
Abstract
The increasingly cost-efficient availability of ‘omics’ data has led to the development of a rich framework for predicting the performance of non-phenotyped selection candidates in recent years. The improvement of phenotypic analyses by using pedigree and/or genomic relationship data has however received much less attention, albeit it has shown large potential for increasing the efficiency of early generation yield trials in some breeding programs. The aim of this study was accordingly to assess the possibility to enhance phenotypic analyses of multi-location field trials with complete relationship information as well as when merely incomplete pedigree and/or genomic relationship information is available for a set of selection candidates. For his purpose, four winter bread wheat trial series conducted in Eastern and Western Europe were used to determine the experimental efficiency and accuracy of different resource allocations with a varying degree of relationship information. The results showed that modelling relationship between the selection candidates in the analyses of multi-location trial series was up to 20% more efficient than employing routine analyses, where genotypes are assumed to be unrelated. The observed decrease in efficiency and accuracy when reducing the testing capacities was furthermore less pronounced when modelling relationship information, even in cases when merely partial pedigree and/or genomic information was available for the phenotypic analyses. Exploiting complete and incomplete relationship information in both preliminary yield trials and multi-location trial series has thus large potential to optimize resource allocations and increase the selection gain in programs that make use of various predictive breeding methods.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The increasing availability of cost-efficient ‘omics’ data has led to the development of a rich framework for predicting genotype performance in recent years (Robertsen et al. 2019; Montesinos-López et al. 2021; Sneller et al. 2021; Bayer et al. 2021). The usage of genome-wide distributed markers for a so-called genomic selection has thereby gained an especially large popularity in many breeding programs (Belamkar et al. 2018; Juliana et al. 2019; Haikka et al. 2020; Raffo et al. 2022). A major goal in the genomic selection framework is given by obtaining as accurate as possible predictions of non-phenotyped selection candidates in early generations for traits like grain yield (Tsai et al. 2020; Borrenpohl et al. 2020), disease resistance (Beukert et al. 2020; Moreno-Amores et al. 2020) as well as costly and laborious to phenotype quality traits (Schmidt et al. 2016; Lado et al. 2018). The improvement of phenotypic analyses by using pedigree and/or genomic relationship data has on the other hand received much less attention (Endelman et al. 2014; Terraillon et al. 2022), albeit it has shown large potential for increasing the efficiency of early generation observation and preliminary yield trials in some breeding programs (Michel et al. 2019; Tsai et al. 2020; Borrenpohl et al. 2020). These studies generally assumed that a particular source of relationship information is fully covering the entire set of selection candidates, which is however not always the case in practice, for example when some of the tested lines were developed by another breeding program. The aim of this study was accordingly to take a step towards generalizing these previous results obtained for preliminary yield trials, and assess the possibility of enhancing phenotypic analyses of multi-location field trials when merely incomplete pedigree and/or genomic relationship information is available for a set of selection candidates.
Materials and methods
Plant material and genotypic data
Four panels of 147–177 recombinant inbred and double haploid breeding lines developed in the winter bread wheat breeding program of Saatzucht Donau GesmbH & CoKG in Austria were analysed in this study. Each panel was phenotyped for grain yield in a different trial series each with four locations in Western Europe in 2015 (151 lines) and 2016 (150 lines) as well as in Eastern Europe in 2015 (177 lines) and 2016 (147 lines). Environmental means were available for each of the locations within the respective trial series (year-by-region combinations), so each line occurred with four replicates (total number of observations) within each of the trial series. The breeding lines were part of 277 different families with a size of 1–13 lines per family and a genealogy of 338 ancestors tracing back up to 8 generations. All lines were genotyped with the DArT genotyping-by-sequencing approach (Diversity Arrays Technology Pty Ltd 2020), and markers with more than 10% missing data and a minor allele frequency smaller than 5% were filtered out. Only one marker of identical marker pairs was furthermore retained for all subsequent analyses. A chromosome-wise imputation of missing data points with the missForest algorithm (Stekhoven and Bühlmann 2012) and after quality filtering the final marker dataset contained 1908 markers, which were used for investigating the population structure (Suppl. Fig. S1).
