
Abstract
Testing for potato viruses is globally very important to prevent a critical shortage of potato supply. In most countries, testing is obligated by law. In Germany, seed potatoes are monitored for six viruses: PLRV, PVY, PVM, PVA, PVX and PVS. They can cause up to 90% loss of potato tubers in the field. Common methods currently used for testing are ELISA and conventional real-time PCR, but both are very time-consuming, and the former needs a high capacity of green houses and human resources, the latter elaborate RNA extraction steps. Recently, we proposed a new method called real-time DiRT-PCR which enables us to test for PLRV, PVY and PVS along with an internal control in three duplex real-time PCR reactions directly on diluted tuber sap. In this study, we describe the first TaqMan® assay for PVM published so far and embed it into a multiplex system to detect the remaining viruses. We are now able to sensitively test for the presence of six viruses in two multiplex reactions using the real-time DiRT-PCR without RNA purification.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Potato viruses can cause a major loss of yield and, thus, are an important problem for seed potato production. For seed certification, the following six viruses are considered as most problematic: Potato Leaf Roll Virus (PLRV) with 20-90% yield loss, Potato Virus Y (PVY) with 50-90% yield loss, Potato Virus S (PVS) with 5-75% yield loss, Potato Virus A (PVA) with up to 40% yield loss, Potato Virus X (PVX) with approximately 10% yield loss and Potato Virus M (PVM) with 20-50% yield loss at least in Germany (Bauch et al. 2012).
Potato plants can be infected by one or several viruses in two ways. Viruses are transmitted to host plant via aphids and then spread to the tubers (Radcliffe and Ragsdale 2002). Alternatively, infected mother plants cause secondary infections in their tuber progeny as virus particles are transported along with metabolites through the stolon into the tubers (Dupuis 2017; Malnoe et al. 1994). It is an important issue for seed potato growers all over the world as vegetative propagation favours secondary infections in the field.
Therefore, growers started potato field observations to find and eliminate virus infected plants very early. After 1959, virus examination for important viruses in potato culture became obligatory in German ministerial seed potato certification (Hepting 2002). In Bavaria, Double Antibody Sandwich-Enzyme Linked Immunosorbent Assay (DAS-ELISA) is currently the standard procedure for virus detection in the seed potato certification process (Gugerli and Gehriger 1980). However, the application of DAS-ELISA on dormant tubers is not reliable (Spiegel and Martin 1993) . Therefore, DAS-ELISA is performed on leaf extracts of four weeks cultivated tuber-eye-cuttings that is called growing-on DAS-ELISA. This tuber cultivation leads to virus enrichment for some virus species (Stammler et al. 2018) but is time-consuming and requires big seasonal greenhouse capacities. Therefore, an assay on dormant potato tubers in order to monitor the quality of seed-potato-lots is more rapid and does not require cultivation of plants in the greenhouse, especially for seed potatoes intended for export. There are already various potato virus diagnostic protocols published which are also reliable for virus detection on dormant tubers (RT-PCR: Singh et al. 2000; RT-qPCR: Hühnlein et al. 2016; Boonham et al. 2009; Agindotan et al. 2007; Mortimer-Jones et al. 2009; Hühnlein et al. 2013; Macroarray: Agindotan and Perry 2007; RT-IC-PCR: Ahouee et al. 2010; NASBA: Leone et al. 1997; LAMP: Ju 2011). One important drawback of all these conventional potato virus detection protocols (CoRT-qPCR) is the requirement of RNA purification for each sample. In Stammler et al. (2018) and Stammler (2020), we described an assay without RNA purification in order to save time and resources called direct reverse transcription quantitative PCR (DiRT-qPCR). This is possible by using a commercially available kit named KAPA3G™ Plant PCR kit (former KAPA Biosystems, USA; now Roche, Switzerland). The KAPA3G™ DNA polymerase was selected for successful amplification in the presence of PCR-inhibitors like polyphenols and polysaccharides. This kit was initially developed for fast amplification of genomic DNA (Schori et al. 2013). By adding a robust reverse transcriptase, we are able to amplify mRNA and viral RNA for diagnosis by using crude potato plant sap (Stammler et al. 2014, 2018; Stammler 2020). However, this method was used for qualitative and not for quantitative comparison. Therefore, the former term DiRT-qPCR (Stammler et al. 2018; Stammler 2020) is in our opinion misleading and, for that reason, we rename it in this paper to real-time DiRT-PCR. As shown by Stammler et al. (2018), the duplex DiRT-qPCR performs well in comparison to DAS-ELISA and a conventional RT-qPCR protocol (Mortimer-Jones et al. 2009) for TaqMan® primer sets to detect PVY (Singh et al. 2013), PVS (Mortimer-Jones et al. 2009) and PLRV (Agindotan et al. 2007) on single tuber heels. For reliability, the duplex detection of cox (cytochrome oxidase) was used as an internal control (Weller et al. 2000). However, three important viruses, PVA, PVX, and PVM, were not considered in the former investigations. One reason is that PVA and PVX are no longer observed frequently in Germany. Additionally, no PVM TaqMan® primer and probe were available in literature. However, in Stammler (2020) all conducted investigations on real-time DiRT-PCR detection of PVA, PVX and PVM are presented. The probably most interesting innovation, the development, selection and validation of the, to our knowledge, first PVM TaqMan® primer set (termed PVM_St15) is presented here to a broader scientific community and directly ties up with experiments published in Stammler et al. (2018) for real-time DiRT-PCR detection of PVY, PLRV and PVS.
In addition, testing for one virus per reaction is still very expensive and time consuming. Therefore, we also developed two real-time DiRT-PCR multiplex reactions, one for PLRV, PVY, PVM and cox and the other for PVS, PVA, PVX and cox on crude potato tuber sap.
Material and methods
Plants and tubers
PVM TaqMan® primer set development and validation
For PVM comparison experiments and PVM TaqMan® primer and probe selection, we used virus infected in vitro potato plants as test plants and/or controls. They were cultivated on MS medium (Murashige and Skoog 1962) supplied with 0.03 g/L chlormequat chloride (CCC720, Bayer CropScience, Monheim, Germany) at a temperature of 22 °C with a photoperiod of 16 h light at 50-70 μmol/m2 s. The PVM in vitro plants were infected with a PVM isolate (sequence not published) holding the internal number 4/007 that was provided by Dr. Petyr Dedič (Potato Research Institute Havlíčkův Brod, Department of Virology, Czech Republic).
In addition, 332 dormant field grown potato tubers of different varieties were sampled as test material for the PVM protocol validation experiment with the standard DAS-ELISA protocol of the Bavarian seed potato agency. The tuber material was harvested and processed in 2015. The virus infection probability of the potato tubers was empirically high owing to their origin.
