
Abstract
A diagnosis of diabetes mellitus and prediabetes has been associated with increased risk of Parkinson’s disease (PD) in several studies, but results have not been entirely consistent. We conducted a systematic review and meta-analysis of cohort studies on diabetes mellitus, prediabetes and the risk of PD to provide an up-to-date assessment of the evidence. PubMed and Embase databases were searched for relevant studies up to 6th of February 2022. Cohort studies reporting adjusted relative risk (RR) estimates and 95% confidence intervals (CIs) for the association between diabetes, prediabetes and Parkinson’s disease were included. Summary RRs (95% CIs) were calculated using a random effects model. Fifteen cohort studies (29.9 million participants, 86,345 cases) were included in the meta-analysis. The summary RR (95% CI) of PD for persons with diabetes compared to persons without diabetes was 1.27 (1.20–1.35, I2 = 82%). There was no indication of publication bias, based on Egger’s test (p = 0.41), Begg’s test (p = 0.99), and inspection of the funnel plot. The association was consistent across geographic regions, by sex, and across several other subgroup and sensitivity analyses. There was some suggestion of a stronger association for diabetes patients reporting diabetes complications than for diabetes patients without complications (RR = 1.54, 1.32–1.80 [n = 3] vs. 1.26, 1.16–1.38 [n = 3]), vs. those without diabetes (pheterogeneity=0.18). The summary RR for prediabetes was 1.04 (95% CI: 1.02–1.07, I2 = 0%, n = 2). Our results suggest that patients with diabetes have a 27% increased relative risk of developing PD compared to persons without diabetes, and persons with prediabetes have a 4% increase in RR compared to persons with normal blood glucose. Further studies are warranted to clarify the specific role age of onset or duration of diabetes, diabetic complications, glycaemic level and its long-term variability and management may play in relation to PD risk.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The prevalence of diabetes mellitus (DM) has increased to epidemic levels over the last decades, concurrent with the increased rates of overweight and obesity globally. The number of adults with diabetes, approximately 90% of whom have the type 2 DM form (T2D), increased from 108 million in 1980 to 422 million in 2014 globally, and the age-standardized diabetes prevalence increased from 4.3 to 9.0% in men and from 5.0 to 7.9% in women in the same period [1]. The number of diabetes patients globally has further increased to 463 million in 2019 [2]. Persons with diabetes are at increased risk of various complications including retinopathy [3], neuropathy [3] and nephropathy [3], as well as a large number of chronic diseases including cardiovascular diseases [4], several cancers [4], and several other diseases across multiple organ systems [4, 5].
A growing body of evidence suggests that diabetes may also significantly increase the risk for Alzheimer’s disease (AD), Parkinson’s disease (PD), and other progressive late onset neurodegenerative diseases [4, 6], with an ever growing number of sufferers, in parallel to increases in life expectancy [7]. Several epidemiological studies have investigated the association between a history of diabetes mellitus and the risk of PD [8,9,10,11,12,13,14,15,16,17,18,19,20,21]; but the results have not been entirely consistent with some studies reporting a positive association [9,10,11, 13,14,15,16,17, 19,20,21], whilst others found no clear association [8, 12, 18]. A previous meta-analysis of seven cohort studies, that included 1,761,632 individuals reported a 38% increased risk of PD among patients with diabetes [22]. An earlier meta-analysis of five cohort studies (reported in four publications and including 2975 PD cases and 483,551 participants) found a similar 37% increase in risk of PD among persons with diabetes [23]. However, results from case-control studies showed a contrasting non-significant reduction in risk [23]. Two more recent meta-analyses including 7 and 10 cohort studies both reported a 29% increase in risk of PD among diabetes patients [24, 25]. Six cohort studies with > 65,000 PD cases and > 20.4 million participants have since been published [13, 16, 17, 19,20,21], and all of these reported increased risk [13, 16, 17, 19,20,21]. A few studies also suggested increased PD risk in persons with prediabetes [21, 26] or diabetes complications [15, 16], although results were not always consistent [10]. To further clarify whether there is an association between DM, prediabetes and PD we therefore conducted a systematic review and meta-analysis of cohort studies to provide a more up-to-date and comprehensive assessment of the evidence, and to evaluate potential sources of heterogeneity between studies.
Materials and methods
Search strategy
We searched PubMed and Embase databases up to 6th of February 2022 for eligible studies. The search terms used are provided in the supplementary text. We followed the PRISMA criteria for reporting of meta-analyses [27]. The reference lists of the included publications were screened for further potentially relevant studies. DA and SS conducted the screening of the literature search independently.