Phenotypic analysis
An across-trial analysis was conducted for each of the four trial series individually by using a linear mixed model of the form:
where \({\text{y}}_{{{\text{jk}}}}\) are the observations for grain yield, \({\upmu }\) is the grand mean, and \({\text{l}}_{{\text{k}}}\) is the effect of the kth location that was modelled as random. The effect of the jth line \({\text{g}}_{{\text{j}}}\) was modeled as random with \({\mathbf{g}} \sim {\text{N}}\left( {{\mathbf{0}}, {\mathbf{I}}{\upsigma }_{{\text{g}}}^{2} } \right)\) to obtain an estimate of the genetic variance and best linear unbiased predictions (BLUP) of the lines’ performances. The effect \({\text{e}}_{{{\text{jk}}}}\) that incorporated both the line-by-trial interaction variance and the residual effect was assumed to be random following a normal distribution with \({\mathbf{e}}{ }\sim {\text{ N}}\left( {{\mathbf{0}},{ }{\mathbf{I}}{\upsigma }_{{\text{e}}}^{2} } \right)\). The entry-mean heritability was subsequently estimated following the suggestion by Cullis et al. (2006):
where \( {\upsigma }_{{\text{g}}}^{2}\) is the genetic variance and \({ }\overline{{{\text{VD}}}}\) the mean variance of a difference of two genotypic BLUPs. All phenotypic analyses were conducted with the package sommer (Covarrubias-Pazaran 2016) for the R statistical environment (R Core Team 2022).
Empirical assessment of the experimental efficiency
Sets of 70 lines, each coming from a different family, were 50 times randomly sampled from each of the four investigated trial series individually. This resulted in 50 unique and different sets per trial series (year-by-region combination) and a total of 200 sets across all trial series. These sets were subsequently analysed separately for assessing the efficiency of several experimental layouts without including relationship information as well as with including complete and incomplete relationship information into the analyses of the phenotypic data. The experimental designs comprised a fully orthogonal testing of the lines across all four locations of a given trials series as well as a reduction of the testing capacities to three or two locations. Furthermore, the merit of allocating lines to the locations according to an incomplete block design was tested by reducing the number of total observations per line from four to two.
The percentage of genotyped lines within these sets was subsequently varied between 0 and 100%, whereas the number of lines with pedigree information was varied between 0, 30, 50, 80, and 100%. The corresponding proportions of lines were thereby sampled randomly, while the sampling of pedigreed and genotyped lines was additionally independent from each other. The lines were in this way allocated to groups for which both genomic and pedigree relationship information was available, only genomic or pedigree relationship information was available, and one group without relationship information (Fig. 1). Model [1] was again used to obtain the mean variance of a difference \(\overline{{{\text{VD}}}}\) of two genotypic BLUPs for each of these scenarios, where the effect of the jth line \({\text{g}}_{{\text{j}}}\) followed in this case \({\mathbf{g}}{ }\sim {\text{ N}}\left( {{\mathbf{0}},{ }{\mathbf{H}}{\upsigma }_{{\text{g}}}^{2} } \right)\), where \({\mathbf{H}}\) was computed as:

The percentage of lines for which no relationship information was available (1st row), only pedigree (2nd row) or genomic relationship information (3rd row) was available, and the percentage of lines for which both pedigree and genomic relationship information was available (4th row) with different proportions of pedigreed and genotyped lines in the randomly sampled subsets
with the genomic relationship matrix \({\mathbf{G}}_{{{\text{adj}}}}\) and the pedigree relationship matrix \({\mathbf{A}}\). The matrix \({\mathbf{A}}_{11}\) contained the pedigree relationship between non-genotyped lines, \({\mathbf{A}}_{22}\) the pedigree relationship between genotyped lines, while \({\mathbf{A}}_{12}\) and \({\mathbf{A}}_{21}\) modelled the pedigree relationship between genotyped and non-genotyped lines. All lines were assumed to have undergone seven cycles of selfing for the computation of \({\mathbf{A}}\), while non-pedigreed lines i.e., lines that were part of above-mentioned group without pedigree information were likewise included into the pedigree relationship matrix but possessed a covariance of zero with all other lines in \({\mathbf{A}}\). The genomic relationship matrix \({\mathbf{G}}\) was computed following Endelman and Jannink (2012):
