quantitatively measured in five different tissues (heart, liver, mammary gland, ovary and muscle) of the three cows used for Western blotting analysis. For the first CpG island, the methylation status varied remarkably in different tissues (Fig. 2a). All the three individuals displayed significantly lower methylation level in mammary gland than in other tissues. The proportion of the methylated alleles in mammary gland was very low with an average of 16%, while it was 44–64% in other tissues (Fig. 2c). However, the methylation levels of the second CpG island were almost the same in all tissues and the proportion of methylated alleles reached nearly 100% (Fig. 2a). The methylation rates for each CpG site of the two CpG islands were also calculated for the three individuals and are displayed in Fig. 2b. The methylation levels in the first CpG island were well in accordance with the expression levels of EEF1D in different tissues, i.e., the lower the methylation level, the higher the protein expression levels, suggesting that the expression of EEF1D is regulated by the first CpG island methylation in its promoter.

Methylation analysis of the two CpG islands in the promoter region of EEF1D in five tissues of three lactating cows. a Methylation patterns of the two CpG islands. Error bars represent the standard deviation. b Methylation profiles of all sites of the two CpG islands. c Methylation status of the 32 sites of the first CpG island in mammary gland (left) and muscle (right) of one cow. Ten independent PCR product clones were demonstrated for each lines. Solid dot, methylated CpG dinucleotide; circle, unmethylated GpG dinucleotide
Analysis the promoter region of RPL8
We also analyzed the protein expression of RPL8 in the five different tissues of the three cows by using Western blotting. We observed three- to seven-fold higher expression of the RPL8 protein in mammary gland than in other tissues (Fig. 1b), which is well in accordance with the mRNA expression of RPL8 in these tissues in our previous study (Jiang et al. 2014).
Just like the situation in EEF1D, no significant SNPs were detected in the coding regions of RPL8 in our previous association studies, we focused our attention on its promoter region again. Only one CpG island (−252 to −41 upstream from ATG) was detected in the promoter region. However, the methylation analysis showed that there was no difference in methylation level among the five tissues. Besides, all the CpG sites of this island were unmethylated in the five different tissues. These results suggest that the differential expression of RPL8 in different tissues is not regulated by DNA methylation of the CpG island in RPL8 promoter in dairy cattle.
Discussion
Following our previous GWAS based on bovine 50k SNP array (Jiang et al. 2010), target region association analysis based on targeted re-sequencing, and mRNA expression analysis of candidate genes, we performed protein expression and methylation analysis for two strong candidate genes, EEF1D and RPL8, in this study. Western blotting analysis showed that the expression of the EEF1D and RPL8 proteins were well in accordance with their mRNA expression, suggesting that EEF1D and RPL8 may play an important role in milk production traits in dairy cattle. Moreover, both of these two genes are located on chromosome 14 and EEF1D gene located a mere 806 kb away from RPL8. According to the gene annotation, RPL8 is a component of the ribosome 60S subunit. EEF1D encodes a subunit of the elongation factor-1 complex, which is responsible for the enzymatic delivery of aminoacyl tRNAs to the ribosome and functions as a guanine nucleotide exchange factor. EEF1D very likely participates in the binding of aminoacyl-tRNA at the ribosomal 60S subunit interface. Thus, there might be an interaction effect between these two genes. In addition, it has been reported that both EEF1D and the ribosome 60S were highly expressed in the mammary tissue of lactating cows (Jiang et al. 2014; Bionaz and Loor 2011). Therefore, both EEF1D and RPL8 play important roles in milk production, either separately or interactively. Considering no significant variants in the coding regions were detected, we focused our attention on their promoter regions, where SNPs with significant effects on milk fat percentage were identified.
DNA methylation status in promoter region of a gene has been proved to play important roles in regulation of gene expression (Zhang et al. 2012; Heyn and Esteller 2012; Heyn et al. 2013). In particular, hypermethylation of the promoter region of a gene could effectively silence its transcription (Jandrig et al. 2004; Yang et al. 2009; Misawa et al. 2013). Therefore, we investigated the methylation patterns of EEF1D and RPL8. For EEF1D, we found two CpG islands in its promoter region, and the methylation level of the first CpG island in mammary gland was much lower than in other tissues, which was well in accordance with the mRNA and protein expression of EEF1D in these tissues, i.e., the higher (lower) expression level was corresponding to the lower (higher) methylation level, while the methylation levels of the second CpG island were almost the same in all tissues. These results suggest that the expression of EEF1D is regulated by the methylation status of the first CpG island in its promoter region. For RPL8, we found only one CpG island in its promoter region. However, no difference in methylation level among different tissues was detected, suggesting that the differential expression of RPL8 in different tissues is not relevant to the methylation status in its promoter region.
