
Abstract
Genotyping by sequencing (GBS) and single nucleotide polymorphism (SNP) chip technologies are the primary SNP genotyping technologies used today. However, these genotyping technologies have some drawbacks that limit their usefulness in analysis. We have developed a robust protocol called genotyping by multiplexed sequencing (GMS) using SNP markers, providing informative genotypic data with greater flexibility. The genotypes derived from direct sequence reads reduce ambiguity in genetic analysis. The advantages of this protocol include: (1) This PCR-based direct sequencing protocol generates information from markers of interest and provides a more streamlined and accurate analysis process, by multiplexing hundreds of informative markers into a single sequencing run. (2) The marker sets are easily customized to the species of interest and can readily be changed. In this study we have taken the GMS protocol developed in wheat and adapted it to barley. We have identified 577 SNP markers that work well using this protocol providing adequate genome coverage for genomic selection and tag 267 QTL’s for genes of interest. Good markers have an adequate read depth of at least 5 amplicons and are reliably present across the population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Barley ranks a distant 4th in grain production worldwide with a total of 140.6 metric tons annually. The United States produced approximately 169.6 million bushels of barley at an estimated price of 761.2 million dollars in 2019 (USDA NASS). Many barley traits contribute to malting quality, as well as food and feed quality. Here we describe a more efficient and adaptable protocol for tracking important barley traits for marker-assisted selection (MAS). Marker-assisted selection provides opportunities for enhancing the response from selection because molecular markers can be applied in early generations, with high precision and reductions in cost (Miedaner and Korzun 2012). Marker assisted selection (MAS) has been an effective tool for ensuring the transfer of quantitative trait loci (QTL) with high effects into elite barley lines. Genotyping by Multiplexed Sequencing (GMS) is a highly adaptable and effective genotyping tool for tracking markers of interest with high accuracy, which can translate to a more efficient and affordable MAS process in future breeding projects. Multiplexing on a small scale has been demonstrated by (Tamang et al. 2019; Poudel et al. 2018; Richards et al. 2016). The primer pools described therein have low marker complexity with approximately 12 markers per pool. The protocol presented here provides an avenue to multiplex more than 300 markers in a single pool.
Many marker systems have been developed over the years in order to detect genetic differences among DNA samples and SNPs have become the most useful in high throughput next generation sequencing (NGS). SNPs are the most commonly used marker in genetic studies because they represent the greatest source of genetic variability in eukaryotic organisms (Agarwal, Shrivastava and Padh 2008; Brookes 1999; Bybee et al. 2011; Campbell et al. 2015; Liu 2015; Onda et al. 2018; Voss-Fels and Snowdon 2014; Ruff et al. 2020). SNPs occur throughout the genome in both genic and non-genic regions, occurring more often in non-genic regions with no phenotypic correlation (Agarwal et al. 2008). SNPs found in genic regions are invaluable to breeders and researchers alike, because they may directly impact an organism’s phenotype (Allen, et al. 2011; Chan 2009) (Ruff et al. 2020). SNPs are generally bi-allelic and have a low mutation rate making them ideal for mapping, genomic studies, and MAS (Syvanen 2001; Batley and Edwards 2007; He et al. 2009; Liu et al. 2014; Poland et al. 2012; Zhao et al. 2011; Bernardo et al. 2015). This protocol includes 267 SNP markers with known QTL associations (Table 2).
GBS and SNP chip technologies are currently the most popular protocols for SNP genotyping to date. However, both technologies have their problems. GBS results can be arduous to analyze if the whole genome is not known, because it produces random DNA sequences within the genome. The random selection and amplification of DNA sequences through the GBS process results in missing data between samples. Therefore, imputation is required, reducing the accuracy of GBS. Currently the 50 k Illumina Infinium iSelect chip is the primary chip used for genotyping in barley; however, a 9 k chip preceded the 50 k chip and was incorporated into the 50 k chip during its development (Bayer et al. 2017). The majority of SNP’s on the 50 k iSelect chip have been derived from variants called in exome capture data of a wide range of European barley germplasm (Bayer et al. 2017). SNP chip technology is based on a hybridization technique which results in a dot plot that may misrepresent true allelic states, particularly at heterozygous loci. Also, once the SNP chip is developed researchers cannot easily add new markers, making adaptation and customization impossible. This is not the case for the GMS protocol which makes adding and customizing the marker pool less difficult. In barley, the GMS protocol produces greater genetic accuracy in the event of a false heterozygote call at a locus which has a paralogue in the genome. The GMS protocol is designed to be used in genomic selection and sequencing may be sequenced on either the Illumina or Ion Torrent NGS platforms (Ruff et al. 2020).