Study selection and inclusion criteria
We included published retrospective and prospective cohort studies and case-control studies nested within cohorts that reported adjusted relative risk (RR) estimates (including hazard ratios, risk ratios and odds ratios) and 95% confidence intervals (CIs) for the association between DM and the risk of PD. When several publications were published from the same cohort we included the publication with the largest sample size, with one exception, where the included study adjusted for several additional confounders [14]. A list of the excluded studies and the corresponding rationale can be found in Supplementary Table 1.
Data extraction
The following data were extracted from each study: The first author’s last name, publication year, country where the study was conducted, the name of the study, study period and duration of follow-up, sample size, sex, age, number of cases, type of diabetes, subgroup, RRs and 95% CIs and variables adjusted for in the analysis. The data extraction was conducted by DA and checked for accuracy by YMS.
Study quality assessment
Study quality was assessed using a modified version of the Newcastle Ottawa Scale (NOS) which rates studies according to selection, comparability and outcome assessment [28]. The version applied include the following modifications that, in our view, address important limitations of NOS [29] and can potentially add value to the estimated score: (1) the point regarding representativeness was removed, as it is not relevant for study quality; (2) scoring 0.25 point per confounding factor adjusted for, up to a maximum of 2 points, instead for giving 2 points for adjustments for two confounders. This was deemed justifiable, as studies with relatively crude adjustment (e.g. for age and sex) could still receive, in the original scale, a maximum score but could still be prone to confounding; and (3) for the outcome assessment, we allocated half a point for record linkage and one point for independent outcome assessment by another physician or healthcare worker or for validated assessment of the outcome. This modified NOS gave a total score range from 0 to 8, instead of the range of 0 to 9 of the original scale.
Statistical methods
We used random effects models to calculate summary RRs (95% CIs) for the association between DM and PD risk [30]. The average of the natural logarithm of the RRs was estimated and the RR from each study was weighted using random effects weights [30]. For studies that reported results stratified by sex or diabetes complications, but not overall, we pooled the results from each subgroup using a fixed effects model before including the study in the overall analysis.
Heterogeneity between studies was evaluated using the Q test and I2 statistics [31]. I2 is a measure of how much of the heterogeneity is due to between study variation rather than chance, ranging from 0 to 100%. We conducted main meta-analyses (all studies combined) and stratified by study characteristics such as sex, duration of follow-up, exclusion of early follow-up, geographic location, number of cases, assessment of Parkinson’s disease diagnosis, type of diabetes, assessment of diabetes, timing of diabetes diagnosis, diabetes duration, presence of diabetes complications (including episodes of hypoglycaemia, retinopathy, peripheral neuropathy and nephropathy), study quality and by adjustment for confounding factors to investigate potential sources of heterogeneity. Furthermore, we calculated E-values for the association between diabetes and PD, to assess the potential impact of unmeasured or uncontrolled confounding [32]. The E-value is defined as the minimum strength that an unmeasured or uncontrolled confounder would have with both the exposure and the outcome to fully explain away the observed association. We used the World Cancer Research Fund criteria for grading the overall evidence regarding DM and PD, with possible gradings rated as convincing, probable, limited-suggestive, limited - no conclusion, or substantial effect on risk unlikely [33]. The criteria are described in more detail in Supplementary Table 2.
Publication bias was assessed using Egger’s test [34] and Begg’s test [35] and by inspection of funnel plots. The statistical analyses were conducted using the software package Stata, version 13.0 (StataCorp, Texas, US).
Results
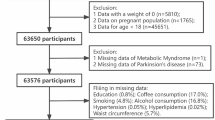
A total of 2660 records were screened and of these we identified 15 population-based cohort studies (15 publications, 14 risk estimates) [8,9,10,11,12,13,14,15,16,17,18,19,20,21] with 29.9 million participants and 86,345 PD cases that were included in the meta-analysis (Fig. 1; Table 1). One publication reported results for two cohort studies combined [8]. One additional publication [26] was included in a subgroup analysis by duration of diabetes as it overlapped with another publication with a larger sample, from the same study [16] that was used for the main analysis, however, it [26] was also included in the analysis of prediabetes and PD. Six of the included studies (five publications) were from the United States (US) [8, 10,11,12, 17], five studies from Europe [9, 13, 15, 20, 21] and four studies from Asia [14, 16, 18, 19] (Table 1). All studies were conducted in adult populations, the age at baseline ranged from 18 to 84 years for the studies that provided an age range. Four studies only used self-reported diabetes [10,11,12, 19], while eleven studies (10 publications) [8, 9, 13,14,15,16,17,18, 20, 21] also included either registry linkage, blood glucose, and/or a validation study of diabetes diagnoses. PD diagnoses were validated or assessed independently by health practitioners in six studies (five publications) [8,9,10,11,12], while nine studies [13,14,15,16,17,18,19,20,21] used a combination of self-report and registry linkage or registry linkage only. The mean (median) study quality scores, using the modified NOS, was 5.9 (5.75) out of 8; corresponding scores for each item across studies are shown in Supplementary Table 3. Lack of reporting on adequacy (completeness) of follow-up was the most common reason for a lower than optimal score. Other relatively common limitations that contributed to a lower than optimal score across multiple studies include using only self-reported data on diabetes, not having an independent or validated outcome assessment and inadequate adjustment for confounders.