Multiplex development and validation
For the multiplex development (MD) we used sap of tubers of virus free seedlings from different breeding programs cultivated in the glasshouse. We further used the PVY (number 2/045; 2/187 for PVY-O; 2/027; 2/110 for PVY-N; 2/193; 2/196; 2/201 for PVY-N-Wilga; 2/044; 2/063; 2/190; 2/191 for PVY-N-NTN), PLRV (number 1/045; 1/047), PVA (number 3/057; 3/027; 3/039), PVX (number 5/005; 5/021; 5/008) and PVS (number 6/270 for PVS-O) infected in vitro plants provided by Dr. Petyr Dedič (Potato Research Institute Havlíčkův Brod, Department of Virology, Czech Republic), cultivated as described above to extract virus RNA (PureLink®). Plants were infected with only one virus strain per plant. The amount of total RNA was measured with nanodrop 2000 and diluted to a starting concentration of 100 ng/μL with ultrapure H2O.
For the multiplex validation (MV), we processed different varieties of field grown potato tubers. Tubers were stored in cool temperatures from September on and used until May.
Protocols for different methods used in this study
Growing-on DAS-ELISA (virus-enrichment)
The tuber dormancy was broken by dipping one bud of the tuber rose-end into 0.0001% (w/v) gibberellic acid (GA3, Carl Roth, Karlsruhe, Germany) solution for 20 min. After air drying, the bud cuttings were cultivated in peat-sand substrate in the greenhouse under artificial light with a photoperiod of 16 h light at 22 °C. After 4-6 weeks cultivation, the third fully developed leaf was sampled for PVM detection using the DAS-ELISA assay.
150 μL of the centrifuged leaf sap were added to antibody coated microtiter plates. Antibodies provided by Bioreba AG (Reinach, Switzerland) were used in the DAS-ELISA assay that was performed according to the manufacturer’s instructions. Para-nitrophenol (pNP) was used as substrate to visualize the assay results by using a spectrophotometer (Power Wave HT, Bio Stack Ready, BioTek Instruments, Winooski, USA) at 405 nm absorbance (A405nm) with a molar extinction coefficient of 18,000 M−1 cm−1.
Ten-fold diluted plant sap stocks derived from virus-free in vitro potato plants were used as negative controls. Extraction buffer without any sample material was added to each DAS-ELISA assay as no template control (NTC).
Method comparison regarding sensitivity: within one biological repetition, three technical repeats have been performed for each ten-fold dilution. Virus-positive results have been accepted when A405nm was at or above the cut-off of 0.1 for at least two of the three technical repeats.
Validation by protocol comparison: one reaction per sample was evaluated. This was according to the standard procedure of the growing-on DAS-ELISA for seed potato certification of the Bavarian seed potato agency, LfL (Freising, Germany).
PVM TaqMan® primer set development and validation
For validation of the newly established PVM TaqMan® primer set, we used the same terms and conditions as in Stammler et al. (2018).
Sensitivity of methods (Method comparison): The sensitivity of three PVM detection methods was measured using dilution series in three biological repetitions. Thereby, DAS-ELISA, one-step duplex real-time CoRT-PCR (a modified protocol of Mortimer-Jones et al. 2009), and one-step duplex real-time DiRT-PCR have been compared. The used plant sap was obtained from in vitro plants for one-step duplex real-time DiRT-PCR and DAS-ELISA and the used RNA template for one-step duplex real-time CoRT-PCR was purified from the same plant sap.
Validation (Protocol comparison): By using field grown potato tubers, the one-step duplex real-time DiRT-PCR protocol on dormant potato tubers was compared with growing-on DAS-ELISA (on the leaves of 4–6-weeks cultivated tuber eye cuttings) with respect to their qualitative results by using field grown potato tubers.
For the development of the PVM TaqMan® real-time PCR detection system, three forward primers (FP), four reverse primers (RP) and five TaqMan® hydrolysis probes (HP) were designed (Primer3Plus Software; Untergasser et al. 2007). The primers are listed in Table 1. Forward and revers primers and untagged oligonucleotide sequences intended for TaqMan® hydrolysis probe selection have been tested in different combinations for PVM detection in a real-time DiRT-PCR system supplemented with SYBR® Green.
The ingredients for the applied one-step real-time DiRT-PCR protocol were: 12.5 μL KAPA3G Direct Plant PCR buffer (2 x Kapa Biosystems/Roche, Basel, Switzerland), 0.3 μM of each respective primer (FP + RP or HP + RP), 2.5 μL SYBRGreen® mix (10 x, Applichem, Darmstadt, Germany), 20 U SuperScript®III reverse transcriptase (Thermo Fisher Scientific, Waltham, USA), 0.5 U KAPA3G Direct Plant DNA polymerase (Kapa Biosystems/Roche), 1.2 M betaine (Sigma-Aldrich, St. Louis, USA) and 2 μL of RNA/plant sap or tuber sap.
The applied temperature schedule included a subsequent melting curve analysis (42 °C for 15 min, 95 °C for 3 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 45 s, afterwards the melting curve analysis was applied from 60 °C to 95 °C for 40 min). The complete thermal cycling protocol took 98 min, and the threshold for quantification was set automatically by the software (CFX Manager 3.1, Bio-Rad, Hercules, USA). The quantitative threshold (Cq) was set at 36 cycles (cut-off) as the limit for discrimination between virus-infected and virus-free potato material. Afterwards, all PCR products were put on an agarose gel (TBE buffer with 1.5% (w/v) agarose powder and 1% (w/v) ethidium bromide (both Applichem).
For validation of the selected PVM TaqMan® primer set a standard curve has been established using cRNA in known concentration. The cRNA-10-fold dilution series starts with 1010 copies per reaction. Therefore, 100 mg potato in vitro plant material infected with PVM was grinded in liquid nitrogen. RNA was extracted as described in the manual of the used PureLink® Plant RNA Reagent (Thermo Fisher Scientific, Waltham, USA). The purified RNA was resolved in ultrapure H2O. Subsequently, the purified RNA was transformed into cDNA. The cDNA was maintained and propagated in E. coli using the plasmid pCR™2.1-TOPO® vector (TOPO® TA Cloning® kit; Thermo Fisher Scientific) according to the manufacturer’s instructions. Afterwards, cRNA synthesis and copy number calculation was conducted according to Fronhoffs et al. (2002).
The DAS-ELISA, the growing-on DAS-ELISA, the real-time CoRT-PCR and the real-time DiRT-PCR comparing PVM detection were conducted according to the comparison experiments of Stammler et al. (2018).
The reaction mix for the real-time one-step duplex DiRT-PCR protocol is as follows: 12.5 μL KAPA3G Direct Plant PCR buffer (2x, Kapa Biosystems), 0.4 μM of each primer/probe of the newly developed PVM TaqMan® primer set, 0.2 μM of the TaqMan® forward and reverse primers of the internal control cox and 0.3 μM of the cox TaqMan® probe, 20 U SuperScript®III reverse transcriptase (Thermo Fisher Scientific), 0.5 U KAPA3G Direct Plant DNA polymerase (Kapa Biosystems), 1.2 M betaine (Sigma-Aldrich, St. Louis, USA) and 2 μL the of plant sap. The reaction volume was set to 25 μL with ultrapure H2O. In addition, the quantitative threshold (Cq) at 36 cycles was set (cut-off) as the limit for discrimination between virus-infected and virus-free potato material.
