
Abstract
Arnica montana is a clonal, self-incompatible herb of economic and intrinsic ecological value which is declining in large parts of its range. With the employment of microsatellite markers, we characterized the population structure and distribution of genetic diversity of 40 populations of A. montana sampled throughout Europe. We detected a clear geographical pattern of isolation and strong population structure, indicating limited gene flow. We also observed a negative latitudinal gradient in genetic diversity. Such patterns can be explained by paleo-historical colonization routes following the last glacial maximum, with regions characterized by higher genetic diversity corresponding to former glacial refugia. We recommend the implementation of conservation measures such as assisted gene flow in the populations characterized by low genetic diversity under consideration of the observed population structure. For the populations where high levels of genetic diversity are still retained, we recommend the maintenance of suitable habitat conditions. Our study emphasises the importance of conducting large-scale population genetic studies that consider postglacial recolonization history when planning active conservation measures such as assisted gene flow.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In Europe, grasslands belong to the most endangered habitat types, with more than half of them threatened to some extent (European Commission 2017) because of human activities resulting in fragmentation, deterioration or even destruction of these habitats. Semi-natural nutrient-poor grasslands, in particular, have been facing a steady decline in the last decades (Peter et al. 2009). These habitats are characterized by high biodiversity levels including several biotic communities and numerous endangered species (Bailly-Maître et al. 2015) and therefore are of high ecological value. Furthermore, they provide important ecosystem services such as water supply and flow regulation, carbon storage, and erosion control (Bengtsson et al. 2019). Land-use change, and in particular abandonment or farming intensification, are threatening these habitats thereby causing the decline of associated species.
One of these species is the perennial herb Arnica montana L., a characteristic species of species-rich Nardus grasslands (habitat type 6230 of the Habitats Directive, Council Directive 92/43/EEC, 21 May 1992), heathlands with moderate mowing or grazing levels (Galvánek et al. 2008), and marginal coniferous forests (Radusiene and Labokas 2007). This plant is an irreplaceable component of the ecosystem, with specialist species depending on its presence for survival (Korneyev et al. 2015). Because of its sensitivity to soil nitrification and the consequent shift in community composition that follows eutrophication, it can be employed as an indicator species for habitat quality (Hollmann et al. 2020). Besides, it is particularly valuable for its use in traditional and modern medicine due to the high content of secondary metabolites, such as sesquiterpenes, flavonoids, and phenolic acids (Petrova et al. 2012).
The distribution range of A. montana is large, spanning from western Norway (up until 63.1°N) at its northern margin, to the Pyrenees at its South-Western margin, and the Carpathians at its eastern margin. It is also found at different altitudes from sea-level lowlands (e.g. Netherlands) to alpine grasslands (up to 3000 m a.s.l.). Although still relatively common in the Alps and in the Carpathians, A. montana is declining rapidly in Central and Northern Europe. For this reason, A. montana has been included in different national red lists (Zarzycki and Szeląg 2006; Rossi et al. 2013; Metzing et al. 2018).
The task of detecting the “main” drivers for this decline is challenging because of the intrinsic complexity of anthropogenic activities and their impact on natural environments. Abandonment (Dullinger et al. 2003), eutrophication (Van der Berg et al. 2003), direct and indirect effects of climate change (Bruelheide 2003; Vikane et al. 2019; Stanik et al. 2020), and invasive species (Bruelheide and Scheidel 1999) are some of the proposed external factors causing population decline in this species. Also, due to its value for medicinal purposes, this species suffered from overexploitation from wild collection and for this reason has been included in the Habitats Directive (Annex V).
Also, genetic factors such as inbreeding, limited gene flow, and genetic erosion negatively affect A. montana populations (Duwe et al. 2017; Van Rossum and Raspé 2018) and are hence potential drivers for the species’ decline. Previous studies generated differing results in patterns of neutral genetic diversity. For example, Maurice et al. (2016) found lower levels of genetic diversity in montane populations compared to colline populations on a rather regional level, comparing populations from Western Germany, Luxembourg, Eastern Belgium as well as populations from the Vosges mountains in Northern France. Duwe et al. (2017), however, identified on a larger scale, covering the Alpine Arch as well as the whole of Germany, the opposite pattern with alpine populations showing slightly higher genetic diversity than central-European lowlands. Both studies described strong population differentiation between different regions and elevations finding also evidence of isolation by distance (IBD). On the contrary, the study from Van Rossum and Raspé (2018) found no evidence of IBD at the regional scale (Southern Belgium and French Ardennes).
Comparing different studies conducted with different genetic markers and at different spatial scales has its limitations and hence, for a better understanding of the population genetic structures of A. montana at regional scales, a wider, range-wide perspective is necessary. This is even more true when it comes to future conservation challenges (e.g. climate change). A clear understanding of the basic population genetic features of A. montana populations, such as distribution of genetic diversity, differentiation of populations, and inbreeding levels, is fundamental for the development of appropriate conservation measures e.g., genetic restoration. Populations located in different regions of the species range can be expected to differ significantly in those population genetic parameters and, therefore, may require specific conservation strategies (Shay et al. 2021). Moreover, under current climate change, wild populations are predicted to shift northward and/or upward given the availability of suitable nearby habitat patches (Kelly and Goulden 2008).
Finally, it is important to ascertain whether genetic patterns arise due to human interference, such as fragmentation, or are the results of natural processes, such as range dynamics. This knowledge can be employed to determine if observed indications of genetic erosion necessitate proactive conservation interventions. It is worth noting that the footprints of natural processes are better evident at larger spatial scales (Hewitt 1999; Trense et al. 2021). Therefore, we argue that for a better view of the genetic features of A. montana populations a comprehensive sampling design is required.
Our study focuses on the range-wide population genetic features of the declining herbaceous plant A. montana. Using microsatellite markers, we aim to characterize (1) whether and how populations are differentiated at this large scale, and (2) how neutral genetic diversity is spatially distributed across the range of A. montana throughout Europe. Furthermore, we discuss the potential implications of our results for the future conservation of this valuable species.
Materials and methods
Sampling
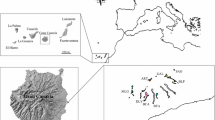
Overall, 43 populations with a total amount of 807 individual plants were randomly sampled throughout Europe (Fig. 1), spanning along the altitudinal, longitudinal and latitudinal gradients. To cover areas with lower geographic coverage, additional data from 90 samples from an earlier study (unpublished data) were included here. The sampling and genotyping methods for these samples are differing from our methods and are therefore described in detail in Appendix S1.