where \({\mathbf{W}}\) is a centered marker matrix of the j lines with \({\text{W}}_{{{\text{jl}}}} = {\text{ Z}}_{{{\text{jm}}}} + 1 - 2{\text{p}}_{{\text{m}}}\) and \({\text{m}}\) being the allele frequency at the mth marker locus. The genomic relationship matrix \({\mathbf{G}}\) was moreover adjusted by solving:
and setting \({\mathbf{G}}_{{{\text{adj}}}} = {\text{a}} + {\text{b}}{\mathbf{G}}\) as suggested by Christensen et al. (2012) before computing \({\mathbf{H}}\), in order to account for the impact of genetic trends across multiple generations and the reduction in genetic variance from the base population to the population of genotyped lines. It should be noticed that in the case all lines possess genotypic information \({\mathbf{H}}\) reduces to \({\mathbf{G}}\), while in the case no genotypic information is available \({\mathbf{H}}\) reduces to \({\mathbf{A}}\), and given no relationship is available \({\mathbf{H}}\) reduces to the identity matrix \({\mathbf{I}}\).
The efficiency modelling complete and incomplete relationship information in the evaluated experimental designs for each sampled set of lines in a given trial series was determined by using the mean variance of a difference analogous to Piepho et al. (2006):
where \(\overline{{{\text{VD}}}}_{{{\text{REF}}}}\) is the mean variance of a difference (squared standard error of a difference) of all pairwise comparisons among the genotypic BLUPs obtained from the analysis of a set of lines that was completely orthogonal tested in all four locations in a given trial series without including any relationship information i.e., \({\mathbf{g}}{ }\sim {\text{ N}}\left( {{\mathbf{0}},{ }{\mathbf{I}}{\upsigma }_{{\text{g}}}^{2} } \right)\). This reference value was compared with \(\overline{{{\text{VD}}}}_{{{\text{HBLUP}}}}\), which is the mean variance of a difference of all pairwise comparisons among the genotypic BLUPs obtained from the multi-location analysis of the different experimental designs that included complete or incomplete pedigree and genomic relationship information as described above.
Simulation layout for assessing the prediction accuracy
Analogous to the empirical study different sets of 70 lines, each coming from a different family, were 50 times randomly sampled each of the four investigated trial series individually in order to assess the prediction accuracy i.e., the correlation of the true genotypic value with the predicted genotypic value in a simulation study. True genotypic values of each line were derived by randomly sampling \({\text{N}}_{{{\text{QTL}}}} = 150\) marker loci as causal variants of a quantitative inherited trait, for which a vector of effects \({{\varvec{\upalpha}}}\) was randomly sampled from a normal distribution with \({{\varvec{\upalpha}}}\sim {\text{N}}\left( {0,1} \right)\):
where \({{\varvec{\upalpha}}}{ }\) is the vector of effects of the causal loci, \({\mathbf{Q}}\) is the marker matrix of the investigated set of lines, and \({\mathbf{L}}_{{{\mathbf{TGV}}}}\) is the vector of their true genotypic values. The observed genotypic values were accordingly computed as:
where the vector of error effects \({\mathbf{e}}\) was randomly sampled from a normal distribution with zero mean and a variance equal to
where \({\upsigma }_{{{\text{TGV}}}}^{2} { }\) is the variance of the true genotypic values, and \({\text{h}}^{2}\) an aspired repeatability of \({\text{h}}^{2} = 0.10\), \({\text{h}}^{2} = 0.30\) and \({\text{h}}^{2} = 0.50\) for a given location. Like in the empirical study, 2–4 locations with completely orthogonal testing or an allocation according to an incomplete block design were simulated and used assess the prediction accuracy. The prediction accuracy was in this case measured as \({\text{r}}\left( {{\mathbf{L}}_{{{\mathbf{PGV}}}} ,{\mathbf{L}}_{{{\mathbf{TGV}}}} } \right)\), where \({\mathbf{L}}_{{{\mathbf{PGV}}}}\) is the predicted line performance when analysing the data with linear mixed models following Eq. (1) including the relationship matrix \({\mathbf{H}}\) described in Eq. (3). The same proportions of complete and incomplete relationship information described above for the empirical study were tested in the simulations. The pedigree relationship matrix \({\mathbf{A}}\) was like in the empirical study based on the original pedigree records, while the genomic relationship matrix \({\mathbf{G}}\) was constructed with random samples of \({\text{N}}_{{{\text{SNP}}}} = 1500\) markers that served as linked loci to the causal variants.