The reaction mix for the real-time one-step duplex CoRT-PCR protocol is based on Mortimer-Jones et al. (2009): 12.5 μL JumpStart™ Taq ReadyMix™ (2x, including 3 mM MgCl2, Sigma-Aldrich), 0.4 μM of each primer/probe of the newly developed PVM TaqMan® primer set, 0.2 μM of the TaqMan® forward and reverse primers of the internal control cox and 0.3 μM of the cox TaqMan® probe, 20 U SuperScript®III reverse transcriptase (Thermo Fisher Scientific), 2.5 mM MgCl2 (Merck, Darmstadt, Germany) and 2 μL of purified total RNA. The reaction volume was set to 25 μL with ultrapure H2O. According to Mortimer-Jones et al. (2009), the Cq cut-off at 30 cycles was set as the limit for discrimination between virus-infected and virus-free potato material.
For verification of each real-time RT-PCR assay, we include a negative control by adding plant sap or purified RNA of virus-free potato material, and a positive control with purified RNA derived from in vitro potato plants infected with the respective virus species. In addition, we used ultrapure H2O as no template control (NTC).
For the development of the cRNA standard curve using PVM_St15 TaqMan® primers, three technical repeats represent one cRNA quantity status (Fig. 1).

Standard curve and preparation procedure of the developed TaqMan® primer set PVM_St15 for PVM detection
For method comparison regarding sensitivity, three technical repeats were performed for each dilution step of the plant sap within one biological repetition. In total, three biological repetitions were performed. For virus-positive result acceptance, at least two of three technical repeats had to be assessed positive.
For validation by protocol comparison, one reaction per sample was evaluated (unclear first results in real-time DiRT-PCR were repeated twice) to adapt the experimental set up of standard growing-on DAS-ELISA for seed potato certification at Bavarian State Research Center for Agriculture (LfL, Freising, Germany).
Multiplex development and validation (MD)
1 g seedling tubers were homogenized in an extraction bag with a membrane (Bioreba) together with 3 mL extraction buffer, transferred to an Eppendorf tube and centrifuged with 10,000×g for 3 min. The supernatant was further diluted 1:10 with extraction buffer. We then produced a dilution series from 10−1 to 10−5 using the sap of these tubers spiked with total RNA (100 ng/μL) isolated from the in vitro plants described above. In the special case of multiplex experiments, we mixed the total RNAs of three in vitro plants each infected with a different virus.
(MV) 90 tubers of one variety were washed together in tap water. Afterwards, approximately 0.2-0.5 g of the heel end of each tuber was homogenized in a Bioreba extraction bag together with 1 mL extraction buffer using a Homex 6 (Bioreba). The sap was transferred into 96 Well plates (Brand, Germany) and centrifuged 30 min on 4000×g at 4 °C. The supernatant was further diluted 1:50 with extraction buffer and 2 μl used for real-time DiRT-PCR.
All primers and TaqMan® probes were ordered from Biomers (Ulm, Germany) except for the minor-grove-binding probe (MGB, Eurofins, Ebersberg, Germany). For the specific sequence, fluorophore and origin see Table 2.
Combinations of viruses in multiplex sets were chosen according to the practical use in routine testing.
The concentrations of primers, probes, MgCl2, polymerase (Kapa Biosystems/ Roche), dNTPs (Genaxxon, Ulm, Germany) and SSIII reverse transcriptase (Sigma Aldrich) were varied extensively until we found the lowest dilution detectable. Final concentrations can be found in Table 3. For duplex reactions, we used the same concentrations as Stammler et al. (2018).
The real-time PCR program was run on a Bio-Rad CFX96 at 42 °C for 20 min, 95 °C for 3 min, followed by 41 cycles of 95 °C for 45 s, 60 °C for 45 s, and 72 °C for 60 s. The four different channels were used with the fluorescence dyes 6 Carboxyfluoresceine (6-Fam), Hexachlorofluoresceine (Hex), Cyanine 3.5 (almost equal to Texas Red in wavelength) and Cyanine 5 (Cy5).
We extended the reverse transcription in (MV) to 1 h and changed the reverse transcriptase from SuperScript®III (Thermo Fisher Scientific) to HighScriber (HighQu, London, GB, 5000 U/25r). The amount of HighScriber (HighQu) was kept as low as possible to optimize for low cost but receive reliable results. The final concentration was 0.28 U/μL for set A and 0.23 U/μL for set B.
For analyzing the multiplex, BioRad CFX™ Manager 3.1 was used with following settings: Baseline subtracted curve fit, fluorescence drift correction and Cq determination mode “single threshold”. Due to the high fluctuation in background fluorescence, the threshold line was set manually above background fluorescence. We excluded samples for which the positive controls showed a fluorescence lower than ~200 RFU. A cut-off- Cq value was empirically set at following Cycles for each primer and probe set in the multiplex separately: PLRV, PVA and PVX: Cycle 36; PVY: Cycle 39; PVM and PVS: Cycle 33.
All validation experiments
Cohens Kappa coefficient (k) was calculated according to the method by Petrie and Sabin (2009). This is commonly used as a measure to rate the inter-method agreement for categorical items. Cohen’s Kappa coefficient (k) ranges between 0 and 1 (Cohen 1960). We also calculated Gwet’s AC1 as a measure of inter-method agreement for experiments with unequal numbers of positive and negative samples (Gwet 2008; Wongpakaran et al. 2013). In addition, we used the rating categories developed by Landis and Koch (1977).
Results
PVM TaqMan® primer set development
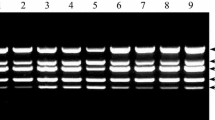
For all PVM infected samples (RNA, leaf sap and tuber sap), the combination of PVM_F_3 + PVM_R_4 is displayed, in both repeats (Supplement 1A: 1st run and 2nd run) with a specific amplified fragment 107 bp in size and PVM negative samples fragments smaller than 100 bp. Primers are listed in Table 1.
In addition, evident temperature differences could be observed in the melting curve analysis between the NTC (no template control), the PVM negative leaf sap control and all PVM positive samples (RNA, leaf sap and tuber sap, Supplement 1A). However, it was not possible to discriminate purified RNA derived from PVM negative potato plants and all PVM positive samples (RNA, leaf sap and tuber sap) by using just PVM_F_3 + PVM_R_4 primer combination for the real-time DiRT-PCR assay. This was because one longer fragment appeared for the PVM negative purified RNA target (Supplement 1A, highlighted by an arrow pointing the DNA fragment and the appropriate melting curve). Still, all samples taken from PVM infected plants were identified by this primer combination including the tuber sap samples comprising low virus titer.
For hydrolysis probe selection (Supplement 1B), the probe and reverse primer combination PVM_P_5 + PVM_R_4 was tested and later selected. Therefore, the unlabeled oligonucleotide sequence of the probe (Supplement 1B) was used as forward primer and combined with the reverse primer. By doing this, the melting peaks of PVM negative samples can be hardly differentiated from PVM positive samples (see Supplement 1B). That means PVM_P_5 is a rather unspecific probe. However, the usage of the selected forward and reverse primers PVM_F_3 + PVM_R_4 in combination with this rather unspecific PVM_P_5 TaqMan® hydrolysis probe empirically leads to a convincing PVM real-time TaqMan® DiRT-PCR system (termed PVM_St15, see Table 1, Supplement 1).