Map of the sampling locations. Each of the 40 populations sampled across the A. montana range is represented by a white dot. Greyscale represents the altitudinal gradient
Coordinates of A. montana stands were obtained from publicly available datasets (via gbif.org), local nature agencies and collaborators. Locations were visited in 2018 and 2019 during the flowering/fruiting season, from June to August depending on the locality. Within each population, 20 plants were randomly sampled at a minimum distance of 1 m from each other in order to avoid the collection of ramets of the same clone. For each plant, two leaves were collected and stored in silica gel. Upon arrival in the laboratory, leaves were dried for 24 h in a drying cabinet at 40 °C and stored at room temperature. The graphical output for the sampling map was produced with QGIS V3.18.3 (QGIS Development Team 2022). DEM profiles were obtained from the WorldClim dataset (Fick and Hijmans 2017).
DNA isolation and genotyping
DNA was extracted from dry leaves ground to a fine powder using a bead mill homogenizer (Omni Bead Ruptor 24, Biovendis, Mannheim, Germany). A CTAB protocol (Appendix, S2) modified from the original developed by Barbier et al. (2019) for extraction-recalcitrant tissues was employed to extract DNA. The working solution DNA yield for each sample was estimated with a spectrophotometer (NanoDrop 1000, Thermo Scientific, Waltham, Massachusetts) and diluted to 1 ng/µl to minimize interference with PCR reaction observed at higher sample concentrations. PCR was conducted by multiplexing ten nuclear microsatellite loci previously developed by Duwe et al. (2015) (Appendix, S3), using the Type-it Microsatellite PCR Kit DNA (Qiagen, Hilden, Germany). Multiplexed PCR reactions (Appendix, S4) were developed taking into account possible dimer formation between primers by using AmplifX (Jullien 2019). Different parameter settings were used in the multiplexed set of markers during PCR amplification (Appendix, S4). Amplification products were analysed by capillary electrophoresis with a 3500 Genetic Analyzer using GeneScan ™ 600 LIZ ™ as internal size standard (Applied Biosystems, Waltham, Massachusetts). Genemarker V2.6.7 (SoftGenetics, Hulce et al. 2011) was used for allele scoring.
Population genetic analyses
First, the dataset was scanned for identical multi-locus genotypes (MLGs) with Genalex V6.51b2 plugin (Peakall and Smouse 2012) and manually checked for allele-scoring errors and missing data by searching for outliers (by applying column filters in Excel). In case of multiple ramets per genotype, only one ramet was randomly chosen and kept for further analyses. Furthermore, only populations with at least ten different MLGs were retained for the following analyses. Departure from Hardy Weinberg Equilibrium (HWE) was tested with the “exact HW test” (Haldane 1954) with the online software Genepop On the Web (Rousset 2008). For populations showing deviation from HWE, heterozygote deficiency accounting for null allele presence was tested with a complete enumeration method (Dempster et al. 1977) with Genepop. Bonferroni sequential p-value correction was applied for accounting for multiple testing (Holm 1979).
In detail, unbiased expected heterozygosity (uHe), observed heterozygosity (Ho), genetic distance (FST) averaged across all loci were calculated with Genalex plugin. Inbreeding coefficient (FIS) with corresponding 95% confidence interval (bootstrapped) for each population was calculated with R (“diveRsity” package) (Keenan et al. 2013). Adjusted allelic richness was assessed with the rarefaction method implemented in HP-Rare v1.0 (Kalinowski 2005) with a sample size of n = 10. In order to test how genetic parameters are distributed across space, the influence of geographical variables (elevation, latitude, longitude) on adjusted allelic richness (Ar), unbiased heterozygosity corrected for sample size (uHe) and inbreeding coefficient (FIS), were tested using linear regression analysis in R (R Core Team 2022) with a backward stepwise selection of explanatory variables (LAT, LON, ALT). The collinearity between altitude and latitude, which is an intrinsic characteristic of European geography, was taken into account by deleting the variable with the lowest R2 value between collinear variables. Results were plotted with the R package “ggplot2” (Wickham 2016).
Genetic differentiation within and among populations was calculated from a pairwise genetic distance (FST) matrix corrected for null alleles with the software FreeNa (Chapuis and Estoup 2006) and its significance was tested with a hierarchical analysis of molecular variance (poppr.amova,1000 permutations, ~ Regions/Populations), as implemented in the ”poppr” R package (Kamvar & Grünwald 2015). Regions were defined on the basis of the first hierarchical level of the Structure analysis, differentiating between the detected clusters at K = 2. Furthermore, to assess limitations in gene flow, isolation by distance (IBD) was assessed with a Mantel test on R (“vegan” package, Oksanen et al. 2022) by testing the relation between genetic distances (FST/(1- FST) corrected for null alleles and geographic distances of populations. Additional Mantel tests were performed separately for the two main genetic clusters detected in the populations structure analysis.
Population genetic structure
The presence of genetic clusters (k) in A. montana populations was tested by performing a model-based Bayesian cluster analysis as implemented in the software Structure V2.3.4 (Pritchard et al. 2000) coupled with the parallel computation tool StrAuto (Chhatre and Emerson 2017) for optimizing computing time. An admixture model with correlated allele frequencies and without prior information about the regional membership of the populations was considered (100,000 burn-in period; 100,000 Markov chain Monte Carlo (MCMC), 15 independent runs for each K, for K = 1–40). Estimation of K was calculated using Structure Harvester online tool based on Evanno’s ΔK approach (Evanno et al. 2005) (Appendix S5 and S6). The same procedure was repeated another time with a second hierarchy analysis, by running separately the two main groups (K = 2) detected with the first run. Populations were assigned to one of these main groups based on their proportion of membership with one of the two clusters, with 50% considered as a threshold. CLUMPAK online tool (Kopelman et al. 2015) was used for aligning results from Q-matrices obtained for each run (including DISTRUCT pipeline). Graphical output for each K was produced with QGIS V3.18.3(QGIS Development Team 2022), CLUMPAK and R.