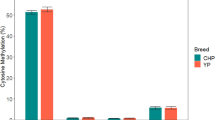
To further confirm the regulation effect of methylation at the first CpG island in the promoter region of EEF1D on its expression, we collected blood samples of another three cows at early stage of lactation (15 days in milk) as well as in dry period (30 days before calving) and analyzed the mRNA expression of EEF1D and the methylation status of this CpG island in blood in the two periods. As a result, the mRNA expression in the dry period was higher than that at the early stage of lactation in all the three cows (Fig. 3a), while the methylation level in the dry period was lower than that at the early stage of lactation (Fig. 3b). These results support the methylation is negative association with the expression of EEF1D.

Relative mRNA expression (a) and methylation level (b) in blood of three cows at dry (blue) and lactating (red) periods. Error bars represent the standard deviation
The function of a gene is closely related to its specifically spatial and temporal expression. Many previous studies reported that a large amount of genes were down-regulated in lactation stage compared with in non-lactation period in bovine mammary gland tissue (Finucane et al. 2008; Gao et al. 2013). In our previous study (Yang et al. 2016), we detected differential expression of genes by using RNA-seq in milk samples of two groups of cows with extremely high and low 305-day milk yield, milk fat yield and milk protein yield, respectively, at day 10 (early stage of lactation) and day 70 (peak stage of lactation) after calving. Our results indicated that most of the differentially expressed genes showed lower expression in the cows of group of high yield as well as at the peak stage of lactation. This implied the high milk production is associated with down-regulation of majority of genes. The expression of EEF1D in blood of cows in dry period and at the early stage of lactation showed the similar pattern, i.e., higher expression in dry period than at the early stage of lactation. It is reasonable to expect that this pattern would also be observed in mammary gland. This suggests that the expression of EEF1D is also related to the on-set of lactation and is regulated by the methylation at its first CpG island.
Some previous studies reported that epigenetic regulation might be involved in oocyte development, spermatogenesis and fat deposition (Diederich et al. 2012; Luo et al. 2013; Liu et al. 2011; Magee et al. 2010). Our data implied that epigenetic modification changed the expression of some important genes, which were significantly associated with milk production trait. Currently, genomic selection (GS) is the widely used in dairy cattle breeding. The theory of GS assumes that differences in DNA sequence lead to genetic differences between animals. However, there is evidence for epigenetic differences between individuals. These epigenetic differences are associated with changes in the expression of genes. In cattle, it has been reported that the additive effects of SNPs only explain 32–80% of genetic variance (Goddard ME and Whitelaw 2014). Although it is difficult to answer the relationship between methylation status and SNPs, it would be worthwhile to know more epigenetic information of major genes in cattle genome and to include these information in genetic evaluation approaches in the future to increase selection efficiency in cattle populations.
Conclusion
In mammals, tissue-specific DNA methylation patterns were established only in pigs (Li et al. 2012a, b). In this study, we found that the DNA methylation of EEF1D likely plays an important role in its transcriptional regulation and may have a severe effect on milk production traits in dairy cattle. Our results contribute to the knowledge of epigenetic effects in dairy cattle and promotes a better understanding of the global genetic architecture of milk production traits.