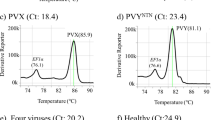
This real-time TaqMan® DiRT-PCR system using the primer set PVM_St15, was validated by obtaining a standard curve (Fig. 1). The regression analysis of the PVM_St15 standard curve using the cRNA copy numbers 1000 - 10,000,000,000 gave a good linearity of 0.9834. The efficiency of the standard curve was 100% with a slope of −3.3114 for real-time DiRT-PCR (Fig. 1). The lowestcRNA copy number concentration (1 × 103) of the standard curve displayed in Fig. 1 is not indicating the limit of detection but the limit of linearity.
On the left, the established standard curve is shown using a dilution series of prepared cRNA with calculated copy numbers (103-1010). On the right, the preparation procedure of the standard curve is roughly displayed.
The sensitivity of both real-time RT-PCR methods was inconsistent. The sensitivity of real-time DiRT-PCR was 100,000,000-fold higher than that of the immunological method DAS-ELISA (Fig. 2A, Cq-values in Supplement 2). In contrast, the real-time CoRT-PCR assay for PVM that was conducted according to the protocol of Mortimer-Jones et al. (2009) was only 10-fold more sensitive than DAS-ELISA. On average, real-time DiRT-PCR was 10,000,000-fold more sensitive than real-time CoRT-PCR. However, the no template control (NTC, water) used in real-time DiRT-PCR archived in each of the three biological repeats a relative fluorescence signal before the cut-off (cycle 36). But the negative control using virus-free plant sap remained negative (NC, Cq-values in Supplement 2) in all assays for PVM detection.

Method (A) and protocol (B) comparison of PVM detection
Validation by protocol comparison for PVM detection (growing-on DAS-ELISA vs. real-time DiRT-PCR on dormant tubers)
The potato virus detection protocol using the real-time DiRT-PCR method was compared with the standard virus detection protocol using growing-on DAS-ELISA on dormant potato tubers that were grown in the field, harvested and processed in 2015. We used both protocols for PVM detection to test 332 tubers and compared them in terms of high-throughput ability and result agreement.
The qualitative protocol comparison of growing-on DAS-ELISA using 4–6-week-old leaves of tuber eye cuttings to validate the established real-time DiRT-PCR protocol for PVM detection using dormant tubers without RNA purification, revealed high agreement of 93.1% (Fig. 2B). A Cohens Kappa (k) value of 0.86 was calculated, and thus, according to Landis and Koch (1977), an almost perfect agreement was achieved. However, 0.6% of total sample size of 332 tubers showed a positive result for PVM detection by real-time DiRT-PCR but not by DAS-ELISA. Merely, 6.3% of the specimens were indicated PVM positive by DAS-ELISA and PVM negative by real-time DiRT-PCR (Fig. 2B).
A: The sensitivity of double-antibody sandwich (DAS-) ELISA, real time Direct Reverse Transcription PCR (DiRT-PCR), and real-time Conventional Reverse Transcription PCR (CoRT-PCR) was compared (Sensitivity of methods). On the left, sensitivity values are shown with three biological repeats using plant sap derived from PVM-infected in vitro potato plants in a ten-fold (10−1 to 10−10) dilution series. The dark lines indicate the average sensitivity differences. Each colored square represents at least two virus positive technical repeats out of three (see Supplement 2). On the upper right, the sampling procedure is roughly described for the sensitivity of methods experiment. For real-time CoRT-PCR, RNA was purified, and the quantity was measured in μg/μL. For all three biological repeats the RNA quantity is displayed in the lower right quadrant. B: The agreement of the validation by protocol comparison experiment between DAS-ELISA on leaves of 4–6-weeks cultivated tuber eye cutting and real-time DiRT-PCR using dormant potato tubers, of the same 332 field-grown tubers is displayed on the left. For 93.1% of the tuber samples both protocols gave the same result (51.8% PVM positive and 41.3% PVM-negative). The Cohens Kappa (k) coefficient gave almost perfect agreement (0.86). Top right, the sampling procedure is roughly described for the protocol comparison experiment (validation).
Multiplex development
To compare the sensitivity of the duplex (Stammler et al. 2018; this study) and multiplex (this study) reaction mixtures, we created a 10-fold dilution series of RNA derived of virus-infected in vitro plants. We used 100 ng/μL total RNA of up to 3 different plants infected with different viruses as a starting concentration for dilution with either water or tuber sap. The dilution range was 10−1 to 10−5. When comparing the duplex mixtures in water versus diluted tuber sap, the detection limit was the same in both test conditions (Fig. 3). Specifically, the detection limit was reached for the reaction mixtures of PLRV, PVY and PVX at a dilution of 10−5 and at a dilution of 10−4 for the reaction mixtures of PVM, PVS and PVA (Fig. 3).

Detection of different potato viruses in a 10-fold dilution series from 10−1 to 10−5 of total RNA from virus infected in vitro plants in either water or diluted tuber sap. The total RNA dilution series contained either single or three pooled RNAs from plants each infected with one of three different viruses. It was tested by a duplex (cox + virus) or multiplex assay (cox + three viruses) shown on the left. A colored field marks two of two positive detections, hatched fields mark one of two positive detections and white fields negative detections
For the developed PLRV/PVY/PVM multiplex reaction mixture, the sensitivity was equal to the duplex mixtures on tuber sap for PLRV (10−5) but slightly reduced for PVY (10−4) and PVM (10−3). However, one of two technical replicates still showed a positive Cq at a dilution of 10−5 for PVY and 10−4 for PVM (Fig. 3, Supplement 3). The developed PVS/PVA/PVX multiplex reaction mixture detected each virus as sensitively as the duplex reactions, that is 10−4 for PVS and PVA and 10−5 for PVX (Fig. 3, Supplement 3).
Multiplex validation
In Fig. 4, we compare test results from DAS-ELISA on leaves and the above-described multiplex real-time DiRT-PCR mixtures for PLRV/PVY/PVM and PVS/PVA/PVX on heel-ends of ~2000-3000 single tubers of different potato varieties. We tested tuber batches with a high virus content simulating a seed certification process.