Furthermore, a principal coordinate analysis (PCoA) on pairwise genetic distances of individuals was conducted within Genalex and plotted with ggplot2. A discriminant analysis of principal components (DAPC) was conducted with R (“Adegenet” package) (Jombart 2008) as a complement to the Bayesian analysis and the PCoA. The analysis was conducted using populations as a-priori groups and the number of PCA axes and DA axes to be retained was judged using the explorative tools present in Adegenet (60 PC axes and 8 DA axes retained, automatic selection of the axes). In this multivariate method, differences between groups are maximised while minute differences within the groups are overlooked. PCoA and DAPC do not need to fit the assumptions of Hardy-Weinberg equilibrium and linkage disequilibrium (Miller et al. 2020) and thus might be more suitable to describe the population structure in case some assumptions are not met.
Results
Genotyping
All the selected loci were polymorphic, spanning from 5 alleles in Armo3 to 26 alleles in Arm6. A few clones were present in the dataset despite the sampling design. Out of 807 samples, 76 individuals showed to share the same multilocus genotype (MLG) with at least another individual. In one specific case, a Norwegian population (not included in the present dataset) was composed of only 4 genets out of 20 sampled individuals. After discarding all but one ramet per MLG from the data set, overall 749 samples were left. Furthermore, three populations had less than ten different genets and were thus deleted from the dataset. The final filtered dataset contained 712 individuals and 40 populations (Table 1), with the smallest (ITA_03) containing 12 individuals. Out of the 10 SSR loci, two of them (Arm7 and Arm10) showed evidence of deviation from HWE after sequential Bonferroni correction. Subsequent analysis with Genepop using the EM algorithm (Rousset 2008), found that heterozygote deficiency explained the deviation, likely caused by null alleles. Moreover, in one case (ROM_01) we found evidence for a complete allelic dropout at Arm7 locus. Hence, the main analyses (Genalex genetic parameters, AMOVA, Structure, PCoA, DAPC) were conducted on two datasets. One including all markers and the other excluding Arm07 and Arm10. No significant differences were detected between the full dataset and the dataset without Arm7/Arm10. Due to the stability of the overall analysis despite the limited presence of null alleles, no markers were discarded for the final analysis, as the drawbacks of deleting them outbalanced the benefits of having more statistical power.
Population structure
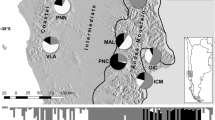
Results of the Bayesian clustering analysis conducted with Structure showed that, by following the Evanno-∆K approach, in the first hierarchy analysis, two main clusters were detected (Fig. 2, but see also Fig. S7, H = 1). The two clusters are separated by the Alpine arc and Carpathian mountains, with all the populations north of the Alps belonging to one cluster, with the exception of Lithuanian and Czech populations, which are assigned to the southern cluster. This separation reflects the uppermost hierarchical level of genetic structure at a wider geographical scale. Targeting higher levels of K, populations as well as individuals were consistently assigned to distinct clusters pointing to further hierarchies, even down to the level of K = 40 (Fig. 2). Analysis at the second hierarchy analysis (Fig. S7, H = 2) identified two clusters in the northern cluster (K = 2), the “continental” group composed of Central European populations and the “Scandinavian” group composed of the most northern populations. In the southern cluster, four main groups (K = 4) were identified: Western Alps, Central Alps, Austrian Alps & White Carpathians and finally Eastern European populations (Lithuanian populations and Romanian Apuseni).

Population genetic structure of 40 A. montana populations based on microsatellite data. The graphs show the results of the Bayesian cluster analysis (first hierarchy). Each bar chart specifies a different number of clusters assumed (K = 2, 4, 6, 8, 16, 24, 40). Vertical bars display individuals grouped within populations. Different colours indicate different genetic clusters. On top of the graph geographical regions and assignment of populations to clusters (northern vs. southern) for K = 2 are specified. Barcharts are produced with CLUMPAK online tool
Similarly to the Bayesian clustering results, in DAPC (Appendix S8) and PCoA (Appendix S9) the separation of K = 2 (North-South) of the first hierarchy level of the Structure analysis is described by the first PC axis (PC1) with the Lithuanian and the Pyrenean populations (LIT_01, LIT_02, FRA_01) taking an intermediate position between these groups. At the same time, the further separation described by higher values of k (k > 2) are better described by the second PC axis (PC2), especially in the PCoA. Hierarchical AMOVA (1000 permutations) showed high differentiation between populations, with 29.5% of the genetic variation found among populations: 13.55% between regions (p < .001), 15.97% between populations within regions (p < .001) and 70.5% within populations. Genetic differentiation (FST) across loci averaged 0.37. Furthermore, the Mantel test (Fig. 3) showed a strong (r = 0.64) correlation between genetic distance and geographic distance. When Mantel tests were conducted separately on the “Northern” and Southern” clusters, similar results were obtained (r = 0.19 and r = 0.20, respectively, Appendix, S10 and S11).

Mantel correlation between genetic distances (FST/(1- FST)) and geographical distances (in km). Black circles display pairwise differences of A. montana populations. *** p ≤ 0.001
Patterns of genetic diversity

Relationship of latitude with unbiased heterozygosity (uHe) (upper), allelic richness (Ar) (middle) and inbreeding (Fis) (lower) in A. montana. Black circles represent A. montana populations. The red dotted line represents the regression line. *** (p ≤ 0.001); ns (non-significant)
Ho averaged 0.42 and was highest in ITA_01 (0.68) and lowest in NOR_02 (0.14). Unbiased genetic diversity (uHe) averaged 0.44 and was generally higher in southern populations with the highest values found in ITA_08 (0.65) and lowest in SWE_04 (0.20). Most populations of A. montana showed inbreeding coefficient values (FIS) close to zero (averaging 0.03), with extremes represented by NOR_02 (0.38) and GER_06 (-0.19). Allelic richness (Ar) spanned between 1.80 (NOR_02) and 5.80 (AUS_05) (Table 1). Regression analysis displayed a statistically significant linear relationship between uHe and latitude (F3,36=18.4, R2 = 0.52, p = < 0.001; Fig. 4, upper). Additionally, between allelic richness (Ar) and latitude, there was a statistically significant linear relationship (F1,38=30.6, R2 = 0.43, p = < 0.001; Fig. 4, middle). On the other hand, the inbreeding coefficient (FIS) was not correlated with any of the geographical variables (Fig. 4, lower).