Materials and methods
Barley markers for this project were selected due to informative nature and chromosome coverage for use in genomic selection. They were derived from markers developed with the 50 k barley SNP chip (Bayer et al. 2017). To ensure performance when multiplexing, five hundred markers were selected for each of three pools for a total of 1500 barley markers. Primers were then designed from sequence data using Sequenom MassArray Design 4.0 (Sequenom, San Diego, CA.), which reduces the chance of primer dimers and standardizes the annealing temperature. These three marker pools were then multiplexed on 25 lines of the Oregon Wolfe Barley (OWB) bi-parental population. The OWB population was developed by Dr. Bob Wolfe by systematically crossing recessive alleles into one parent and dominant alleles into the other. The population consists of 175 barley doubled haploid progeny (Costa et al. 2001). Figure 1 provides an overview of the PCR based, direct sequence read GMS protocol. Due to duplicate markers with different but synonymous names the total resulting markers in all pools were reduced to 830 unique markers. To ensure data quality duplicate samples were genotyped to confirm output calls. The majority of cleaning is completed in the analysis pipeline which includes throwing out calls with inadequate read depth below 5 amplicons per sample. The protocol used here has been adapted to barley from the GMS protocol used in wheat and published earlier in 2020 (Ruff et al. 2020).

Summary of the GMS process
PCR 1
The first PCR produces the target amplicons using a 10 µl reaction. Locus-specific forward primers have an M13 tail attached to the 5′ end. Reagents include 2.35 µl H2O, 1 µl 10 × MCLAB Taq PCR Buffer with 20 mM MgCl2 (MCLAB, San Francisco, CA.), 0.45 µl MgCl2 at 1.125 mM, 1 µl dNTP’s at 100 mM, 1 µl primer pool at 125 nM, 4 µl sample DNA at 6.6 ng/µl, and 0.2 µl HOTaq DNA Polymerase (MCLAB, San Francisco, CA.) at 5 unit/µl. Thermocycler conditions were: 10 min at 94 °C for initial denaturation, then cycle 35 times at 94 °C denaturation for 20 sec, 56 °C annealing for 2 min, and 68 °C extension for 30 sec; followed by final 3 min elongation at 72 °C.
PCR 2
PCR 1 amplicons are diluted 1:1 and used as template DNA in the second PCR. PCR 2 adds a unique barcode and standard Ion A Adapter to the M13 tail of the forward primer, which then becomes the forward primer in successive amplicons. Barcodes are used for sample identification and the standard Ion A Adapter is used for attachment to the Ion Proton chip for sequencing. Reagents of PCR 2 include 0.875 µl H2O, 0.5 µl 10 × MCLAB Taq PCR Buffer with 20 mM MgCl2 (MCLAB, San Francisco, CA.), 0.2 µl MgCl2 at 25 nM, 0.025 µl dNTP’s at 100 mM, 0.2 µl P1B Primers at 10 µM, 2 µl unique barcoded adapters at 2 µM, 2 µl diluted PCR #1 product, and 0.2 µl HOTaq DNA Polymerase (MCLAB, San Francisco, CA.) at 5 unit/ µl. Thermocycler conditions: 94 °C 10 min for initial denaturation, then cycle 15 times at 94 °C denaturation for 20 sec, 60 °C annealing for 30 s, and 72 °C extension for 1 min, followed by final 3 min elongation at 72 °C.