The genomic relationship matrix \({\mathbf{G}}\) was computed with sommer (Covarrubias-Pazaran 2016), the pedigree relationship matrix \({\mathbf{A}}\) was obtain with the package pedigreeTools (Vazquez et al. 2018), the combined relationship matrix \({\mathbf{H}}\) was derived with the package AGHmatrix (Amadeu et al. 2016), and the incomplete bock designs were randomized with the package crossdes (Sailer 2022) for the R statistical environment (R Core Team 2022). All models for assessing the experimental efficiency and prediction accuracy were fitted with the R package sommer (Covarrubias-Pazaran 2016). An example dataset and accompanied R Code are available as supplemental material to illustrate the utilized models.
Results
The different trial series conducted in Eastern and Western Europe in 2015–2016 showed a substantial genotype-by-environment interaction exemplified by an average correlation of r = 0.18–0.27 between the series-specific locations. Nevertheless, a broad genetic variation was observed in each trial series for which the estimated entry-mean heritability varied between h2 = 0.45 and h2 = 0.58, which suggested that the dataset at hand was suitable for investigating the experimental efficiency with complete and incomplete relationship information as well as varying resource allocations (Table 1).
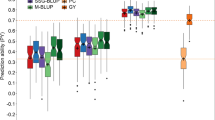
The empirical assessment of the experimental efficiency with different sets of randomly sampled lines revealed that a reduction in the number of test locations resulted on average in a 10–20% loss in efficiency in comparison to a completely orthogonal testing in four locations (Fig. 2). This reduction in efficiency was however much less pronounced if pedigree and/or genomic relationship was integrated into the phenotypic analysis of the data. Modelling the relationship between the lines by \({\mathbf{H}} = {\mathbf{G}}\), i.e., \({\mathbf{g}}{ }\sim {\text{ N}}\left( {{\mathbf{0}},{ }{\mathbf{G}}{\upsigma }_{{\text{g}}}^{2} } \right)\) and \({\mathbf{g}}{ }\sim {\text{ N}}\left( {0,{ }{\mathbf{H}}{\upsigma }_{{\text{g}}}^{2} } \right)\), was in fact up to 20% more efficient than modelling lines independent in a routine analysis with \({\mathbf{I}}\), i.e., \({\mathbf{g}}{ }\sim {\text{ N}}\left( {{\mathbf{0}},{ }{\mathbf{I}}{\upsigma }_{{\text{g}}}^{2} } \right)\), in the investigated resource allocations. A similar observation was made for cases in which merely incomplete genomic and/or pedigree relationship was available, where an increase in the availability of relationship information steadily increased the experimental efficiency. Interestingly, decreasing the testing capacities by up to one quarter appeared to be feasible in all investigated trial series without losing any efficiency in the case that most of the lines possessed genotypic data (Suppl. Figs. S2–S5).