Agreement of protocol comparison (validation) between DAS-ELISA on leaves of 4-6 weeks old tuber eye cuttings and real-time DiRT-PCR multiplex on dormant tubers of the same field grown tuber samples. The stacked columns represent, for each virus (x-axis), the % (y-axis) of the total number of tubers (n, table below) tested negative in green and tested positive in red by both methods, in orange tested only positive by multiplex real-time DiRT-PCR and negative by DAS-ELISA and blue tested only positive by DAS- ELISA and negative by real-time DiRT-PCR. For numbers see table below graphic. The % of agreement between the two methods is shown between the arrows left to the columns for each virus. Cohen’s kappa and Gwet’s AC1 as measures of agreement are the last numbers below each column
For PLRV, there was a large general agreement of 97.6% between DAS-ELISA and multiplex real-time DiRT-PCR, with 96.1% negative and 1.5% positive in both methods. 2.1% of samples where solely positive with the multiplex real-time DiRT-PCR and 0.3% were solely positive in DAS-ELISA. Interestingly, Cohen’s kappa showed a moderate agreement of 0.54. This is due to the generally high number of negative samples. In contrast to Cohen’s kappa, Gwet’s AC1 does not take into acount the unequal number of positiv an negative samples. Accordingly, Gwet’s AC1 assumes a value of 0.97, which is now an almost perfect agreement.
When comparing the results for PVY, we found 83.9% agreement overall, where 64.5% of samples were tested negative and 19.5% tested positive in both methods, 5% were solely positive in real-time DiRT-PCR and 11% solely in DAS-ELISA. Cohen’s kappa was 0.6, which is on the upper end of a moderate agreement and Gwet’s AC1 is already within the range of a substantial agreement with 0.73.
For PVM we found 78.8% general agreement with 65.2% of the samples being positive and 12.8% being negative, 15.3% were solely positive with DAS-ELISA and 6.7% were solely positive with real-time DiRT-PCR. Still, the Cohen’s kappa is 0.4, which is on the upper end of a fair agreement. In contrast, the calculation of Gwet’s AC1 showed a substantial agreement with 0.65. Consequently, we also calculated Gwet’s AC1 for duplex real-time DiRT-PCR above. There was no difference here compared to Cohen’s kappa with both taking a value of 0.86.
Further, there is a 90.2% overlap for PVS in both methods, with 80.8% positive and 9.4% negative agreement. 5.6% were solely positive with DAS-ELISA and 4.3% solely with real-time DiRT-PCR. Cohen’s kappa is on the upper end of a moderate agreement with 0.6 and Gwet’s AC1 is in the range of an almost perfect agreement with 0.87.
Notably, all tubers were negative for PVA and PVX with both methods. As there were no positive samples, we did not calculate Cohen’s kappa or do any further analysis with these viruses.
Discussion
Since direct virus detection by solely DAS-ELISA on dormant potato tubers is not reliable (Gugerli and Gehriger 1980; Spiegel and Martin 1993) the standardized procedure for virus detection during seed potato certification in Bavaria (Germany) is growing on DAS- ELISA. Growing on DAS-ELISA is based on leaf extracts of 4-6-week-old glasshouse-cultivated tuber eye cuttings. This phase of virus-enrichment is time consuming and requires large, seasonal glasshouse capacities. Therefore, other certification agencies in Germany and Europe are using different protocols of real-time CoRT-PCR (Schumpp et al. 2016; Agroscope video: https://www.youtube.com/watch?v=o2jXuCzVdYk) that requires expensive RNA extraction. For that reason, we directed our research efforts into making real-time PCR a cost-effective alternative for high throughput testing. Thus, we first developed the real-time DiRT-PCR for single virus detection in duplex application for PLRV, PVY and PVS (Stammler et al. 2018). In this study, we present a single virus detection for PVM and two multiplex applications for PLRV, PVY, PVM, PVS, PVA and PVX. Once this new method will be embedded into a pool testing scheme, the advantage of this new method over DAS-ELISA and conventional qPCR methods lies mainly in saving costs for extraction or glasshouse keeping and testing the three most important viruses (PLRV, PVY and PVM) in one reaction.
PVM TaqMan® primer set development
Potato Virus M reduces crop yield up to 45% (Bauch et al. 2012) and, thus, is part of the seed potato certification process for most European countries. There are primers published for PVM detection by conventional RT-PCR using agarose gel electrophoresis (Zhang et al. 2017; Xu et al. 2010). While there are a lot of TaqMan® primers and probes published for detection of other potato viruses (Boonham et al. 2009; Agindotan et al. 2007; Mortimer-Jones et al. 2009), it is striking, that there has not yet been a published TaqMan® primer and probe set to detect PVM. Reasons for that might be poor prerequisites in conserved sequence regions of the known PVM isolates.
However, in this study, it was possible to establish primer and probe candidates for TaqMan® based PVM detection. Yet, most of these candidates gave poor results regarding sensitivity and specificity or both (Data not shown). Solely, the forward and reverse primer combination PVM_F_3 + PVM_R_4 was sensitive and specific enough to reduce the tendency to obtain unspecific binding by one PVM probe, namely PVM_P_5 (Table 1).
To our knowledge, this is the first published TaqMan® primer and probe set specific to Potato Virus M. It performs reliably on direct potato tuber sap in duplex reactions using 6-carboxyfluorescein (6-FAM) as a fluorophore in combination with the internal control cox (cytochrome oxidase, Weller et al. 2000) using hexachlorofluorescein (HEX). The results are directly comparable to duplex potato virus detection of PVY, PLRV and PVS by real-time DiRT-PCR published by Stammler et al. (2018) and Stammler (2020). One disadvantage of these PVM detecting TaqMan® primer and probe set is that an assay using just water as no template control (NTC) tends to result in fluorescence signal before 36 cycles, discussed later. However, the negative control always stays clear (this study and Stammler 2020).
Standard curves for PVM detection by one step duplex real-time DiRT-PCR
The obtained standard curves for the newly established PVM primer set in one step duplex real-time DiRT-PCR. led to an efficiency of 100% and a regression (R2) value of 0.9834. Thus, the developed TaqMan® PVM primer and probe combination is acceptable for diagnostics (Kavanagh et al. 2011). However, during establishment of cRNA standard curves, the no reverse transcription control (NRTC) showed Cq-values within a range for potato virus positive evaluation. We do not know if that is based on insufficient DNAse treatment or coming from background signals as also experienced for PVM-negative RNA and water (NTC) during primer selection and further experiments like sensitivity of methods, later discussed.
Multiplex real-time DiRT-PCR development
Our one-step real-time multiplex DiRT-PCR assay is the first multiplex for potato viruses, which detects four targets in one reaction and runs directly and sensitively on diluted tuber sap. To save time and money, sensitive one-step multiplex real-time PCRs on extracted RNA from potatoes have found increased popularity in past years and consequently primers and probes have been developed for several potato viruses (Agindotan et al. 2007; Boonham et al. 2009; Singh et al. 2013; Ali et al. 2010; Mortimer-Jones et al. 2009). Also, a two-step multiplex was developed for diluted crude tuber sap (Agindotan et al. 2007). Our multiplex assay consists of primers and probes for PVY, PLRV, PVS, PVA and PVX from those publications and we added our newly developed PVM primer and probe set. Stammler et al. (2018) and Stammler (2020) showed that these primers and probes can be used to successfully detect viruses directly on tuber sap in a qualitative manner. Thus, in this study, we confirm that detection is extremely sensitive for the suggested combinations of published primer and probes even on such PCR-inhibitory material as tuber sap. When combining our new primer/probe set for PVM with those for PLRV and PVY however, sensitivity to detect PVM was decreased. Nevertheless, this combination of multiplex sets was preferable and sensitive enough for our routine testing, but it remains to be tested if sensitivity increases again in other combinations, i.e., with PVA, PVS or PVX.