Discussion
Geographical patterns of genetic variation
Based on AMOVA and Mantel test results, very high differentiation exists between A. montana populations and strong isolation by distance (IBD) patterns are present throughout the species range. The present study displays at a larger scale what was found by Duwe et al. (2017) with the same microsatellite loci. We obtained comparable results in terms of differentiation between populations from the AMOVA (27% in Duwe et al. 2017 vs. 29.5% in our study, respectively), average FST values (0.27 vs. 0.37) and Mantel test correlation coefficient (0.43 vs. 0.64).
Overall, we conclude that gene flow across long distances is very limited in A. montana populations, explained most likely by poor dispersal ability. Evidence of limited seed dispersal in A. montana at the local scale is present in the literature (Luijten et al. 1996). In particular, Strykstra et al. (1998) found that seeds disperse for a few meters only and fine-scale genetic structure detected by Luijten et al. (1996) with rosettes (genets) in the vicinity of each other sharing similar genotypes is indicative, too. Furthermore, the main pollinators of this species are solitary hymenopterans, syrphids and lepidopterans (Van Rossum and Raspé 2018) and hence restricted pollen dispersal can be expected, too.
In addition, the integrity of many natural habitats is affected by human activities, causing their fragmentation, with long-lasting consequences e.g. biodiversity loss (Haddad et al. 2015; Rogan and Lacher 2018). In a meta-analysis of 50 plant species, a large negative effect of fragmentation on pollination and reproductive success was found (Aguilar et al. 2006), which was stronger in self-incompatible species. Self-incompatible species are obligate outbreeders and therefore are more dependent on pollination that occurs between genetically different (and thus more distant) individuals. Consequently, A. montana as a self-incompatible species with very limited seed and pollen dispersal is particularly susceptible to habitat fragmentation. We underline therefore the importance of considering the aforementioned aspects when planning conservation measures.
The highly differentiated A. montana populations were hierarchically clustered into geographically concise groups, as shown by the Bayesian analysis and confirmed by the PCoA and DAPC. The observed pattern is likely to be a result of the climatic cycles of the quaternary, which have shaped the genetic structure of many species (Hewitt 1996). Thus, the geographically concise genetic clusters might either originate from distinct glacial refugia, which had been isolated during the glaciation or might result from barriers that have established after recolonization. Anyway, with our current coverage of the range, it is difficult to evaluate whether the “northern” cluster arose from a refugium outside of the Alpine region or directly from the “southern” cluster, which would mean that the differentiation is due to isolation after migrating north. The separation within the southern cluster seems more likely to originate from separate refugia because no apparent barriers or distribution gaps between East and West are evident and a parallel colonisation of the Alps from several refugia seems plausible as was observed also for other species (e.g. Wagner et al. 2015). When the population structure was analysed at even higher resolution, we observed that more, separated genetic groups could be defined. Individuals were consistently assigned to geographically concise genetic groups and, even at the highest possible K value (K = 40, Fig. 2), well defined and almost “pure” genetic clusters can still be observed in some regions, further confirming the strong population differentiation detected by the AMOVA.
Besides a strong hierarchical genetic structure, we observed a strong geographical cline of genetic diversity, with both heterozygosity (uHe) and allelic richness (Ar) decreasing from the Alpine region towards Scandinavia. Based on our range-wide sampling we could show that the major driver of genetic diversity is latitude, which may coincide with altitude in large parts of the range. However, at smaller geographic scales other factors may modulate genetic diversity (Maurice et al. 2016). Due to our large-scale study, we can now re-assess the distribution of genetic diversity in A. montana and put into context the previous studies conducted on the population genetic features of this species. In detail, Duwe et al. (2017) found an altitudinal gradient in genetic diversity (He) with populations at lower altitudes showing slightly lower levels of genetic diversity compared to populations at higher altitudes. Altitude was interpreted as the major factor modulating genetic diversity. Additionally, increased anthropogenic activities associated with lower altitudes (either due to eutrophication or habitat destruction) were considered the main responsible factors for lower genetic diversity in Central Europe. The finding that decreased habitat quality and habitat loss can cause a reduction in genetic diversity due to genetic erosion has been described in the scientific literature as a main result of increased fragmentation and consequent reduction in population sizes (Pitra et al. 2011; Schlaepfer et al. 2018). Hence, the loss of genetic diversity caused by human activities is subject of concern and the conservation of adequate levels of genetic diversity is fundamental for the preservation of natural habitats (Salo and Gustafsson 2016). However, by following the hypothesis that anthropogenic activities explain the latitudinal gradient in genetic diversity, the distribution of A. montana’s genetic diversity should be associated with habitat quality and/or habitat loss. That is, the areas of lowest genetic diversity (i.e. Scandinavia) would match geographically the regions where anthropogenic pressure on natural habitats is the highest. This is not the case, since habitat quality is generally higher in Scandinavia than Central Europe (Haberl et al. 2014). As such, low levels of genetic diversity are not necessarily a consequence of human activities but can also be footprints of natural phenomena, such as past founder events or isolation caused by natural barriers. Although we did not test directly for correlations with habitat availability or quality, we claim that, based on our results, the anthropogenic activities are unlikely to be driving the distribution of genetic diversity at this scale in A. montana.
Instead, the observed gradual decrease in genetic diversity is in line with the previously analysed patterns (differentiation, gene flow, population structure) explained by migration events. Such migratory processes are likely to have led to recurrent founder events and population bottlenecks. As such, our results are interpretable in the context of past colonization routes following the end of the last ice age. Postglacial migration events resulted in a progressive loss of neutral genetic diversity from south to north in various European species (Excoffier et al. 2009; Sebasky et al. 2016; Daco et al. 2022). The direct consequence of this gradual genetic diversity loss is that most of the genetic variation present in A. montana is stored in the former glacial refugia while the northern parts of the distribution range exhibit much less variation. The strong geographical pattern in genetic structure and differentiation we find, further support our interpretation of the genetic diversity gradient in A. montana being the product of postglacial colonization routes from south to north. Other studies reached similar conclusions. For instance, the study by Daco et al. (2022) on the herb Anthyllis vulneraria conducted on a comparable scale detected a latitudinal gradient in genetic diversity, which was best explained by past founder events during postglacial colonization (and not population size or habitat quality). Furthermore, in several pan-European species differing in life-history traits, a comparable S-N latitudinal gradient in genetic diversity was observed and interpreted as a footprint of postglacial colonization: Silene vulgaris (Sebasky et al. 2016), Rhinanthus angustifolius (Strykstra et al. 1996) and more (Pellissier et al. 2016).