PCR clean up and amplicon quantification
After PCR 2 approximately 6 µl are taken from each well of the plate and combined into a single 1.5 mL Eppendorf tube. The final combined PCR product is purified using the QIAquick PCR Purification Kit (Qiagen Hilden Germany). The PCR product is further purified using Agencourt AMPure XP beads (BeckmanCoulter, Indianapolis, IN) using 0.8 µl of beads per 1 µl of PCR product. Then size selection is done with 4% E-Gel SizeSelect Gel (Life Technologies, Carlsbad, CA), selecting for products between 144 and 233 bp. further gel purification is performed with a QIAquick Gel Extraction Kit (Qiagen) resulting in an elution at 30 µl EB. The final size selection is accomplished on a 2% E-Gel Size Select Gel (Life Technologies, Carlsbad, CA) targeting products between 140 and 250 bp and followed by a final purification with the QIAquick PCR Purification Kit. The purified and size-selected product was quantified using Qubit dsDNA HS assay kit (Life Technologies, Carlsbad, CA), which provided an initial DNA concentration. The final quantification is performed with the Agilent DNA 7500 Kit (Agilent, Santa Clara, CA), which allows for the final dilution to a minimum concentration of 80 pmol/L and have amplicon sizes between 185 and 260 bp. The GMS product is spiked with a compatible and diverse NGS library for sequence complexity. The GMS library must then be loaded onto the Ion Proton chip for sequencing. GBS libraries are loaded onto the Ion Proton chip using the Ion Chef (Life Technologies, Carlsbad, CA) with the Ion PI™ Hi-Q Sequencing 200 and the Ion PI™ Chip Kit v3. The loaded chips are then sequenced on an Ion Torrent Proton NGS platform (Life Technologies, Carlsbad, CA).
Sequence analysis pipeline
Individual OWB lines were de-multiplexed from raw sequence data using the unique barcode attached in PCR 2. Then raw sequence data from each OWB line are analyzed with a custom Python script. The first step uses cd-hit-est which collapses the raw sequence files into approximately similar sequence reads (Li and Godzik 2006). Each one of these clusters of unique sequence reads are considered a bin. The bins are then aligned to target sequence keyfiles with Muscle (Edgar 2004) at 100% homology excluding the SNP of interest. This output is organized in a FASTA file which notes the number of reads per sample and percent match to the marker key-file using a custom Python script. These results can then be looked into individually for further analysis within each line and marker for genotyping clean up if genotyping ambiguity exists.
Results and discussion
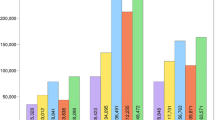
Duplicate samples of OWB DNA were analyzed for each of the 25 OWB samples to provide redundancy on ambiguous calls and provide confidence in analysis. During the development of this protocol it was discovered that adding sequence diversity resulted in higher quality sequence reads. To test the accuracy of this new genotyping protocol in barley, our results were compared to the published 50 k Illumina iSelect genotyping array results of the OWB population. The results matched 99.998% at each locus across all 25 OWB lines sequenced with the GMS protocol. Out of the 4,824 data points only one was called differently than the published data, excluding missing calls. A sample of 201 barley markers were selected for comparison which had the greatest polymorphic information content (PIC) and least missing data. The average missing data for each marker across all 25 OWB lines was 5%, with the largest being 12%. Of the 577 barley markers; 77 map to chromosome 1H, 85 to 2H, 67 to 3H, 66 to 4H, 143 to 5H, 48 to 6H, and 91 to 7H (Fig. 1). Marker chromosome and map location was derived from the BOPA consensus map (Close et al. 2009). Marker distribution across the genome has an average cM distance less than 3.03 per chromosome with an average of 82 markers per chromosome (Fig. 2)(Table 1). Polymorphic marker content was 47% on our panel of 25 OWB lines. The markers used in this study are not all informative, as many markers were selected to maximize genome coverage for use in genomic selection.

Heat map of marker coverage across all 7 barley chromosomes
The first two pools of 500 markers were selected as markers of interest and markers with a known function. The third pool was selected to optimize chromosome coverage and fill in chromosomal gaps. This resulted in a total of three barley marker pools for genotyping. Chromosomal coverage is fair with few gaps larger than 10 cM (Fig. 2). The average map distance across all chromosomes is 3.03 cM, so any QTL would theoretically be in linkage disequilibrium with at least one marker. Average linkage disequilibrium was observed to decay below a critical level (r2-value 0.2) within a map distance of 5–10 cM (Pasam et al. 2012). There is at least one example of cM distance greater than 10 on each chromosome, chromosome 6H has 4 chromosomal gaps larger than 10 cM.