Validation process
In contrast to comparison of sensitivity (method comparison) of PVY, PVS and PLRV detection in Stammler et al. (2018), the PVM detection sensitivity differed extremely between the two different RT-PCR assays. While real-time DiRT-PCR was up to 100,000,000-fold more sensitive than DAS-ELISA, real-time CoRT-PCR was only up to 10-fold more sensitive. Up to now, it is unclear if these results are due to false-positive or true-positive results of real-time DiRT-PCR. False-positive detection could happen here due to a potential predisposition of the polymerase to hydrolyze the probe in PVM detection without previous amplification in a blank and poor environment. Therefore, high dilutions might lead to unspecific reactions when either no target or no plant sap matrix is available. This would explain the obtained false-positive results of the developed PVM TaqMan® primer set PVM_St15 for the NTC (water) and PVM-free RNA diluted in nuclease free water (see Supplement 2, Cqs), but the same assay remained true negative for the virus free plant sap target on the same reaction plate.
Also, Hadersdorfer et al. (2011) found greater sensitivity for purified RNA than for crude plant sap within one nucleic acid-based virus detection method in plums. Because of the highly concentrated total RNA in comparison to the unprocessed crude plant sap, we generally expected greater sensitivity when comparing real-time DiRT-PCR with real-time CoRT-PCR. Nevertheless, further experiments gave hints approving this PVM primer set in real-time DiRT-PCR for high detection sensitivity at low virus titers.
We compared different protocols but also the sampling of different parts of the same tuber, namely rose-end and heel-end. For the real-time DiRT-PCR protocol, the heel-end of the dormant tuber was directly used whereas for the growing-on DAS-ELISA protocol the rose-end of the same dormant tuber was further cultivated. After 4-6 weeks cultivation, leaves of these plants were used in the DAS-ELISA assay. Different biological prerequisites were present in different potato tuber parts of the same sampled tuber that may lead to non-homogeneous virus distribution/titer in both samples (Dupuis 2017). This could have led to virus exclusion for one or the other protocol. Additionally, virus replication also differs by virus-type due to differing virus-enrichment capacity across the virus species and isolates during enrichment phase for DAS-ELISA.
In addition, results might be affected by altered inhibitor concentrations in different potato varieties, because the used KAPA3G™ Direct Plant DNA polymerase is referred to be robust but not immune against existing PCR inhibitors (Gallup 2011; Schori et al. 2013).
The protocol agreement for PVM detection using the newly developed TaqMan® primer set gave “almost perfect” agreement (protocol agreement: 93.1%, Cohens Kappa (k): 0.86) according to Landis and Koch (1977). However, the growing-on DAS-ELISA protocol was slightly more sensitive compared to the real-time DiRT-PCR protocol (6,3%). Assuming no false-positive and false-negative results, the coverage of all PVM isolates of the used primer set and considering the extreme sensitivity of real-time DiRT-PCR in the sensitivity comparison of methods (this study), we propose for PVM high natural viral replication frequency during virus enrichment phase of the growing-on DAS-ELISA protocol that filled the sensitivity gap obtained between real-time DiRT-PCR and DAS-ELISA to obtain high agreement between both protocols.
For all viruses tested, real-time DiRT-PCR multiplex shows a fair to an almost perfect protocol agreement with DAS-ELISA, depending on the measures applied. Interestingly, Cohen’s kappa and Gwet’s AC1 differ for each virus, especially for PLRV. This is due to the fact that Cohen’s kappa is influenced by the ratio of negative and positive samples, which in our case is greater for negative samples (Wongpakaran et al. 2013; Gwet 2008). Therefore, we chose Gwet’s AC1 (Gwet 2008) as a second measure of agreement. It has been used frequently in clinical studies including comparison experiments with DAS-ELISA (e.g. Petzold et al. 2010).
Looking at the Gwet’s AC1 values, the multiplex results largely resemble the results for the duplex protocol comparison (Stammler et al. 2018) in terms of the percentages of overlap and agreement coefficents for PLRV (duplex: 92.8%, 0.84; muliplex: 97.6%, 0.97), PVY (duplex: 83.8%, 0.67; muliplex: 83.9%, 0.73) and PVS (duplex: 82.3%, 0.62; muliplex: 90.2%, 0.87). This underlines, that our designed multiplex real-time DiRT-PCR is a sensitive tool for direct potato virus detection.
In detail, the multiplex real-time DiRT-PCR detected more samples PLRV positive than DAS-ELISA and for PVY slightly less samples positive in comparison to DAS-ELISA. The PVY primer and probe used for our experiment was developed but also compared to DAS-ELISA by Singh et al.(2013). They compared their established real-time CoRT-PCR protocol using dormant tubers to that with growing-on DAS-ELISA (Singh et al. 2013) using leaves of 6–8-week old progeny plants of the same tubers. Additionally, the tuber sprouts were tested by DAS-ELISA before planting. Their results also showed the highest sensitivity for growing-on DAS-ELISA. Their real-time CoRT-PCR approach ranked on second position comparable to our validation experiment by protocol comparison of real-time DiRT-PCR, whereas DAS-ELISA on tuber sprouts was scored lowest. In addition, high virus multiplication ability for PVY isolates (Stare et al. 2015), PLRV isolates (Stammler et al. 2018) and possibly also PVM (this study and Stammler 2020) in the first 4-6 weeks of cultivation is extremely high and therefore detection with DAS-ELISA seems much more sensitive.
One of several possible reasons for discrepancies in this validation by protocol comparison could be that PLRV is also more concentrated in the heel end of the tuber whereas PVY is distributed unequally throughout the tuber phloem and in some varieties it can even be found only in some eyes and not in others (Vetten et al. 1983; Dupuis 2017, personal communication with O. Schumpp, Agroscope, Switzerland).
The detection sensitivity for PVM is particularly reduced compared to single detection (duplex: 93.1%, 0.86; muliplex: 78.8%, 0.66). This reflects the results in the multiplex development phase above. Reasons for this reduction might prevalently be the strong interaction between primers and probes in the multiplex (Pan et al. 2018). Furthermore, our samples were thawed and frozen several times due to limited time and workforce. Many freezing and thawing cycles evidently affect RNA stability (Wang et al. 2015) and might affect viruses like PVM more than others (personal communication with Bioreba AG, Switzerland), especially combined with free enzymes and inhibitors present in potato tuber sap. Therefore, the agreement between DAS-ELISA and real-time DiRT-PCR might be improved when tubers are freshly processed and directly put into real-time DiRT-PCR.
These factors all account for additional loss of detection sensitivity. Nonetheless, detection sensitivity is still strong enough to produce very high agreement scores for three of the viruses tested by our multiplex sets. Thus, this multiplex real-time DiRT-PCR is a very good candidate for pool testing and will be less costly but still sensitive enough for routine procedures.