Flow-chart of study selection
The summary RR for PD was 1.27 (95% CI: 1.20–1.35, I2 = 82.3%, pheterogeneity<0.0001) for persons with diabetes vs. persons without diabetes (Fig. 2). There was no evidence of publication bias with Egger’s test (p = 0.41), Begg’s test (p = 0.99), or by visual inspection of the funnel plot (Supplementary Fig. 1). The summary RR ranged from 1.25 (95% CI: 1.18–1.33) when excluding the study by Lee et al. et al [16] to 1.29 (95% CI: 1.22–1.37) when excluding the study by Kummer et al. [17] (Supplementary Fig. 2). The E-value for the summary RR was 1.86 (lower CI: 1.69), which is the minimum strength that an unmeasured confounder would have with both DM and PD to fully explain away the observed association.

Diabetes mellitus and Parkinson’s disease
Two studies [21, 26] with 11,547,811 participants and 45,292 cases were included in the analysis of prediabetes and risk of PD. The summary RR was 1.04 (95% CI: 1.02–1.07, I2 = 0%, pheterogeneity=0.44) (Supplementary Fig. 3). The E-value for the summary RR was 1.24 (lower CI: 1.16).
Subgroup analyses
Positive associations were consistently observed between DM and PD across most subgroup analyses including analyses stratified by sex, duration of follow-up, exclusion of early follow-up, geographic location, number of cases, type of diabetes, assessment of Parkinson’s disease diagnosis, assessment of diabetes, timing of diabetes diagnosis, diabetes duration, diabetes complications, study quality, and adjustment for confounding factors including age, socioeconomic status, alcohol intake, smoking, and BMI, however, there were few studies in some subgroups (Table 2). There was some indication of a stronger association among diabetes patients with history of complications than for patients without complications (summary RR, 95% CI: 1.54, 1.32–1.80 vs. 1.26, 1.16–1.38), although the test for heterogeneity between subgroups was not significant (p = 0.18). There was relatively high heterogeneity in the overall analysis (I2 = 81.8%), but lower or moderate heterogeneity was observed among men, US studies, studies using both prevalent and incident DM cases, and in studies that adjusted for hypertension and serum cholesterol levels (Table 2).
Grading of evidence
We considered the overall grading of the evidence to be supportive of a probably causal relationship between DM and PD. This was based on the following criteria which we considered as met: (1) evidence from at least two independent cohort studies (15 cohort studies included); (2) no substantial unexplained heterogeneity relating to the presence or absence of an association, or direction of effect, as the vast majority of studies reported positive associations, no studies reported significantly reduced risk, and there was lower heterogeneity in some subgroup analyses; (3) good quality studies to exclude with confidence the possibility that the observed association results from random or systematic error, including confounding, measurement error and selection bias (i.e. there was little difference in the association in subgroup analyses stratified by risk of bias of the studies, by various adjustments, and the association persisted among studies with more comprehensive assessment of diabetes status vs. only self-reported diabetes status); and (4) evidence for biological plausibility (see section on mechanisms in the discussion).
Discussion
This systematic review and meta-analysis of fifteen cohort studies with 86,345 PD cases and 29.9 million participants, provides strong epidemiological evidence of a 27% increase in the relative risk of developing PD among DM patients, compared to persons without DM. Persons with prediabetes had a 4% increase in risk, though this latter finding was based on two studies only. The increased risk of PD in persons with DM was observed in men and women, across geographic regions, and strata of other study characteristics (number of cases, type of diabetes, assessment of diabetes, timing of diabetes diagnosis, study quality, and adjustment for most confounding factors).