Concluding, by conducting a large-scale investigation that included populations from potential former glacial refugia (e.g. Alps, Pyrenees, Carpathians) as well as populations from the range margins of the species’ distribution, we were able to recognise the traces of postglacial colonization events. Moreover, the scale of the study enabled us to define how much differentiation exists between populations located in different biogeographical regions at the continental scale and thus to identify the presence of distinct genetic groups, which can be later acknowledged for conservation purposes (Moritz 1994; Picó and Van Groenendael 2007). Indeed, conservation studies greatly benefit from inclusive sampling designs, because understanding where genetic diversity sources are present in a certain species can help planning conservation actions such as ex-situ conservation, assisted gene flow and reintroductions (Picó and Van Groenendael 2007; Van Rossum et al. 2020).
Persistence of A. montana and conservation aspects
While genetic diversity is rather low in some parts of the distribution, there is no evidence of widespread inbreeding at the genetic level, despite the acknowledged population decline occurring in this species. This is not surprising considering that A. montana is self-incompatible and partially clonal. Self-incompatibility prevents mating between closely related individuals with the same S-allele, a strategy known as inbreeding avoidance (Furstenau and Cartwright 2017). Consequently, populations displaying inbreeding at the genetic level are probably rather rare in A. montana (Luijten et al. 2000). Moreover, in a partially clonal species, even small and isolated populations could maintain their genetic diversity temporarily by shifting to predominantly clonal reproduction. In our study, we did not compare populations for clonal diversity because our sampling design was not appropriate in this context. However, further studies aiming at evaluating the extent and features of clonality at range-wide level in this species might reveal important aspects about the consequences of reductions in population sizes and genetic diversity in a self-incompatible and clonal species like A. montana. In this context, we highlight the importance of monitoring shifts toward predominantly asexual reproduction, as observed in one case (Norwegian population excluded from the dataset for this reason). In fact, an increased level of clonality could make populations more susceptible to future threats, like climate change or diseases, because of lower genetic variation and potentially inbreeding following the breakdown of the self-incompatibility system (Navascués et al. 2010). In fact, in a study conducted by Luijten et al. (2000), hand-selfing resulted in the germination in up to 45% of the seeds (although in a limited number of individuals), which could indicate a partial breakdown of the self-incompatibility system. Therefore, further studies are necessary to unveil the mechanism behind sexual strategies in this species, considering the potential consequences from the conservation perspective.
Most of the genetic variation in A. montana is retained within in the Alps. This region is generally recognised as a hotspot of biodiversity (Petit et al. 2003). However, suitable habitats are disappearing also from the Alps (Dirnböck et al. 2003) where populations retain high levels of genetic diversity. The main reason is a shift in floristic associations caused by the abandonment of traditional pasture. Such deleterious changes can happen as fast as 30 years (Korzeniak 2016). Furthermore, it is likely that A. montana populations in these regions will undergo contractions because of unsuitable climate (La Sorte and Jetz 2010; Stanik et al. 2020) and limited habitat patches available as they migrate toward higher elevations (Hampe and Petit 2005). Hence, habitat-focused conservation becomes particularly important in those areas where high levels of genetic diversity are still retained. In particular, the conservation of managed alpine meadows is necessary for enabling the survival of established individuals and the recruiting and development of new rosettes in available habitat patches at higher altitudes.
On the other hand, populations situated at the geographical centre of the distribution (Central-European lowlands and low-altitude montane areas) have lower neutral genetic variation and are likely experiencing non-optimal habitat quality due to higher human population density (Picó and Van Groenendael 2007) and climate change (Stanik et al. 2020). In addition, such populations have limited possibility of shifts toward higher latitudes/elevations and less available habitats for colonization, as they become more fragmented within patches of non-suitable habitats. Finally, while some northern European species are predicted to expand further north as the climate shifts (van der Meer et al. 2016), some are constrained by climate shifts in unsuitable directions (Vikane et al. 2019), limited genetic diversity and small population sizes (Vucetich and Waite 2003). Populations of A. montana situated at the northern edges are predicted to suffer from increased extinction risk and susceptibility to increasingly unsuitable climate (e.g. increased temperatures and rainfalls) (Vikane et al. 2019). Moreover, as we can see from our data, genetic variability in northern populations is heavily reduced, likely due to serial founder effects. Although the link between adaptive and neutral genetic variation is under discussion in the scientific community (García-Dorado and Caballero 2021), we can assume that adaptive diversity was also severely reduced due to founder effects. Therefore, northern populations are likely more susceptible to diseases and environmental change due to a lack of adaptive variation. These populations with increased extinction risk due to low genetic diversity, small population size might benefit from conservation practices such as genetic restoration and the following monitoring of genetic parameters (Van Rossum and Raspé 2018; Van Rossum et al. 2020). However, geographically distinct genetic groups were recognised in our study. Consequently, the indicated genetic clusters should be considered in the context of conservation practices such as relocations and assisted gene flow (Moritz 1994). Successful translocations of individuals for genetic restoration purposes have been reported by Van Rossum et al. (2020). It must be mentioned, though, that the source and target populations were all from the same region and had much lower differentiation levels than the ones recorded in our study.
Thus, we strongly suggest conducting further studies at larger scales on the adaptive genetic diversity of A. montana to indicate the best strategy (e.g. assisted gene flow) for supporting the survival and expansion toward new habitat patches of those northern low-diversity populations.
Conclusions
Our results show strong geographical patterns in differentiation, genetic diversity and genetic clustering in the endangered plant Arnica montana. Spatially-limited gene flow is present among populations, as shown by the isolation by distance (IBD) pattern. We interpret such patterns as the genetic signature of postglacial colonization events combined with limited dispersal ability, which are still visible in the extant genetic structure and distribution of genetic diversity. These patterns have important implications for the future conservation of this valuable species. Since climate change is expected to force A. montana populations to shift toward higher altitudes and latitudes, studies focusing on the potential responses to climate change in relation to genetic variation could help conservation programmes at maximising survival to climate change. Where neutral genetic diversity is low, and especially in Scandinavia, populations must be closely monitored for signs of reduced recruitment and the consequent population decline. Where population decline is caused (or followed) by a limited S-allele set and the consequent interruption of sexual reproduction, the introduction of new individuals from more diverse populations should be considered. Here, the genetic clusters we identified might help as a first starting point to define distinct evolutionary units for conservation practices such as relocations and assisted gene flow. Also, studies focusing on adaptive genetic variation could help conservation programmes at maximising survival to environmental change (e.g. global warming) in translocated and resident populations. At the same time, where genetic diversity is higher (e.g. Alps), habitat-focused conservation will be fundamental for the survival of this species in the medium and long term. In the meanwhile, ex-situ conservation of A. montana genetic heritage, such as activities aimed at sharing genetic material between botanical gardens and research facilities, could be the last resort for this species in some regions where assisted gene flow is impractical or economically unsustainable.