Average efficiency in the empirical study across all four investigated trial series, expressed relatively to a completely orthogonal testing in all four locations in these trial series without including any relationship information. The investigated experimental designs included a fully orthogonal testing of the lines across two to four locations as well as allocating lines to the locations according to an incomplete block design by reducing the number of total observations per line in a given trial series from four to two. The percentage of genotyped lines was additionally varied between 0 and 100%, while the number of lines with pedigree information was varied between 0, 30, 50, 80, and 100%
The simulations showed furthermore that modelling relationship can result in a higher prediction accuracy (Fig. 3), which was in this study defined as the correlation between the true genotypic value with the predicted genotypic value obtained in the phenotypic analyses of the data. The results of the simulations generally followed the same pattern that has been observed for the experimental efficiency in the empirical investigations, with some advantage of genomic over pedigree relationship information. A gradual increase in prediction accuracy was accordingly observed when modelling pedigree and/or genomic relationship between the lines in comparison to a baseline analysis that assumed independence between the lines. The advantage of modelling relationship information in comparison to this baseline model diminished however with an increase in the repeatability of the individual trials at each location, as did the superiority of genomic over pedigree relationship information (Suppl. Figs. S6–S7).

Average prediction accuracy in the simulation study with a repeatability of h2 = 0.10 at each trial location. The investigated experimental designs included a fully orthogonal testing of the lines across two to four locations as well as allocating lines to the locations according to an incomplete block design by reducing the number of total observations per line in a given trial series from four to two. The percentage of genotyped lines was additionally varied between 0 and 100%, while the number of lines with pedigree information was varied between 0, 30, 50, 80, and 100%. The horizontal black lines correspond to the prediction accuracy of a completely orthogonal testing in all four locations of these trial series without including any relationship information
Discussion
Modelling genetic relationship as an additional source of information in phenotypic analyses has shown promising results for determining the performance of genotypes both in simulation (Bauer et al. 2006; Möhring et al. 2014; Selle et al. 2019; Terraillon et al. 2022) and empirical studies (Moreau et al. 1999; Oakey et al. 2007; Endelman et al. 2014). Although the estimated experimental efficiency and accuracy was highest with complete genomic relationship information in the study at hand, a marked advantage was likewise observed when utilizing pedigree records for modelling relationship. The latter resulted in lower accuracies in comparison to a genomic prediction of non-phenotyped individuals (Auinger et al. 2016; Cericola et al. 2017) as the Mendelian sampling term i.e., segregation within families cannot be addressed in such a case, but pedigree best linear unbiased predictions can readily distinguish between family members if phenotypic observations are already available for them (Michel et al. 2020).
This issue renders pedigree records a relatively cost-efficient alternative to genomic data, depending on the strategy of a program that employs predictive breeding methods for an array of various target traits. Nevertheless, this assumes generally an ideal case where a particular source of relationship information is fully available for the entire set of selection candidates. However, pedigree and genomic data might not be available for every breeding line, especially when it was developed by another breeding program and is tested together with ‘in-house’ developed material in the framework of the breeders’ exemption and bilateral germplasm exchange. Hence, some lines have to be assumed independent in the phenotypic analysis since no relationship information is available for them, even though it might be desirable to integrate such information into the analysis. Employing the single-step framework developed in animal breeding for combining different relationship matrices into a common matrix \({\mathbf{H}}\) (Legarra et al. 2009; Christensen and Lund 2010) enabled to commonly rank lines with and without relationship information as well as simultaneously improving the ranking between lines with different types i.e., pedigree and/or genomic relationship information in study at hand. This step towards generalization of modelling relationships in phenotypic analyses led to a considerable increase both in the experimental efficiency and accuracy. The simulations suggested the largest benefit for traits with a low to medium heritability like grain yield or protein yield in winter bread wheat, whereas this advantage appeared to be rather marginal for traits with a high heritability like plant height or flowering date. This observation was in line with previous reports, which stated that the relative advantage of modelling relationship information in the phenotypic analysis depends on the heritability as well as the testing intensity of field trials (Bauer et al. 2006; Endelman et al. 2014; Terraillon et al. 2022).