These findings are in line with previous meta-analyses of five and seven cohort studies, of much smaller sample sizes, which reported a 37–38% increase in risk of PD among diabetes patients [22, 23], and two more recent meta-analyses including 7 and 10 cohort studies which both reported a 29% increase in risk of PD among diabetes patients [24, 25]. Therefore, our findings provide more robust epidemiological evidence on this association, as our analysis was based on roughly twice the number of studies and a much larger sample size and number of PD cases. The findings are also consistent with a recent Mendelian randomization study which found an increased risk of Parkinson’s disease with genetically determined diabetes mellitus [24], providing some support for causality. Our results are not concordant with the findings of case-control studies [23], which are known to be more prone to survival, recall and selection biases, as well as reverse causality. Increased mid-life and early late-life mortality of DM patients may, indeed, underpin the discrepancies between case-control and cohort studies. A “reverse causation” bias may occur in observational studies, related to the long prodromal period of PD, potentially spanning over two decades. This period is known to be associated with progressive accumulation of PD neuropathology, including inclusions of Lewy bodies (LB) and dopaminergic nigrostriatal neuronal loss, known to potentially affect glycaemic control [10], as well as the occurrence of subtle motor and non-motor clinical manifestations that may alter physical activity and diet, both key lifestyle factors in the management of DM. The observation of considerable weight loss in PD patients, seen several years before diagnosis and persisting for several years after diagnosis [36], may also impact diabetes risk favorably, and could potentially explain the observed protective association of DM on PD in case-control studies. Interestingly, in the current analysis based on cohort studies, the positive association between DM and PD persisted in studies with ≥ 15 or ≥ 20 years of follow-up, and was if anything slightly stronger among studies with long durations of follow-up than short follow-up, suggesting that reverse causation is less likely to explain the findings.
The main limitations of studies employing meta-analysis methods involve factors that may potentially affect results, such as potentially unaccounted and residual confounders, sub-optimal study quality, heterogeneity, misclassification or misdiagnosis of diabetes status, as well as publication bias. For instance, pesticide exposure and head trauma are rarely accounted for, in observational cohort studies; additionally diabetes patients have a higher prevalence of overweight/obesity and smoking, and lower levels of physical activity [37], compared to persons without diabetes; therefore, confounding from some of these factors could have impacted the observed results. In our stratified analyses, the associations persisted among studies which adjusted for these and several other factors, making this a less likely explanation for the observed results. In addition, there was no evidence of heterogeneity between the subgroups stratified by various adjustments with meta-regression analyses, however, given the few studies in some of the subgroups it is also possible that we may have been underpowered to detect significant differences between some subgroups. The calculated E-values suggest that such a confounder would have to be relatively strongly associated (RR = 1.86, lower CI: 1.69) with both DM and PD to fully explain away the observed association. However, the E-value for the association between prediabetes and PD was weaker (RR = 1.24, lower CI: 1.16), suggesting the association may have been more vulnerable to confounding.
In terms of study quality, although the mean study quality across studies was graded as moderate, the summary estimates were similar to the subgroup of studies with high study quality. One common contributor to lower than optimal study quality scores was a lack of reporting on adequacy or completeness of follow-up. However, a low score on this point may not necessarily have been a source of bias, but rather due to poor reporting, because many studies have nearly complete follow-up data, facilitated by linkages to health or mortality registries. Other contributors to non-optimal quality scores included features such as non-optimal assessment of exposure and outcome (e.g. using only self-report or registry linkages without independent assessment or validation) and inadequate adjustment for confounders. Nevertheless, the summary estimates persisted in most subgroup analyses stratified by adjustment for various confounding factors.
Although there was high heterogeneity in the overall analysis as measured by the I2-value, 12 of 14 risk estimates were in the direction of increased risk and 11 of the risk estimates were statistically significant (95% CIs excluding the null value). Moreover, none of the cohort studies reported a statistically significant reduction in relative risk. Hence, the observed heterogeneity is more likely driven by differences in the strength of the association, than by differences in the direction of the association. Also, several of the included studies were very large and had rather narrow 95% CIs around the risk estimates, but with different sizes of risk estimates, and thus, the 95% CIs for the different studies did not always overlap. This likely explains the high I2-value in spite of the relative consistency of the direction of the association, thus less problematic than the presence of heterogeneity related to the direction of an association. In our subgroup analyses, there was less heterogeneity in studies of men or men and women combined, and among US studies.