Data Availability
All the datasets mentioned in the manuscript are available for the corresponding author on request.
References
Aguilar R, Ashworth L, Galetto L, Aizen MA (2006) Plant reproductive susceptibility to habitat fragmentation: review and synthesis through a meta-analysis. Ecol Lett 9:968–980. https://doi.org/10.1111/j.1461-0248.2006.00927.x
Bailly-Maître J, Condé S, Delbaere B (2015) EU 2010 biodiversity baseline: adapted to the MAES typology (2015). EEA Technical report, No 9/2015, European Environment Agency, Luxembourg. https://doi.org/10.2800/382317
Barbier FF, Chabikwa TG, Ahsan MU et al (2019) A phenol/chloroform-free method to extract nucleic acids from recalcitrant, woody tropical species for gene expression and sequencing. Plant Methods 15:62. https://doi.org/10.1186/s13007-019-0447-3
Bengtsson J, Bullock JM, Egoh B et al (2019) Grasslands—more important for ecosystem services than you might think. Ecosphere 10:e02582. https://doi.org/10.1002/ecs2.2582
Bruelheide H (2003) Translocation of a montane meadow to simulate the potential impact of climate change. Appl Veg Sci 6:23–34. https://doi.org/10.1111/j.1654-109X.2003.tb00561.x
Bruelheide H, Scheidel U (1999) Slug herbivory as a limiting factor for the geographical range of Arnica montana. J Ecol 87:839–848. https://doi.org/10.1046/j.1365-2745.1999.00403.x
Chapuis M-P, Estoup A (2006) Microsatellite null alleles and estimation of Population differentiation. Mol Biol Evol 24:621–631. https://doi.org/10.1093/molbev/msl191
Chhatre VE, Emerson KJ (2017) StrAuto: automation and parallelization of STRUCTURE analysis. BMC Bioinformatics 18:192. https://doi.org/10.1186/s12859-017-1593-0
R Core Team (2022) R: A language and environment for statistical computing. R Found Stat Comput Vienna, Austria. https://www.R-project.org/
Council Directive 92/43/EEC of 21 May 1992 on the conservation of natural habitats and of wild fauna and flora (OJ L 206 22.07.1992 p. 7). 2006. In Sands P, Galizzi P (eds), Documents in European Community Environmental Law (pp. 568–583). Cambridge: Cambridge University Press. https://doi.org/10.1017/CBO9780511610851.039
Daco L, Matthies D, Hermant S, Colling G (2022) Genetic diversity and differentiation of populations of Anthyllis vulneraria along elevational and latitudinal gradients. Ecol Evol 12:e9167. https://doi.org/10.1002/ece3.9167
Dempster AP, Laird NM, Rubin DB (1977) J R Stat Soc Ser B Methodol 39:1–38. https://doi.org/10.1111/j.2517-6161.1977.tb01600.x. Maximum Likelihood from Incomplete Data via the EM Algorithm
Dirnböck T, Dullinger S, Grabherr G (2003) A regional impact assessment of climate and land-use change on alpine vegetation. J Biogeogr 30:401–417. https://doi.org/10.1046/j.1365-2699.2003.00839.x
Dullinger S, Dirnböck T, Greimler J, Grabherr G (2003) A resampling approach for evaluating effects of pasture abandonment on subalpine plant species diversity. J Veg Sci 14:243–252. https://doi.org/10.1111/j.1654-1103.2003.tb02149.x
Duwe VK, Ismail SA, Buser A et al (2015) Fourteen polymorphic microsatellite markers for the threatened Arnica montana (Asteraceae). Appl Plant Sci 3:1400091. https://doi.org/10.3732/apps.1400091
Duwe VK, Muller LAH, Borsch T, Ismail SA (2017) Pervasive genetic differentiation among central european populations of the threatened Arnica montana L. and genetic erosion at lower elevations. Perspect Plant Ecol Evol Syst 27:45–56. https://doi.org/10.1016/j.ppees.2017.02.003
European, Commission (2017) Directorate-General for Environment. In: Tsiripidis I, Piernik A, Janssen J et al (eds) European red list of habitats. Part 2, terrestrial and freshwater habitats. European Union, Luxembourg. https://doi.org/10.2779/091372
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Foll M, Petit RJ (2009) Genetic consequences of Range Expansions. Annu Rev Ecol Evol Syst 40:481–501. https://doi.org/10.1146/annurev.ecolsys.39.110707.173414
Fick SE, Hijmans RJ (2017) WorldClim 2: new 1km spatial resolution climate surfaces for global land areas. Int J Climatol 37(12):4302–4315. https://doi.org/10.1002/joc.5086A
Furstenau TN, Cartwright RA (2017) The impact of self-incompatibility systems on the prevention of biparental inbreeding. PeerJ 5:e4085. https://doi.org/10.7717/peerj.4085
Galvánek D, Janák M, European Commission (2008) Management of Natura 2000 habitats. 6230 *Species-rich Nardus grasslands. ISBN 978-92-79-08336-5
García-Dorado A, Caballero A (2021) Neutral genetic diversity as a useful tool for conservation biology. Conserv Genet 22:541–545. https://doi.org/10.1007/s10592-021-01384-9
GBIF.org (2019) GBIF Occurrence. Download at https://doi.org/10.15468/dl.hwgutf Accessed 19 Mar 2019
Haberl H, Erb K-H, Krausmann F (2014) Human appropriation of net primary production: patterns, Trends, and Planetary Boundaries. Annu Rev Environ Resour 39:363–391. https://doi.org/10.1146/annurev-environ-121912-094620
Haddad NM, Brudvig LA, Clobert J et al (2015) Habitat fragmentation and its lasting impact on Earth’s ecosystems. Sci Adv 1:e1500052. https://doi.org/10.1126/sciadv.1500052
Haldane JBS (1954) An exact test for randomness of mating. J Genet 52:631–635. https://doi.org/10.1007/BF02981502
Hampe A, Petit RJ (2005) Conserving biodiversity under climate change: the rear edge matters. Ecol Lett 8:461–467. https://doi.org/10.1111/j.1461-0248.2005.00739.x