The results of the empirical and simulation study showed moreover that a reduction in testing capacities appears to be feasible in multi-location trials when the pedigree and/or genomic data are integrated into the phenotypic analysis. Although resource allocations that follow an incomplete block (Montesinos‐Lopez et al. 2022) or augmented design (Lell et al. 2021) have shown promise in the genomic prediction framework, the logistically easiest option for reducing the testing capacities is given by reducing the number of test locations. The feasibility of this option might however be strongly dependant on the target population of environments of a breeding program. Reducing the number of test locations might for example be readily feasible for regional breeding program with a strong focus on local adaptation, whereas it is probably less suitable for a breeding program that targets many different environments. Resource allocations with sparse testing strategies might thus be more suitable for the latter (Jarquin et al. 2020; Atanda et al. 2022), while including relationship information both into the randomization of specific trial designs as well as the subsequent analyses has shown large merit to increase the experimental efficiency in general (Cullis et al. 2020).
Conclusions
The usage of complete or incomplete relationship information has the potential to render phenotypic analyses more efficient. Such an application has been primarily suggested for preliminary yield trials (Endelman et al. 2014) and applied in practical breeding programs (Michel et al. 2019; Tsai et al. 2020; Borrenpohl et al. 2020), but can also have some merit for multi-location trials in advanced generations. A strong increase of efficiency and accuracy was accordingly observed within the four investigated trials series. The usage of relationship information might furthermore benefit the analysis across multiple trial series especially if their connectivity by commonly tested genotypes is low. Hence, exploiting complete and incomplete relationship information in both preliminary yield trials and multi-location trial series has a large potential to further optimize resource allocations and increase the yearly selection gain in programs that make use of various predictive breeding methods.
Data availability
An example dataset and accompanied R Code are available as supplemental material.
References
Amadeu RR, Cellon C, Olmstead JW et al (2016) AGHmatrix: R package to construct relationship matrices for autotetraploid and diploid species: a blueberry example. Plant Genome 9:1–10. https://doi.org/10.3835/plantgenome2016.01.0009
Atanda SA, Govindan V, Singh R et al (2022) Sparse testing using genomic prediction improves selection for breeding targets in elite spring wheat. Theor Appl Genet. https://doi.org/10.1007/s00122-022-04085-0
Auinger H-J, Schönleben M, Lehermeier C et al (2016) Model training across multiple breeding cycles significantly improves genomic prediction accuracy in rye (Secale cereale L.). Theor Appl Genet 129:2043–2053. https://doi.org/10.1007/s00122-016-2756-5
Bauer AM, Reetz TC, Léon J (2006) Estimation of breeding values of inbred lines using best linear unbiased prediction (BLUP) and genetic similarities. Crop Sci 46:2685–2691. https://doi.org/10.2135/cropsci2006.01.0019
Bayer PE, Petereit J, Danilevicz MF et al (2021) The application of pangenomics and machine learning in genomic selection in plants. Plant Genome 14:1–9. https://doi.org/10.1002/tpg2.20112
Belamkar V, Guttieri MJ, Hussain W et al (2018) Genomic selection in preliminary yield trials in a winter wheat breeding program. G3 Genes Genomes Genet 8:2735–2747. https://doi.org/10.1534/g3.118.200415
Beukert U, Thorwarth P, Zhao Y et al (2020) Comparing the potential of marker-assisted selection and genomic prediction for improving rust resistance in hybrid wheat. Front Plant Sci 11:1–11. https://doi.org/10.3389/fpls.2020.594113
Borrenpohl D, Huang M, Olson E, Sneller C (2020) The value of early-stage phenotyping for wheat breeding in the age of genomic selection. Theor Appl Genet 133:2499–2520. https://doi.org/10.1007/s00122-020-03613-0