Genetic maps for all 7 barley chromosomes were generated from the chromosomal position identified in the 50 k iSelect SNP Chip consensus map (Fig. 3). Of the 468 markers with a known function or associated QTL present in the three pools, 267 produced genotypic data (Table 2). Of the remaining 213 markers with known function that did not work only 32 amplified the loci, but did not produce results because they did not match the sequence keyfile used for genotyping. This failure was largely due to primer dimers. When comparing the most robust genetic variability from this protocol matches the OWB consensus genotype at 99.998% which provides strong evidence for the efficacy of this protocol. This protocol is robust and capable of being customized to any genome or marker of interest, which may be very useful in marker-assisted selection and association mapping. Due to these markers being selected from known markers in the 50 k iSelect chip, which were derived from a wide range of European barley germplasm, there could be ascertainment bias as winter and facultative barley are under-represented in the 50 k chip.

Genetic map of barley markers. Barley marker location was determined from the BOPA consensus map
References
Agarwal M, Shrivastava N, Padh H (2008) Advances in molecular marker techniques and their applicaitons in plant sciences. Plant Cell Rep 27(4):617–631
Allen A, Barker G, Berry S, Coghill J, Gwilliam R, Kirby S, . . . Edwards K (2011) Transcript-specific, single nucleotide polymorphism discovery and linkage analysis in hexaploid bread wheat (Triticum aestivum L.). Plant Biotech J, 9(9), 1086–1099
Batley J, Edwards D (2007) SNP applications in plants. Assoc Mapp Plants, 95–102
Bayer MM, Rapazote-Flores P, Ganal ME, Pete H, Macaulay M, Plieske J, . . . Waugh R (2017) Development and evaluation of a barley 50k iSelect SNP array. Front Plant Sci, 8, 1792. Retrieved from https://doi.org/10.3389/fpls.2017.01792
Bernardo A, Wang S, Amand P, Bai G (2015) Using next generation sequencing for multiplexed trait-linked markers in wheat.Plos One
Brookes AJ (1999) The essence of SNPs. Gene 234(2):177–186
Bybee M, Bracken-Grissom H, Haynes DB, Hermansen RA, Byers RL, Clement MJ, . . . Crandall KA (2011) Targeted amplicon sequencing (TAS): a scalable next-gen approach to multilocus, multitaxa phylogenics. Genome Bio Evol 3(0), 1312–1323
Campbell NR, Harmon SA, Narum SR (2015) Genotyping_in_Thousands by sequencing (GT-seq): a cost Effective SNP genotyping method based on custom amplicon sequencing. MolEcolResour 15(4):855–867
Chan E (2009) Single nucleotide polymorphisms: methods and protocols. Single Nucleotide Polymorph 2(578):95–111
Close T, Bhat P, Lonardi S, Wu Y, Rostoks N, Ramsay L, . . . Waugh R (2009) Development and implementation of high-throughput SNP genotyping in barley. BMC Genom 10(582)
Costa JM, Corey A, Hayes PM, Joblet C, Kleinhofs A, Kopisch-Obusch A, . . . Wolfe RI (2001) Molecular mapping of the Oregon Wolfe Barleys: a phenotypically polymorphic doubled-haploid population. Theor Appl Gen 103, 415-424
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797
He XY, He ZH, Morris CF, Xia XC (2009) Cloning and phylogenic analysis of polyphenol oxidase genes in common wheat and related species. Genetic ResCrop Evol 311–321
Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22(13):1658–1659
Liu, S. (2015). Genotyping by multiplexing amplicon sequencing. pp 10–13. Retrieved from http://schnablelab.plantgenomics.iastate.edu/docs/resources/protocols/pdf/GBMAS-20150706.pdf
Liu S, Yang X, Zhang D, Bai G, Chao S, Bockus W (2014) Genome-wide association analysis identified SNPs closely linked to a gene resistant to Soil-borne wheat mosaic virus. Theor Appl Genet 1039–1047
Miedaner T, Korzun V (2012) Marker-assisted selection for disease resistance in wheat and barley breeding. Am Phytopathological Soc, 560–566.