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58:247–276. https://doi.org/10.1111/j.1095-8312.1996.tb01434.x
Hewitt GM (1999) Post-glacial re-colonization of european biota. Biol J Linn Soc 68:87–112. https://doi.org/10.1111/j.1095-8312.1999.tb01160.x
Hollmann V, Donath TW, Grammel F et al (2020) From nutrients to competition processes: Habitat specific threats to Arnica montana L. populations in Hesse, Germany. PLoS ONE 15:e0233709. https://doi.org/10.1371/journal.pone.0233709
Holm S (1979) A simple sequentially rejective multiple Test Procedure. Scand J Stat 6:65–70. https://doi.org/10.2307/4615733
Hulce D, Li X, Snyder-Leiby T, Johathan Liu CS (2011) GeneMarker genotyping Software: Tools to increase the statistical power of DNA fragment analysis. J Biomol Tech JBT 22:S35–S36 PMCID: PMC3186482
Jombart T (2008) Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Jullien N (2019) AmplifX v2.0.7; Aix-Marseille Univ, CNRS, INP, Inst Neurophysiopathol, Marseille, France https://inp.univ-amu.fr/en/amplifx-manage-test-and-design-your-primers-for-pcr. Accessed 20 July 2019
Kalinowski S (2005) Hp-rare 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5:187–189. https://doi.org/10.1111/j.1471-8286.2004.00845.x
Kamvar ZN, Brooks JC, Grünwald NJ (2015) Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front Genet 6:208. https://doi.org/10.3389/fgene.2015.00208
Keenan K, McGinnity P, Cross TF et al (2013) diveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788. https://doi.org/10.1111/2041-210X.12067
Kelly AE, Goulden ML (2008) Rapid shifts in plant distribution with recent climate change. Proc Natl Acad Sci 105:11823–11826. https://doi.org/10.1073/pnas.0802891105
Kopelman NM, Mayzel J, Jakobsson M et al (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191. https://doi.org/10.1111/1755-0998.12387
Korneyev SV, Khaghaninia S, Namin SM, Zarghani E (2015) Palearctic species of the genus Tephritis (Diptera, Tephritidae) associated with plants of the tribe Senecioneae (Asteraceae). Zootaxa 4007:207–216. https://doi.org/10.11646/zootaxa.4007.2.4
Korzeniak J (2016) Mountain Nardus stricta grasslands as a relic of past farming – the effects of grazing abandonment in relation to elevation and spatial scale. Folia Geobot 51:93–113. https://doi.org/10.1007/s12224-016-9246-z
La Sorte FA, Jetz W (2010) Projected range contractions of montane biodiversity under global warming. Proc Biol Sci 277:3401–3410. https://doi.org/10.1098/rspb.2010.0612
Luijten SH, Oostermeijer JGB, Leeuwen NC, Nijs HCM (1996) Reproductive success and clonal genetic structure of the rare Arnica montana (Compositae) in the Netherlands. Plant Syst Evol 201:15–30. https://doi.org/10.1007/BF00989049
Luijten SH, Dierick A, Gerard J, Oostermeijer B, Raijmann L, Den Nijs HCM (2000) Population Size, Genetic Variation, and Reproductive Success in a Rapidly Declining, Self-Incompatible Perennial (Arnica montana) in The Netherlands. Conserv Biol. 2000;14(6):1776–1787. doi:https://doi.org/10.1111/j.1523-1739.2000.99345.x
Maurice T, Matthies D, Muller S, Colling G (2016) Genetic structure of colline and montane populations of an endangered plant species. AoB Plants 8:plw057. https://doi.org/10.1093/aobpla/plw057
Metzing D, Hofbauer N, Ludwig D, Matzke-Hajek D (2018) Roten listen gefährdeter Tiere, Pflanzen und Pilze Deutschlands. Bundesamt fur Naturschutz, Bonn-Bad Godesberg. ISBN 978-3-7843-5612-9
Miller JM, Cullingham CI, Peery RM (2020) The influence of a priori grouping on inference of genetic clusters: simulation study and literature review of the DAPC method. Heredity 125:269–280. https://doi.org/10.1038/s41437-020-0348-2
Moritz C (1994) Defining ‘Evolutionarily significant units’ for conservation. Trends Ecol Evol 9:373–375. https://doi.org/10.1016/0169-5347(94)90057-4
Navascués M, Stoeckel S, Mariette S (2010) Genetic diversity and fitness in small populations of partially asexual, self-incompatible plants. Heredity 104:482–492. https://doi.org/10.1038/hdy.2009.159
Oksanen J, Blanchet FG, Kindt R et al (2022) vegan: Community Ecology Package. R package version 2.6-4. https://CRAN.R-project.org/package=vegan
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research–an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Pellissier L, Eidesen PB, Ehrich D et al (2016) Past climate-driven range shifts and population genetic diversity in arctic plants. J Biogeogr 43:461–470. https://doi.org/10.1111/jbi.12657
Peter M, Gigon A, Edwards PJ et al (2009) Changes over three decades in the floristic composition of nutrient-poor grasslands in the Swiss Alps. Biodivers Conserv 18:547–567. https://doi.org/10.1007/s10531-008-9521-2
Petit RJ, Aguinagalde I, de Beaulieu J-L et al (2003) Glacial Refugia: hotspots but not melting pots of genetic diversity. Science 300:1563–1565. https://doi.org/10.1126/science.1083264
Petrova M, Zayova E, Vassilevska-Ivanova R, Vlahova M (2012) Biotechnological approaches for cultivation and enhancement of secondary metabolites in Arnica montana L. Acta Physiol Plant 34:1597–1606. https://doi.org/10.1007/s11738-012-0987-x
Picó FX, Van Groenendael J (2007) Large-scale plant conservation in european semi-natural grasslands: a population genetic perspective. Divers Distrib 13:920–926. https://doi.org/10.1111/j.1472-4642.2007.00349.x
Pitra C, Suárez-Seoane S, Martín CA et al (2011) Linking habitat quality with genetic diversity: a lesson from great bustards in Spain. Eur J Wildl Res 57:411–419. https://doi.org/10.1007/s10344-010-0447-0