Cericola F, Jahoor A, Orabi J et al (2017) Optimizing training population size and genotyping strategy for genomic prediction using association study results and pedigree information. A case of study in advanced wheat breeding lines. PLoS ONE 12:e0169606. https://doi.org/10.1371/journal.pone.0169606
Christensen O, Lund MS (2010) Genomic relationship matrix when some animals are not genotyped. Genet Sel Evol 42:1–8. https://doi.org/10.1186/1297-9686-42-2
Christensen O, Madsen P, Nielsen B et al (2012) Single-step methods for genomic evaluation in pigs. Animal 6:1565–1571. https://doi.org/10.1017/S1751731112000742
Covarrubias-Pazaran G (2016) Genome-assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11:e0156744. https://doi.org/10.1371/journal.pone.0156744
Cullis BR, Smith AB, Coombes NE (2006) On the design of early generation variety trials with correlated data. J Agric Biol Environ Stat 11:381–393. https://doi.org/10.1198/108571106X154443
Cullis BR, Smith AB, Cocks NA, Butler DG (2020) The design of early-stage plant breeding trials using genetic relatedness. J Agric Biol Environ Stat 25:553–578. https://doi.org/10.1007/s13253-020-00403-5
Diversity Arrays Technology Pty Ltd. (2020) DArT P/L. https://www.diversityarrays.com. Accessed 17 Dec 2022
Endelman JB, Jannink J-L (2012) Shrinkage estimation of the realized relationship matrix. G3 Genes Genomes Genet 2:1405–1413. https://doi.org/10.1534/g3.112.004259
Endelman JB, Atlin GN, Beyene Y et al (2014) Optimal design of preliminary yield trials with genome-wide markers. Crop Sci 54:48–59. https://doi.org/10.2135/cropsci2013.03.0154
Haikka H, Knürr T, Manninen O et al (2020) Genomic prediction of grain yield in commercial Finnish oat (Avena sativa) and barley (Hordeum vulgare) breeding programmes. Plant Breed 139:550–561. https://doi.org/10.1111/pbr.12807
Jarquin D, Howard R, Crossa J et al (2020) Genomic prediction enhanced sparse testing for multi-environment trials. G3 Genes Genomes Genet 10:2725–2739. https://doi.org/10.1534/g3.120.401349
Juliana P, Poland J, Huerta-Espino J et al (2019) Improving grain yield, stress resilience and quality of bread wheat using large-scale genomics. Nat Genet 51:1530–1539. https://doi.org/10.1038/s41588-019-0496-6
Lado B, Vázquez D, Quincke M et al (2018) Resource allocation optimization with multi-trait genomic prediction for bread wheat (Triticum aestivum L.) baking quality. Theor Appl Genet 131:2719–2731. https://doi.org/10.1007/s00122-018-3186-3
Legarra A, Aguilar I, Misztal I (2009) A relationship matrix including full pedigree and genomic information. J Dairy Sci 92:4656–4663. https://doi.org/10.3168/jds.2009-2061
Lell M, Reif J, Zhao Y (2021) Optimizing the setup of multienvironmental hybrid wheat yield trials for boosting the selection capability. Plant Genome 14:1–13. https://doi.org/10.1002/tpg2.20150
Michel S, Löschenberger F, Ametz C et al (2019) Simultaneous selection for grain yield and protein content in genomics-assisted wheat breeding. Theor Appl Genet 132:1745–1760. https://doi.org/10.1007/s00122-019-03312-5
Michel S, Löschenberger F, Sparry E et al (2020) Multi-year dynamics of single-step genomic prediction in an applied wheat breeding program. Agronomy 10:1591. https://doi.org/10.3390/agronomy10101591
Möhring J, Williams ER, Piepho HP (2014) Efficiency of augmented p-rep designs in multi-environmental trials. Theor Appl Genet 127:1049–1060. https://doi.org/10.1007/s00122-014-2278-y
Montesinos-López OA, Montesinos-López A, Pérez-Rodríguez P et al (2021) A review of deep learning applications for genomic selection. BMC Genomics 22:1–23. https://doi.org/10.1186/s12864-020-07319-x
Montesinos-Lopez OA, Montesinos-Lopez A, Acosta R et al (2022) Using an incomplete block design to allocate lines to environments improves sparse genome-based prediction in plant breeding. Plant Genome. https://doi.org/10.1002/tpg2.20194
Moreau L, Monod H, Charcosset A, Gallais A (1999) Marker-assisted selection with spatial analysis of unreplicated field trials. Theor Appl Genet 98:234–242. https://doi.org/10.1007/s001220051063