Abbreviations
- EEF1D:
-
Eukaryotic translation elongation factor 1 delta
- RPL8:
-
60S ribosomal protein L8
- GWAS:
-
Genome-wide association study
- NGS:
-
Next generation sequencing
- SNP:
-
Single nucleotide polymorphism
References
Ashwell MS, Heyen DW, Sonstegard TS, Van Tassell CP, Da Y, VanRaden PM, Ron M, Weller JI, Lewin HA (2004) Detection of quantitative trait loci affecting milk production, health, and reproductive traits in Holstein cattle. J Dairy Sci 87(2):468–475
Bionaz M, Loor JJ (2011) Gene networks driving bovine mammary protein synthesis during the lactation cycle. Bioinf Biol Insights 5:83–98
Cai M, Tian J, Zhao GH, Luo W, Zhang BR (2011) Study of methylation levels of parkin gene promoter in Parkinson’s disease patients. Int J Neurosci 121(9):497–502
Chellappan P, Xia J, Zhou X, Gao S, Zhang X, Coutino G, Vazquez F, Zhang W, Jin H (2010) siRNAs from miRNA sites mediate DNA methylation of target genes. Nucleic Acids Res 38(20):6883–6894
Cole JB, Wiggans GR, Ma L, Sonstegard TS, Lawlor TJ Jr, Crooker BA, Van Tassell CP, Yang J, Wang S, Matukumalli LK et al (2011) Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary U.S. Holstein cows. BMC Genomics 12:408
Connor EE, Baldwin RLt, Li CJ, Li RW, Chung H (2013) Gene expression in bovine rumen epithelium during weaning identifies molecular regulators of rumen development and growth. Funct Integr Genom 13(1):133–142
Coppieters W, Riquet J, Arranz JJ, Berzi P, Cambisano N, Grisart B, Karim L, Marcq F, Moreau L, Nezer C et al (1998) A QTL with major effect on milk yield and composition maps to bovine chromosome 14. Mamm Genome Off J Int Mamm Genome Soc 9(7):540–544
Cui X, Hou Y, Yang S, Xie Y, Zhang S, Zhang Y, Zhang Q, Lu X, Liu GE, Sun D (2014) Transcriptional profiling of mammary gland in Holstein cows with extremely different milk protein and fat percentage using RNA sequencing. BMC Genom 15:226
Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S, Coarfa C, Harris RA, Milosavljevic A, Troakes C et al (2012) Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol 13(6):R43
Day K, Waite LL, Thalacker-Mercer A, West A, Bamman MM, Brooks JD, Myers RM, Absher D (2013) Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol 14(9):R102
Dedeurwaerder S, Desmedt C, Calonne E, Singhal SK, Haibe-Kains B, Defrance M, Michiels S, Volkmar M, Deplus R, Luciani J et al (2011) DNA methylation profiling reveals a predominant immune component in breast cancers. EMBO Mol Med 3(12):726–741
Diederich M, Hansmann T, Heinzmann J, Barg-Kues B, Herrmann D, Aldag P, Baulain U, Reinhard R, Kues W, Weissgerber C et al (2012) DNA methylation and mRNA expression profiles in bovine oocytes derived from prepubertal and adult donors. Reproduction 144(3):319–330
Farnir F, Grisart B, Coppieters W, Riquet J, Berzi P, Cambisano N, Karim L, Mni M, Moisio S, Simon P et al (2002) Simultaneous mining of linkage and linkage disequilibrium to fine map quantitative trait loci in outbred half-sib pedigrees: revisiting the location of a quantitative trait locus with major effect on milk production on bovine chromosome 14. Genetics 161(1):275–287
Finucane KA, McFadden TB, Bond JP, Kennelly JJ, Zhao FQ (2008) Onset of lactation in the bovine mammary gland: gene expression profiling indicates a strong inhibition of gene expression in cell proliferation. Funct Integr Genom 8(3):251–264
Gao Y, Lin X, Shi K, Yan Z, Wang Z (2013) Bovine mammary gene expression profiling during the onset of lactation. PloS One 8(8):e70393
Goddard ME, Whitelaw E (2014) The use of epigenetic phenomena for the improvement of sheep and cattle. Front Genet 5:247
Herman JG, Baylin SB (2003) Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 349(21):2042–2054
Heyn H, Esteller M (2012) DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet 13(10):679–692