Conclusion
In this study, we introduced and validated the first TaqMan® primers and probes for PVM detection in real-time DiRT-PCR. We further converted this method into a multiplex system to detect PLRV, PVY, PVM, PVS, PVA and PVX using crude tuber sap. However, detection of PVX and PVA was possible in crude sap of in-vitro plants, but no field grown tubers were available for evaluation. The agreement results for real-time DiRT-PCR duplex and multiplex detections of PLRV, PVY, PVM and PVS, on crude sap of field grown tubers in comparison to growing on DAS-ELISA underline that these methods are reliable alternatives to DAS-ELISA and CoRT-PCR in time and cost-effective routine testing for single tubers. We suggest taking this real-time DiRT-PCR one step further into testing tuber pools.
Availability of data and material
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Code availability
Not applicable.
References
Agindotan, B., & Perry, K. L. (2007). Macroarray detection of plant RNA viruses using randomly primed and amplified complementary DNAs from infected plants. Phytopathology, 97(1), 119–127. https://doi.org/10.1094/PHYTO-97-0119
Agindotan, B. O., Shiel, P. J., & Berger, P. H. (2007). Simultaneous detection of potato viruses PLRV, PVA, PVX and PVY from dormant potato tubers by TaqMan real-time RT-PCR. Journal of Virological Methods, 142(1-2), 1–9. https://doi.org/10.1016/j.jviromet.2006.12.012
Ahouee, K. H., Habibi, M. K., & Mosahebi, G. H. (2010). Detection of potato leafroll virus isolated from potato fields in Tehran province in aphids by immunocapture reverse transcription polymerase chain reaction. African Journal of Biotechnology, 9(16), 2349–2352.
Ali, M. C., Maoka, T., Natsuaki, K. T., & Natsuaki, T. (2010). The simultaneous differentiation of potato virus Y strains including the newly described strain PVYNTN-NW by multiplex PCR assay. Journal of Virological Methods, 165(1), 15–20.
Bauch, G.; Steinbach, P. & Thiel, W. (2012). Kleines Handbuch für die Selektion in Pflanzkartoffeln. gefördert mit Mitteln der Landwirtschaftlichen Rentenbank in Zusammenarbeit mit der Union der Deutschen Kartoffelwirtschaft e.V. (UNIKA). Hg. v. Landwirtschaftskammer Niedersachsen, Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischerei MV und Bayerische Landesanstalt für Landwirtschaft (p29-36).
Boonham, N., Laurenson, L., Weekes, R., & Mumford, R. (2009). Direct detection of plant viruses in potato tubers using real-time PCR. Methods in Molecular Biology, 508, 249–258. https://doi.org/10.1007/978-1-59745-062-1_19
Cohen, J. (1960). A coefficient of agreement for nominal scales. Educational and Psychological Measurement, 20(1), 37–46. https://doi.org/10.1177/001316446002000104
Dupuis, B. (2017). The movement of potato virus Y (PVY) in the vascular system of potato plants. European Journal of Plant Pathology, 147(2), 365–373. https://doi.org/10.1007/s10658-016-1008-5
Fronhoffs, S., Totzke, G., Stier, S., Wernert, N., Rothe, M., Brüning, T., Koch, B., Sachinidis, A., Vetter, H., & Ko, Y. (2002). A method for the rapid construction of cRNA standard curves in quantitative real-time reverse transcription polymerase chain reaction. Molecular and Cellular Probes, 16(2), 99–110. https://doi.org/10.1006/mcpr.2002.0405
Gallup, J. M. (2011). In: S. Kennedy and N. Oswald (Eds.), qPCR inhibition and amplification of difficult templates: PCR troubleshooting and optimization: The essential guide. (1. Ed., 23–65). Caister academic press.
Gugerli, P., & Gehriger, W. (1980). Enzyme-linked immunosorbent assay (ELISA) for the detection of potato leafroll virus and potato virus Y in potato tubers after artificial break of dormancy. Potato Research, 23(3), 353–359. https://doi.org/10.1007/BF02360674
Gwet, K. L. (2008). Computing inter-rater reliability and its variance in the presence of high agreement. British Journal of Mathematical and Statistical Psychology, 61(1), 29–48.
Hadersdorfer, J., Neumüller, M., Treutter, D., & Fischer, T. C. (2011). Fast and reliable detection of plum pox virus in woody host plants using the blue LAMP protocol. Annals of Applied Biology, 159(3), 456–466. https://doi.org/10.1111/j.1744-7348.2011.00510.x
Hepting, L. (2002). Pflanzguterzeugung in BAyern. Ein Rückblick über 50 Jahre. Hg. v. Bayerische Landesanstalt für Landwirtschaft.
Hühnlein, A., Drechsler, N., Steinbach, P., Thieme, T., & Schubert, J. (2013). Comparison of three methods for the detection of potato virus Y in seed potato certification. Journal of Plant Diseases and Protection, 120(2), 57–69. https://doi.org/10.1007/BF03356455
Hühnlein, A., Schubert, J., Zahn, V., & Thieme, T. (2016). Examination of an isolate of potato leaf roll virus that does not induce visible symptoms in the greenhouse. European Journal of Plant Pathology, 145(4), 829–845. https://doi.org/10.1007/s10658-016-0872-3
Ju, H.-J. (2011). Simple and rapid detection of potato leafroll virus by reverse transcription loop-mediated isothermal amplification. The Plant Pathology Journal, 27(4), 385–389. https://doi.org/10.5423/PPJ.2011.27.4.385
Kavanagh, I.; Jones, G. & Naveed Nayab, S. N. (2011). In: S. Kennedy and N. Oswald (Eds.), significance of controls and standard curves in PCR: PCR troubleshooting and optimization: The essential guide. (1. Ed., 67–78). Caister academic press.
Landis, J. R., & Koch, G. G. (1977). The measurement of observer agreement for categorical data. Biometrics, 33(1), 159–174.
Leone, G., van Schijndel, H. B., van Genien, B., & Schoen, C. D. (1997). Direct detection of potato leafroll virus in potato tubers by immunocapture and the isothermal nucleic acid amplification method NASBA. Journal of Virological Methods, 66(1), 19–27. https://doi.org/10.1016/S0166-0934(97)02203-9
Malnoe, P., Farinelli, L., Collet, G. F., & Reust, W. (1994). Small-scale field tests with transgenic potato, cv. Bintje, to test resistance to primary and secondary infections with potato virus Y. Plant Molecular Biology, 25(6), 963–975. https://doi.org/10.1007/BF00014670
Mortimer-Jones, S. M., Jones, M. G., Jones, R. A., Thomson, G., & Dwyer, G. I. (2009). A single tube, quantitative real-time RT-PCR assay that detects four potato viruses simultaneously. Journal of Virological Methods, 161(2), 289–296. https://doi.org/10.1016/j.jviromet.2009.06.027
Murashige, T., & Skoog, F. (1962). A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiologia Plantarum, 15(3), 473–497. https://doi.org/10.1111/j.1399-3054.1962.tb08052.x
Pan, P., Jin, W., Li, X., Chen, Y., Jiang, J., Wan, H., & Yu, D. (2018). Optimization of multiplex quantitative polymerase chain reaction based on response surface methodology and an artificial neural network-genetic algorithm approach. PLoS One, 13(7), e0200962. https://doi.org/10.1371/journal.pone.0200962
Petrie, A. & Sabin, C. (2009). Medical statistics at a glance.