Diabetes was self-reported in some studies and this may have led to misclassification of diabetes status that could, potentially, result in an underestimation of the association between DM and PD. However, we found little difference in the summary estimates between subgroups of studies that only relied on self-report and those that used a combination of self-report and linkages to medical records or had a measure of fasting blood glucose at baseline. Subgroup analyses stratified by whether studies only used prevalent (baseline) diabetes cases or also included incident diabetes cases, did not show significant heterogeneity between subgroups, although the summary estimate was slightly higher for the former than the latter (1.30 vs. 1.17). Misclassification of diabetes diagnoses by using only prevalent cases could result in regression dilution bias (bias to the null) as the lack of information on incident diabetes cases could lead to misclassification and potential underestimation of the association. However, this would depend on a recent diabetes diagnosis being most relevant to Parkinson’s disease development. Given that we observed the strongest association in the subgroup using prevalent diabetes cases only, it is possible that long-standing diabetes may be of greater importance. Survival bias is less likely to explain our findings because if such bias was present one would expect a weaker association in the studies using only prevalent diabetes cases, than in those also using incident cases. Although we found no significant difference in the results when stratified by the reported diabetes duration, this analysis was based on only two studies. The summary estimates were somewhat stronger for participants with diabetes complications than among those without such complications (summary RR, 95% CI = 1.54, 1.32–1.80 vs. 1.26, 1.16–1.38). As diabetes complications typically occur in patients with longer disease duration or poor glycaemic control, it is plausible that a longer disease duration and poor diabetes management are key driving factors for the stronger association with PD risk. Given the limited number of studies in these subgroup analyses and the lack of consistent cut-offs reported for diabetes duration, further epidemiological and mechanistic studies are warranted to elucidate the precise role of disease duration and glycaemic control on PD risk. In addition, the available studies did not investigate the role of specific diabetes medications on the observed risk. The majority of the cohort studies included in our meta-analysis only reported on DM overall, three studies reported on type 2 diabetes (T2D) only and none on type 1 diabetes. Given that the vast majority (over 90%) of diabetes cases are of type 2 (1), the current findings likely reflect the impact of insulin resistance and T2D. Lastly, we explored potential publication bias, but found no evidence of such bias with the statistical tests or by inspection of the funnel plot.
Several biological mechanisms may contribute to the increased risk of PD in patients with diabetes. Hyperglycemia, resulting from hypoinsulinaemia in type 1 diabetes or insulin resistance (IR) in T2D, exposes neurons to increased metabolic stress, neuronal dysfunction and death, thus directly contributing to PD pathogenesis [38]. Experiments in diabetic mice showed reduced dopamine transporters [39] and dopamine levels in the striatum [40], thus increasing the vulnerability of nigro-striatal neurons. Recent experiments, using diabetes-induced MitoPark mice, showed that the acquisition of IR phenotype in these animals results in mitochondrial dysfunction by suppressing PGC-1α expression, promoting the upregulated ROS production and oxidative stress, as well as the upregulated expression of phosphorylated α-synuclein (SNCA) [41], a key constituent of Lewy bodies (LB) [42]. Mitochondrial dysfunction, leading to neuronal death was also a main finding of studies using knockout of insulin receptor (NIRKO) mice [43] and diabetic db/db mice [44]. Increased accumulation and phosphorylation of α-synuclein was also observed within the cortex, pre-commissural putamen and dopaminergic neurons in the substantia nigra of cynomolgus monkeys, with spontaneous T2D-like pathology [45]. Of note, abnormal SNCA and LB burden are key pathological features in PD.
A direct effect of hyperglycemia is the increase of advanced glycation end-products (AGEs) and glycation agents, such as the highly reactive methylglyoxal (MGO), that may reach particularly high levels in the substantia nigra [46]. The interactions of AGEs with their receptors (RAGE) may lead to oxidative stress, inflammation and cell death [47]. AGEs have been found in LBs and have been shown, in vitro studies, to cross-link with SNCA to induce its aggregation and formation of SNCA oligomers, of higher neurotoxicity [48]. Furthermore, MGO may inhibit SNCA degradation and increase its accumulation and also react with dopamine to form 1-acetyl-6,7 dihysroxy-1.2.3.4-tetrahysroisoquinoline (ADTIQ), that may further contribute to dopaminergic degeneration [48].
In view of the potential increase of PD risk in diabetes patients, repurposing of antidiabetic medications for the treatment of PD is gaining increasing research interest, in the absence of effective disease modifying therapies. Recently, Mor et al. have demonstrated the neurotoxic effect of bcat-1 knockdown, in an animal model known to recapitulate PD features [49]. Neurotoxicity was found to be mediated through increased mitochondrial respiration (or “hyperactivity”) and oxidative damage. The authors showed that administration of metformin, a first- line anti-T2D medication, reduced mitochondrial respiration to control levels and significantly improved both motor function and neuronal viability [49]. Metformin was also shown to improve motor functions in 6-hydroxydopamine (6-OHDA)-lesioned mice, by activating the AMPK and BDNF signaling pathways and regulating or suppressing genes in reactive astrocytes [50]. In human trials, a double blind placebo-controlled study of Exenatide, a GLP-1 receptor agonist, showed a beneficial effect in PD patients [51]. In a subsequent study, evaluating target engagement through neuronal derived exosomal vesicles (NEVs) isolated from serum samples, it has been shown that patients with exenatide had an increased protein activation of Akt and mTOR cascades, compared to placebo [50].
Strengths of this meta-analysis include the population-based cohort design of the included studies, mitigating the potential for survival bias, recall bias, selection bias, and reverse causation, which may affect case-control studies to a larger degree; secondly it’s large sample size of 29.9 million participants, including over 86,000 PD cases, providing statistical power to detect a moderate association; finally the robustness of the findings in multiple subgroup and sensitivity analyses.