Moreno-Amores J, Michel S, Löschenberger F, Buerstmayr H (2020) Dissecting the contribution of environmental influences, plant phenology, and disease resistance to improving genomic predictions for fusarium head blight resistance in wheat. Agronomy 10:2008. https://doi.org/10.3390/agronomy10122008
Oakey H, Verbyla AP, Cullis BR et al (2007) Joint modeling of additive and non-additive (genetic line) effects in single field trials. Theor Appl Genet 114:1319–1332. https://doi.org/10.1007/s00122-007-0515-3
Piepho HP, Büchse A, Truberg B (2006) On the use of multiple lattice designs and alpha-designs in plant breeding trials. Plant Breed 125:523–528. https://doi.org/10.1111/j.1439-0523.2006.01267.x
R Core Team (2022) R: a language and environment for statistical computing. R Foundation for Statistical Computing. https://www.r-project.org. Accessed 17 Dec 2022
Raffo MA, Sarup P, Guo X et al (2022) Improvement of genomic prediction in advanced wheat breeding lines by including additive-by-additive epistasis. Theor Appl Genet 135:965–978. https://doi.org/10.1007/s00122-021-04009-4
Robertsen C, Hjortshøj R, Janss L (2019) Genomic selection in cereal breeding. Agronomy 9:95. https://doi.org/10.3390/agronomy9020095
Sailer MO (2022) Crossdes: construction of crossover designs, R package v1.1-2. https://cran.r-project.org/package=crossdes. Accessed 17 Dec 2022
Schmidt M, Kollers S, Maasberg-Prelle A et al (2016) Prediction of malting quality traits in barley based on genome-wide marker data to assess the potential of genomic selection. Theor Appl Genet 129:203–213. https://doi.org/10.1007/s00122-015-2639-1
Selle ML, Steinsland I, Hickey JM, Gorjanc G (2019) Flexible modelling of spatial variation in agricultural field trials with the R package INLA. Theor Appl Genet 132:3277–3293. https://doi.org/10.1007/s00122-019-03424-y
Sneller C, Ignacio C, Ward B et al (2021) Using genomic selection to leverage resources among breeding programs: consortium-based breeding. Agronomy 11:1555. https://doi.org/10.3390/agronomy11081555
Stekhoven DJ, Bühlmann P (2012) Missforest-non-parametric missing value imputation for mixed-type data. Bioinformatics 28:112–118. https://doi.org/10.1093/bioinformatics/btr597
Terraillon J, Frisch M, Falke KC et al (2022) Genomic prediction can provide precise estimates of the genotypic value of barley lines evaluated in unreplicated trials. Front Plant Sci 13:1–10. https://doi.org/10.3389/fpls.2022.735256
Tsai H, Cericola F, Edriss V et al (2020) Use of multiple traits genomic prediction, genotype by environment interactions and spatial effect to improve prediction accuracy in yield data. PLoS ONE. https://doi.org/10.1371/journal.pone.0232665
Vazquez AI, Bates D, Siddharth A, Perez P (2018) pedigree tools: versatile functions for working with pedigrees. https://github.com/Rpedigree/pedigreeTools. Accessed 17 Dec 2022
Acknowledgements
We like to thank Maria Bürstmayr and her team for the tremendous work when extracting the DNA of several hundred wheat lines each year as well as Barbara Steiner for many fruitful discussions when conducting this study. We finally like to thank the anonymous reviewers for their comments and suggestions for improving the manuscript.
Funding
Open access funding provided by University of Natural Resources and Life Sciences Vienna (BOKU). This research was funded by the EU Eurostars project “E! 8959 Genomic selection for nitrogen use efficiency in wheat” and the “Frontrunner” FFG project TRIBIO (35412407). Open access funding was provided by the BOKU Vienna Open Access Publishing Fund.
Author information
Authors and Affiliations
Contributions
SM wrote the manuscript and conducted the empirical and simulation studies. CA supported in the statistical analysis. FL and HB initiated and guided through the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest. The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
The authors declare that the experiments comply with the current laws of Austria.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Michel, S., Löschenberger, F., Ametz, C. et al. Improving the efficiency of multi-location field trials with complete and incomplete relationship information. Euphytica 219, 10 (2023). https://doi.org/10.1007/s10681-022-03142-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-022-03142-5