Petzold, A., Altintas, A., Andreoni, L., Bartos, A., Berthele, A., Blankenstein, M. A., Buee, L., Castellazzi, M., Cepok, S., & Comabella, M. (2010). Neurofilament ELISA validation. Journal of Immunological Methods, 352(1-2), 23–31.
Radcliffe, E. B., & Ragsdale, D. W. (2002). Aphid-transmitted potato viruses: The importance of understanding vector biology. American Journal of Potato Research, 79(5), 353–386. https://doi.org/10.1007/BF02870173
Schori, M., Appel, M., Kitko, A., & Showalter, A. M. (2013). Engineered DNA polymerase improves PCR results for plastid DNA. Applications in Plant Sciences, 1(2). https://doi.org/10.3732/apps.1200519
Schumpp, O.; Dupuis, B.; Bréchon, A.; Wild, W.; Frei, P.; Pellet, D. & Schaerer, S. (2016). Molekulare Hochleistungsdiagnosik zum Nachweis von Kartoffel-Viren. Agrarforschung Schweiz 2016, 2016 (7 (10)), 456–465.
Singh, M., Singh, R. P., Fageria, M. S., Nie, X., Coffin, R., & Hawkins, G. (2013). Optimization of a real-time RT-PCR assay and its comparison with ELISA, conventional RT-PCR and the grow-out test for large scale diagnosis of potato virus Y in dormant potato tubers. American Journal of Potato Research, 90(1), 43–50. https://doi.org/10.1007/s12230-012-9274-z
Singh, R. P., Nie, X., & Singh, M. (2000). Duplex RT-PCR. Reagent concentrations at reverse transcription stage affect the PCR performance. Journal of Virological Methods, 86(2), 121–129. https://doi.org/10.1016/S0166-0934(00)00138-5
Spiegel, S., & Martin, R. R. (1993). Improved detection of potato leafroll virus in dormant potato tubers and microtubers by the polymerase chain reaction and ELISA. Annals of Applied Biology, 122(3), 493–500.
Stammler, J. (2020). Investigations on potato virus detection for high-throughput application. Dissertation. https://mediatum.ub.tum.de/1600039?show_id=1540145.
Stammler, J.; Hadersdorfer, J.; Neumüller, M.; Kellermann, A. & Treutter, D. (2014). Poster: Ansatz zur Optimierung des molekularen Nachweises von Kartoffelviren (approach for optimization of molecular detection of potato viruses). Julius-Kühn-Archiv (447).
Stammler, J., Oberneder, A., Kellermann, A., & Hadersdorfer, J. (2018). Detecting potato viruses using direct reverse transcription quantitative PCR (DiRT-qPCR) without RNA purification: An alternative to DAS-ELISA. European Journal of Plant Pathology, 152(1), 237–248. https://doi.org/10.1007/s10658-018-1468-x
Stare, T., Ramšak, Ž., Blejec, A., Stare, K., Turnšek, N., Weckwerth, W., Wienkoop, S., Vodnik, D., & Gruden, K. (2015). Bimodal dynamics of primary metabolism-related responses in tolerant potato-potato virus Y interaction. BMC Genomics, 16, 716. https://doi.org/10.1186/s12864-015-1925-2
Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R. & Am Leunissen, J. (2007). Primer3Plus, an enhanced web interface to Primer3. Nucleic acids research 35 (suppl_2), W71-W74.
Vetten, H. J., Ehlers, U., & Paul, H. L. (1983). Detection of potato viruses Y and a in tubers by enzyme-linked immunosorbent assay after natural and artificial break of dormancy. Journal of Phytopathology, 108(1), 41–53. https://doi.org/10.1111/j.1439-0434.1983.tb00562.x
Wang, Y., Zheng, H., Chen, J., Zhong, X., Wang, Y., Wang, Z., & Wang, Y. (2015). The impact of different preservation conditions and freezing-thawing cycles on quality of RNA, DNA, and proteins in Cancer tissue. Biopreservation and Biobanking, 13(5), 335–347. https://doi.org/10.1089/bio.2015.0029
Weller, S. A., Elphinstone, J. G., Smith, N. C., Boonham, N., & Stead, D. E. (2000). Detection of Ralstonia solanacearum strains with a quantitative, multiplex, real-time, fluorogenic PCR (TaqMan) assay. Applied and Environmental Microbiology, 66(7), 2853–2858.
Wongpakaran, N., Wongpakaran, T., Wedding, D., & Gwet, K. L. (2013). A comparison of Cohen's kappa and Gwet's AC1 when calculating inter-rater reliability coefficients: A study conducted with personality disorder samples. BMC Medical Research Methodology, 13, 61. https://doi.org/10.1186/1471-2288-13-61
Xu, H., D'Aubin, J., & Nie, J. (2010). Genomic variability in potato virus M and the development of RT-PCR and RFLP procedures for the detection of this virus in seed potatoes. Virology Journal, 7(1), 25. https://doi.org/10.1186/1743-422X-7-25
Zhang, W., Zhang, Z., Fan, G., Gao, Y., Wen, J., Bai, Y., Qiu, C., Zhang, S., Shen, Y., & Meng, X. (2017). Development and application of a universal and simplified multiplex RT-PCR assay to detect five potato viruses. Journal of General Plant Pathology, 83(1), 33–45.
Acknowledgments
We thank Dr. Petr Dedic, who worked at the Potato Research Institute Havlíčkův Brod (Czech Republic) for providing virus infected in vitro potato plant material for assay validation and assay sensitivity comparison, the technical assistance team of the Bavarian State Research Center for Agriculture (Freising, Germany) for conducting the DAS-ELISA assay.
Many thanks go to the technical assistant Madlen Ludwig, who conducted most of the work in the wet lab for the multiplex development.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was funded by the Bavarian State Ministry for Food, Agriculture and Forestry (Stmelf). Furthermore, there was a financial contribution by the National Association of seed potato growers Bayern e. V. (Landesverband der Saatkartoffel-Erzeuger-Vereinigungen in Bayern e.V.) and by the potato growers of the German Society for Plant Breeding e.V. (Gesellschaft für Pflanzenzüchtung e.V., GPZ).
Author information
Authors and Affiliations
Contributions
Not applicable.
Corresponding authors
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflicts of interest/competing interests
The authors declare no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Prinz, M., Kellermann, A., Bauch, G. et al. Development of the first PVM TaqMan® primer set and a one-step real-time multiplex DiRT-PCR for the detection of PLRV, PVY, PVM, PVS, PVA and PVX in potato tuber sap. Eur J Plant Pathol 162, 807–823 (2022). https://doi.org/10.1007/s10658-021-02436-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-021-02436-z