As the ever increasing numbers of persons with diabetes are projected to reach 700 million by 2025 worldwide [1], increased rates of debilitating late-onset neurodegenerative diseases such as AD, PD, as well as the associated forms of dementia with Lewy bodies, and Parkinson’s disease dementia are set to become another consequence of the diabetes epidemic, adding significant healthcare and socio-economic burden worldwide. Although the observed association between DM and PD is of moderate size, the findings are still likely to have important public health implications because of the large number of persons who live with diabetes worldwide. In this context, our findings strongly support the need for urgent global public health measures to effectively address the diabetes epidemic worldwide, that may have the added significant benefit in preventing PD, AD and related late-onset neurodegenerative diseases.
Conclusion
This systematic review and meta-analysis provides epidemiological evidence that patients with diabetes mellitus have a 27% increase in the relative risk of PD compared to persons without diabetes. The likelihood of causality was graded as probable using WCRF criteria. There was a suggestion of a 4% increase in risk of PD among those with prediabetes. Further studies should aim at elucidating the specific contribution of disease duration and age of diabetes onset, longitudinal glycaemia and its variability, as well as diabetic complications and antidiabetic medications in this association.
References
NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in diabetes since 1980: a pooled analysis of 751 population-based studies with 4.4 million participants. Lancet 2016 Apr 9;387(10027):1513–30.
International Diabetes Federation. IDF diabetes atlas. 9th edition. Brussels: International Diabetes Federation, 2019.
Faselis C, Katsimardou A, Imprialos K, Deligkaris P, Kallistratos M, Dimitriadis K. Microvascular complications of type 2 diabetes Mellitus. Curr Vasc Pharmacol. 2020;18(2):117–24.
Seshasai SR, Kaptoge S, Thompson A, Di AE, Gao P, Sarwar N, Whincup PH, Mukamal KJ, Gillum RF, Holme I, Njolstad I, Fletcher A, et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N Engl J Med. 2011 Mar;3(9):829–41.
Campbell PT, Newton CC, Patel AV, Jacobs EJ, Gapstur SM. Diabetes and cause-specific mortality in a prospective cohort of one million U.S. adults. Diabetes Care. 2012 Sep;35(9):1835–44.
Zheng B, Su B, Price G, Tzoulaki I, hmadi-Abhari S, Middleton L, Glycemic, Control. Diabetic Complications, and Risk of Dementia in Patients With Diabetes: Results From a Large U.K. Cohort Study. Diabetes Care 2021 May 25;44(7):1556–63.
GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990–2016: a systematic analysis for the global burden of Disease Study 2016. Lancet Neurol. 2018 Nov;17(11):939–53.
Simon KC, Chen H, Schwarzschild M, Ascherio A. Hypertension, hypercholesterolemia, diabetes, and risk of Parkinson disease. Neurology 2007 Oct 23;69(17):1688–95.
Hu G, Jousilahti P, Bidel S, Antikainen R, Tuomilehto J. Type 2 diabetes and the risk of Parkinson’s disease.Diabetes Care 2007 Apr;30(4):842–7.
Driver JA, Smith A, Buring JE, Gaziano JM, Kurth T, Logroscino G. Prospective cohort study of type 2 diabetes and the risk of Parkinson’s disease.Diabetes Care 2008 Oct;31(10):2003–5.
Xu Q, Park Y, Huang X, Hollenbeck A, Blair A, Schatzkin A, Chen H. Diabetes and risk of Parkinson’s disease. Diabetes Care. 2011 Apr;34(4):910–5.
Palacios N, Gao X, McCullough ML, Jacobs EJ, Patel AV, Mayo T, Schwarzschild MA, Ascherio A. Obesity, diabetes, and risk of Parkinson’s disease. Mov Disord. 2011 Oct;26(12):2253–9.
Pupillo E, Cricelli C, Mazzoleni F, Cricelli I, Pasqua A, Pecchioli S, Lapi F, Beghi E. Epidemiology of Parkinson’s Disease: a Population-Based study in primary care in Italy. Neuroepidemiology. 2016;47(1):38–45.
Yang YW, Hsieh TF, Li CI, Liu CS, Lin WY, Chiang JH, Li TC, Lin CC. Increased risk of Parkinson disease with diabetes mellitus in a population-based study. Med (Baltim). 2017 Jan;96(3):e5921.
De Pablo-Fernandez E, Goldacre R, Pakpoor J, Noyce AJ, Warner TT. Association between diabetes and subsequent Parkinson disease: A record-linkage cohort study.Neurology 2018 Jul 10;91(2):e139–e142.