Heyn H, Carmona FJ, Gomez A, Ferreira HJ, Bell JT, Sayols S, Ward K, Stefansson OA, Moran S, Sandoval J et al (2013) DNA methylation profiling in breast cancer discordant identical twins identifies DOK7 as novel epigenetic biomarker. Carcinogenesis 34(1):102–108
Hon GC, Rajagopal N, Shen Y, McCleary DF, Yue F, Dang MD, Ren B (2013) Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat Genet 45(10):1198–1206
Jandrig B, Seitz S, Hinzmann B, Arnold W, Micheel B, Koelble K, Siebert R, Schwartz A, Ruecker K, Schlag PM et al (2004) ST18 is a breast cancer tumor suppressor gene at human chromosome 8q11.2. Oncogene 23(57):9295–9302
Jiang L, Liu J, Sun D, Ma P, Ding X, Yu Y, Zhang Q (2010) Genome wide association studies for milk production traits in Chinese Holstein population. PloS one 5(10):e13661
Jiang L, Liu X, Yang J, Wang H, Jiang J, Liu L, He S, Ding X, Liu J, Zhang Q (2014) Targeted resequencing of GWAS loci reveals novel genetic variants for milk production traits. BMC Genom 15:1105
Jones PA, Liang G (2009) Rethinking how DNA methylation patterns are maintained. Nat Rev Genet 10(11):805–811
Kaupe B, Brandt H, Prinzenberg EM, Erhardt G (2007) Joint analysis of the influence of CYP11B1 and DGAT1 genetic variation on milk production, somatic cell score, conformation, reproduction, and productive lifespan in German Holstein cattle. J Anim Sci 85(1):11–21
Li M, Wu H, Luo Z, Xia Y, Guan J, Wang T, Gu Y, Chen L, Zhang K, Ma J et al (2012a) An atlas of DNA methylomes in porcine adipose and muscle tissues. Nat Commun 3:850
Li M, Wang T, Wu H, Zhang J, Zhou C, Jiang A, Li R, Li X (2012b) Genome-wide DNA methylation changes between the superficial and deep backfat tissues of the pig. Int J Mol Sci 13(6):7098–7108
Liu Z, Li Q, Pan Z, Qu X, Zhang C, Xie Z (2011) Comparative analysis on mRNA expression level and methylation status of DAZL gene between cattle-yaks and their parents. Anim Reprod Sci 126(3–4):258–264
Luo H, Zhou Y, Li Y, Li Q (2013) Splice variants and promoter methylation status of the Bovine Vasa Homology (Bvh) gene may be involved in bull spermatogenesis. BMC Genet 14:58
Magee DA, Sikora KM, Berkowicz EW, Berry DP, Howard DJ, Mullen MP, Evans RD, Spillane C, MacHugh DE (2010) DNA sequence polymorphisms in a panel of eight candidate bovine imprinted genes and their association with performance traits in Irish Holstein-Friesian cattle. BMC Genet 11:93
Mai MD, Sahana G, Christiansen FB, Guldbrandtsen B (2010) A genome-wide association study for milk production traits in Danish Jersey cattle using a 50 K single nucleotide polymorphism chip. J Anim Sci 88(11):3522–3528
Meredith BK, Kearney FJ, Finlay EK, Bradley DG, Fahey AG, Berry DP, Lynn DJ (2012) Genome-wide associations for milk production and somatic cell score in Holstein–Friesian cattle in Ireland. BMC Genet 13:21
Misawa K, Kanazawa T, Misawa Y, Imai A, Uehara T, Mochizuki D, Endo S, Takahashi G, Mineta H (2013) Frequent promoter hypermethylation of tachykinin-1 and tachykinin receptor type 1 is a potential biomarker for head and neck cancer. J Cancer Res Clin Oncol 139(5):879–889
Ogawa K, Utsunomiya T, Mimori K, Tanaka Y, Tanaka F, Inoue H, Murayama S, Mori M (2004) Clinical significance of elongation factor-1 delta mRNA expression in oesophageal carcinoma. Br J Cancer 91(2):282–286
Pai AA, Bell JT, Marioni JC, Pritchard JK, Gilad Y (2011) A genome-wide study of DNA methylation patterns and gene expression levels in multiple human and chimpanzee tissues. PLoS Genet 7(2):e1001316
Park SY, Kwon HJ, Lee HE, Ryu HS, Kim SW, Kim JH, Kim IA, Jung N, Cho NY, Kang GH (2011) Promoter CpG island hypermethylation during breast cancer progression. Virchows Archiv Int J Pathol 458(1):73–84
Ronneberg JA, Fleischer T, Solvang HK, Nordgard SH, Edvardsen H, Potapenko I, Nebdal D, Daviaud C, Gut I, Bukholm I et al (2011) Methylation profiling with a panel of cancer related genes: association with estrogen receptor, TP53 mutation status and expression subtypes in sporadic breast cancer. Mol Oncol 5(1):61–76