Onda Y, Takahagi K, Shimizu M, Inoue K, Mochida K (2018) Multiplex PCR targeted amplicon sequencing (MTA-Seq): simple, flexable, and versitile snp genotyping by highly multiplexed PCR amplicon sequencing. Frontiers Plant Sci
Pasam R, Sharma R, Malosetti M, Eeuwijk F, Haseneyer G, Kilian B, Graner A (2012) Genome-wide association studies for agronomical traits in a world wide spring barley collection. BMC Plant Bio
Poland J, Brown P, Sorrells M, Jannink J (2012) Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequenceing approach. Plos One
Poudel RS, Al-Hashel AF, Gross T, Gross P, Brueggeman R (2018) Pyramiding rpg4- and Rpg1-Mediated stem rust resistance in barley requires the Rrr1 gene for both to function. Frontiers Plant Sci
Richards J, Chao S, Friesen T, Brueggeman R (2016) Fine mapping of the barley chromosome 6H net form net blotch sussceptibility locus. Genes Genomes Gene
Ruff TM, Marston EJ, Eagle JD, Sthapit SR, Hooker MA, Skinner DZ, See DR (2020) Genotyping by multiplexed sequencing (GMS): a customizable platform for genomic selection. Plos One
Syvanen AC (2001) Accessing genetic variation: genotyping single nucleotide polymorphisms. Nat Rev Genet 390–942
Tamang P, Richards JK, Alhashal A, Poudel RS, Horsley RD, Friesen TL, Brueggeman RS (2019) Mapping of barley susceptibility/resistance QTL against spot form net blotch caused by Pyrenophora teres f. maculata using RIL populations. Theoretic Appl Genetics
Voss-Fels K, Snowdon RJ (2014) Understanding and utilizing crop genome diversity via high-resolution genotyping. Plant Biotechnol J 14(4):1086–1094
Zhao K, Tung C, Eizenga G, Wright M, Ali M, Price A, . . . McCouch S (2011) Genome-wide association mapping reveals a rich genetic archetechture of complex traits in Oryza sativa. Nat Commun 476
Acknowledgements
We would like to thank the USDA-ARS for their invaluable assistance and funding for this research project, as well as our colleagues Dr. Jamie Sherman and Dr. Pat Hayes for their assistance in selecting markers for the pools and refinement of the manuscript. Finally, I would like to thank Washington State University, the Department of Plant Pathology, and the ARCS fellowship program for their financial assistance, education, and professional development.
Author information
Authors and Affiliations
Contributions
Jonathan Eagle was the lead researcher overseeing the advancement of the research specifically pertaining to barley protocol development. Travis Ruff, USDA Biologial Science Technician overseeing a similar project in wheat and offering assistance and advice for protocol optimization and manuscript revision. Marcus Hooker assisted in the development and optimization of the analysis pipeline and manuscript revision. Sajal Sthapit, fellow graduate student who assisted in research and manuscript revision. Elliott Marston, fellow graduate student who assisted in research and manuscript revision. Karol Marlowe, USDA Biological Science Technician who assisted in sequencing, protocol optimization, and manuscript revision. Dolores Covarrubias, former WSU lab technician who assisted in sequencing and protocol optimization. Daniel Skinner assisted in early development of the analysis pipeline. Patrick Hayes, collaborator assisting in marker selection for traits of interest and manuscript revision. Jamie Sherman, fellow collaborator in marker selection for traits of interest and manuscript revision. Deven See, USDA ARS WRSGGL lab director providing funding, mentorship, and specialized knowledge for the development of the GMS protocol.
Ethics declarations
Conflict of interest
There are no relevant financial or non-financial competing interests to report.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Eagle, J., Ruff, T., Hooker, M. et al. Genotyping by Multiplexed Sequencing (GMS) protocol in Barley. Euphytica 217, 77 (2021). https://doi.org/10.1007/s10681-021-02811-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-021-02811-1