Lee SE, Han K, Baek JY, Ko KS, Lee KU, Koh EH. Association Between Diabetic Retinopathy and Parkinson Disease: The Korean National Health Insurance Service Database. J Clin Endocrinol Metab 2018 Sep 1;103(9):3231–8.
Kummer BR, Diaz I, Wu X, Aaroe AE, Chen ML, Iadecola C, Kamel H, Navi BB. Associations between cerebrovascular risk factors and parkinson disease. Ann Neurol. 2019 Oct;86(4):572–81.
Kizza J, Lewington S, Mappin-Kasirer B, Turnbull I, Guo Y, Bian Z, Chen Y, Yang L, Chen Z, Clarke R. Cardiovascular risk factors and Parkinson’s disease in 500,000 chinese adults. Ann Clin Transl Neurol. 2019 Apr;6(4):624–32.
Azizova TV, Bannikova MV, Grigoryeva ES, Rybkina VL, Hamada N. Occupational exposure to chronic ionizing radiation increases risk of Parkinson’s disease incidence in Russian Mayak workers. Int J Epidemiol 2020 Apr 1;49(2):435 – 47.
Jacobs BM, Belete D, Bestwick J, Blauwendraat C, Bandres-Ciga S, Heilbron K, Dobson R, Nalls MA, Singleton A, Hardy J, Giovannoni G, Lees AJ, et al. Parkinson’s disease determinants, prediction and gene-environment interactions in the UK Biobank. J Neurol Neurosurg Psychiatry. 2020 Oct;91(10):1046–54.
Sanchez-Gomez A, Diaz Y, Duarte-Salles T, Compta Y, Marti MJ. Prediabetes, type 2 diabetes mellitus and risk of Parkinson’s disease: A population-based cohort study. Parkinsonism Relat Disord 2021 Aug;89:22–7.
Yue X, Li H, Yan H, Zhang P, Chang L, Li T. Risk of Parkinson Disease in Diabetes Mellitus: an updated Meta-analysis of Population-Based Cohort Studies. Med (Baltim). 2016 May;95(18):e3549.
Cereda E, Barichella M, Pedrolli C, Klersy C, Cassani E, Caccialanza R, Pezzoli G. Diabetes and risk of Parkinson’s disease: a systematic review and meta-analysis. Diabetes Care. 2011 Dec;34(12):2614–23.
Chohan H, Senkevich K, Patel RK, Bestwick JP, Jacobs BM, Bandres CS, Gan-Or Z, Noyce AJ. Type 2 diabetes as a determinant of Parkinson’s Disease Risk and Progression. Mov Disord. 2021 Jun;36(6):1420–9.
Liu W, Tang J. Association between diabetes mellitus and risk of Parkinson’s disease: a prisma-compliant meta-analysis. Brain Behav. 2021 Aug;11(8):e02082.
Rhee SY, Han KD, Kwon H, Park SE, Park YG, Kim YH, Yoo SJ, Rhee EJ, Lee WY. Association between Glycemic Status and the risk of Parkinson Disease: a Nationwide Population-Based study. Diabetes Care. 2020 Sep;43(9):2169–75.
Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ. 2009;339:b2535.
Wells G, Shea B, O’Connell D, Peterson J, Welch V, Losos M, Tugwell P. The Newcastle-Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta-analyses. http://www.ohrica/programs/clinical_epidemiology/oxfordasp, Accessed 15.09.2022.
Stang A. Critical evaluation of the Newcastle-Ottawa scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur J Epidemiol. 2010 Sep;25(9):603–5.
DerSimonian R, Laird N. Meta-analysis in clinical trials.Control Clin Trials 1986 Sep;7(3):177–88.
Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med 2002 Jun 15;21(11):1539-58.
VanderWeele TJ, Ding P. Sensitivity Analysis in Observational Research: Introducing the E-Value.Ann Intern Med 2017 Aug 15;167(4):268–74.
World Cancer Research Fund/American Insitute for Cancer Research. Food, Nutrition, Physical Activity and the Prevention of Cancer: a global perspective. Washington DC: AICR; 2007.
Egger M, Davey SG, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ 1997 Sep 13;315(7109):629–34.
Begg CB, Mazumdar M. Operating characteristics of a rank correlation test for publication bias.Biometrics 1994 Dec;50(4):1088–101.
Chen H, Zhang SM, Hernan MA, Willett WC, Ascherio A. Weight loss in Parkinson’s disease. Ann Neurol. 2003 May;53(5):676–9.
Hu FB, Manson JE, Stampfer MJ, Colditz G, Liu S, Solomon CG, Willett WC. Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. N Engl J Med 2001 Sep 13;345(11):790-7.