Singh M, Thomson PC, Sheehy PA, Raadsma HW (2013) Comparative transcriptome analyses reveal conserved and distinct mechanisms in ovine and bovine lactation. Funct Integr Genom 13(1):115–131
Slieker RC, Bos SD, Goeman JJ, Bovee JV, Talens RP, van der Breggen R, Suchiman HE, Lameijer EW, Putter H, van den Akker EB et al (2013) Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenet Chromatin 6(1):26
Smallwood SA, Tomizawa S, Krueger F, Ruf N, Carli N, Segonds-Pichon A, Sato S, Hata K, Andrews SR, Kelsey G (2011) Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat Genet 43(8):811–814
Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, Meissner A (2012) A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 484(7394):339–344
Viitala S, Szyda J, Blott S, Schulman N, Lidauer M, Maki-Tanila A, Georges M, Vilkki J (2006) The role of the bovine growth hormone receptor and prolactin receptor genes in milk, fat and protein production in Finnish Ayrshire dairy cattle. Genetics 173(4):2151–2164
Wang H, Jiang L, Liu X, Yang J, Wei J, Xu J, Zhang Q, Liu JF (2013) A post-GWAS replication study confirming the PTK2 gene associated with milk production traits in Chinese Holstein. PloS One 8(12):e83625
Wickramasinghe S, Rincon G, Islas-Trejo A, Medrano JF (2012) Transcriptional profiling of bovine milk using RNA sequencing. BMC Genom 13:45
Winter A, Kramer W, Werner FA, Kollers S, Kata S, Durstewitz G, Buitkamp J, Womack JE, Thaller G, Fries R (2002) Association of a lysine-232/alanine polymorphism in a bovine gene encoding acyl-CoA:diacylglycerol acyltransferase (DGAT1) with variation at a quantitative trait locus for milk fat content. Proc Natl Acad Sci USA 99(14):9300–9305
Yang N, Eijsink JJ, Lendvai A, Volders HH, Klip H, Buikema HJ, van Hemel BM, Schuuring E, van der Zee AG, Wisman GB (2009) Methylation markers for CCNA1 and C13ORF18 are strongly associated with high-grade cervical intraepithelial neoplasia and cervical cancer in cervical scrapings. Cancer epidemiology, biomarkers and prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive. Int Soc Cell 18(11):3000–3007
Yang J, Jiang J, Liu X, Wang H, Guo G, Zhang Q, Jiang L (2016) Differential expression of genes in milk of dairy cattle during lactation. Anim Genet 47(2):174–180
Zhang Q, Boichard D, Hoeschele I, Ernst C, Eggen A, Murkve B, Pfister-Genskow M, Witte LA, Grignola FE, Uimari P et al (1998) Mapping quantitative trait loci for milk production and health of dairy cattle in a large outbred pedigree. Genetics 149(4):1959–1973
Zhang W, Qu L, Xu G, Lian L, Zheng J, Yang N (2012) Hypomethylation upregulates the expression of CD30 in lymphoma induced by Marek’s disease virus. Poult Sci 91(7):1610–1618
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was supported by a grant from the National Natural Science Foundations of China (31201772), Chinese Universities Scientific Fund (2014XJ003) and the Program for Changjiang Scholars and Innovative Research Team in University (IRT_15R62).
Conflict of interest
The authors declare that they have no conflict of interests in this work.
Ethics approval
The whole procedure for collection of the tissue samples of all animals was carried out in strict accordance with the protocol approved by the Animal Welfare Committee of China Agricultural University (Permit number: DK996).
Author contributions
XL and LJ carried out the experiment and drafted the manuscript, LJ and QZ participated in its design and coordination, and provided editorial suggestions and revisions. JY contributed materials. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Liu, X., Yang, J., Zhang, Q. et al. Regulation of DNA methylation on EEF1D and RPL8 expression in cattle. Genetica 145, 387–395 (2017). https://doi.org/10.1007/s10709-017-9974-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-017-9974-x