Morsi M, Maher A, Aboelmagd O, Johar D, Bernstein L. A shared comparison of diabetes mellitus and neurodegenerative disorders. J Cell Biochem. 2018 Feb;119(2):1249–56.
Jones KT, Woods C, Zhen J, Antonio T, Carr KD, Reith ME. Effects of diet and insulin on dopamine transporter activity and expression in rat caudate-putamen, nucleus accumbens, and midbrain. J Neurochem. 2017 Mar;140(5):728–40.
Perez-Taboada I, Alberquilla S, Martin ED, Anand R, Vietti-Michelina S, Tebeka NN, Cantley J, Cragg SJ, Moratalla R, Vallejo M. Diabetes causes dysfunctional dopamine neurotransmission favoring Nigrostriatal Degeneration in mice. Mov Disord. 2020 Sep;35(9):1636–48.
Hong CT, Chen KY, Wang W, Chiu JY, Wu D, Chao TY, Hu CJ, Chau KD, Bamodu OA. Insulin Resistance Promotes Parkinson’s Disease through Aberrant Expression of a-Synuclein, Mitochondrial Dysfunction, and Deregulation of the Polo-Like Kinase 2 Signaling. Cells 2020 Mar 17;9(3):740.
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature 1997 Aug 28;388(6645):839–40.
Kleinridders A, Cai W, Cappellucci L, Ghazarian A, Collins WR, Vienberg SG, Pothos EN, Kahn CR. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc Natl Acad Sci U S A. 2015 Mar;17(11):3463–8.
Khang R, Park C, Shin JH. Dysregulation of parkin in the substantia nigra of db/db and high-fat diet mice. Neuroscience 2015 May 21;294:182–92.
Sun Y, Guo C, Yuan L, Li W, Wang ZY, Yue F, Li JY. Cynomolgus Monkeys with spontaneous Type-2-diabetes-mellitus-like Pathology develop alpha-synuclein alterations reminiscent of Prodromal Parkinson’s Disease and Related Diseases. Front Neurosci. 2020;14:63.
Uchiki T, Weikel KA, Jiao W, Shang F, Caceres A, Pawlak D, Handa JT, Brownlee M, Nagaraj R, Taylor A. Glycation-altered proteolysis as a pathobiologic mechanism that links dietary glycemic index, aging, and age-related disease (in nondiabetics). Aging Cell. 2012 Feb;11(1):1–13.
Cheong JLY, De Pablo-Fernandez E, Foltynie T, Noyce AJ. The Association between type 2 diabetes Mellitus and Parkinson’s Disease. J Parkinsons Dis. 2020;10(3):775–89.
Vicente Miranda H, Cassio R, Correia-Guedes L, Gomes MA, Chegao A, Miranda E, Soares T, Coelho M, Rosa MM, Ferreira JJ, Outeiro TF. Posttranslational modifications of blood-derived alpha-synuclein as biochemical markers for Parkinson’s disease. Sci Rep 2017 Oct 20;7(1):13713.
Mor DE, Sohrabi S, Kaletsky R, Keyes W, Tartici A, Kalia V, Miller GW, Murphy CT. Metformin rescues Parkinson’s disease phenotypes caused by hyperactive mitochondria. Proc Natl Acad Sci USA 2020 Oct 20;117(42):26438–47.
Ryu YK, Go J, Park HY, Choi YK, Seo YJ, Choi JH, Rhee M, Lee TG, Lee CH, Kim KS. Metformin regulates astrocyte reactivity in Parkinson’s disease and normal aging. Neuropharmacology 2020 Sep 15;175:108173.
Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, Hibbert S, Budnik N, Zampedri L, Dickson J, Li Y, viles-Olmos I et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet 2017 Oct 7;390(10103):1664–75.
Funding
This work was funded by the Imperial College School of Public Health and the South-East Regional Health Authority of Norway.
Author information
Authors and Affiliations
Contributions
DA designed the research, conducted the literature search, extracted data and analyses and wrote the first draft of the paper. DA and SS conducted the literature screening, and YMS checked the data extractions for accuracy. DA, SS, YMS, BZ, CTUM, LM interpreted the data, revised the subsequent drafts for important intellectual content, read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no duality of interest associated with this manuscript.
Disclaimer
Where authors are identified as personnel of the International Agency for Research on Cancer / World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the International Agency for Research on Cancer / World Health Organization.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aune, D., Schlesinger, S., Mahamat-Saleh, Y. et al. Diabetes mellitus, prediabetes and the risk of Parkinson’s disease: a systematic review and meta-analysis of 15 cohort studies with 29.9 million participants and 86,345 cases. Eur J Epidemiol 38, 591–604 (2023). https://doi.org/10.1007/s10654-023-00970-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10654-023-00970-0