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1093/genetics/155.2.945
QGIS Development Team (2022) QGIS Geographic Information System. Open Source Geospatial Foundation Project. http://www.qgis.org
Radusiene J, Labokas J (2007) Population performance of Arnica montana L. in different habitats. In: Maxted N, Ford-Lloyd BV, Kell SP et al (eds) Crop wild relative conservation and use. CABI, Wallingford, pp 380–388
Rogan JE, Lacher TE Impacts of Habitat Loss and Fragmentation on Terrestrial Biodiversity. In: Reference Module in Earth Systems and Environmental Sciences., Elsevier (2018) https://doi.org/10.1016/B978-0-12-409548-9.10913-3
Rossi G, Montagnani C, Gargano D, Peruzzi L et al (2013) Lista Rossa della Flora Italiana 1 policy species e altre specie minacciate. Comitato Italiano IUCN e Ministero dell’Ambiente e della Tutela del Territorio e del mare. Roma, pp 1–58. http://www.iucn.it/pdf/Comitato_IUCN_Lista_Rossa_della_flora_italiana_policy_species.pdf
Rousset F (2008) Genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Salo T, Gustafsson C (2016) The effect of genetic diversity on Ecosystem Functioning in Vegetated Coastal Ecosystems. Ecosystems 19:1429–1444. https://doi.org/10.1007/s10021-016-0014-y
Schlaepfer DR, Braschler B, Rusterholz H-P, Baur B (2018) Genetic effects of anthropogenic habitat fragmentation on remnant animal and plant populations: a meta-analysis. Ecosphere 9:e02488. https://doi.org/10.1002/ecs2.2488
Sebasky ME, Keller SR, Taylor DR (2016) Investigating past range dynamics for a weed of cultivation, Silene vulgaris. Ecol Evol 6:4800–4811. https://doi.org/10.1002/ece3.2250
Shay JE, Pennington LK, Mandussi Montiel-Molina JA et al (2021) Rules of Plant Species ranges: applications for conservation strategies. Front Ecol Evol 9:700962. https://doi.org/10.3389/fevo.2021.700962
Stanik N, Lampei C, Rosenthal G (2020) Summer aridity rather than management shapes fitness-related functional traits of the threatened mountain plant Arnica montana. Ecol Evol 10:5069–5078. https://doi.org/10.1002/ece3.6259
Strykstra R, Bekker RM, Verweij GL (1996) Establishment of Rhinanthus angustifolius in a successional hayfield after seed dispersal by mowing machinery. Acta Bot Neerlandica 45:557–562. https://doi.org/10.1111/j.1438-8677.1996.tb00810.x
Strykstra R, Pegtel D, Bergsma A (1998) Dispersal distance and achene quality of the rare anemochorous species Arnica montana L.: meeting on re-introduction of endangered plant species. Acta Bot Neerlandica 47:45–56
Trense D, Hoffmann AA, Fischer K (2021) Large- and small-scale geographic structures affecting genetic patterns across populations of an Alpine butterfly. Ecol Evol 11:14697–14714. https://doi.org/10.1002/ece3.8157
Van der Berg LJL, Vergeer P, Roelofs JGM (2003) Heathland restoration in the Netherlands: Effects of turf cutting depth on germination of Arnica montana. Appl Veg Sci 6:117–124. https://doi.org/10.1111/j.1654-109X.2003.tb00571.x
Van der Meer S, Jacquemyn H, Carey PD, Jongejans E (2016) Recent range expansion of a terrestrial orchid corresponds with climate-driven variation in its population dynamics. Oecologia 181:435–448. https://doi.org/10.1007/s00442-016-3592-7
Van Rossum F, Raspé O (2018) Contribution of genetics for implementing population translocation of the threatened Arnica montana. Conserv Genet 19:1185–1198. https://doi.org/10.1007/s10592-018-1087-2
Van Rossum F, Hardy OJ, Le Pajolec S, Raspé O (2020) Genetic monitoring of translocated plant populations in practice. Mol Ecol 29:4040–4058. https://doi.org/10.1111/mec.15550
Vikane JH, Rydgren K, Jongejans E, Vandvik V (2019) Rainfall and temperature change drive Arnica montana population dynamics at the Northern distribution edge. Oecologia 191:565–578. https://doi.org/10.1007/s00442-019-04519-5
Vucetich JA, Waite TA (2003) Spatial patterns of demography and genetic processes across the species’ range: null hypotheses for landscape conservation genetics. Conserv Genet 4:639–645. https://doi.org/10.1023/A:1025671831349
Wagner S, Litt T, Sánchez-Goñi M-F, Petit RJ (2015) History of Larix decidua Mill. (european larch) since 130 ka. Quat Sci Rev 124:224–247. https://doi.org/10.1016/j.quascirev.2015.07.002
Wickham H (2016) ggplot2: Elegant Graphics for Data Analysis. Springer: New York, NY, USA, 2016; ISBN 9783319242750
Zarzycki K, Szeląg Z (2006) Red list of the vascular plants in Poland. Red list plants Fungi Pol. W. Szafer Institute of Botany, Polish Academy of Sciences, Kraków, pp 9–20
Acknowledgements
We thank Leoni Illner, Christina Mengel and Ulrike Hüwe for their assistance with the wet-lab and genotyping work. We are particularly grateful to everyone that helped during the sampling season with active sampling and/or provided logistical support in-field: Vera Wersebeckmann, Mindaugas Lapele, Silvia Oevermann, Michel Krol & Mark Hilboezen, Henning Fjord Aaser, Erik Dalsgaard, Jan Hornik, Giuliana Pincelli, Guy Rochat & Elisa Di Marco, Kirsten Myhr, Harald Bratli, Guido Teppa, Max Schröder, Anne Steen, Ellen Johanne Svalheim, Sven Nilsson, Birgit Kantner, Florin Pacurar, Katia Diadema, Lutz Eckstein, Andreas Bohner, Jörg Weise, Quentin Lambert, Claudia Plank and Massimo Vaira. We thank all the authorities who kindly provided the necessary documentation for the collection of the samples and access to the sampling sites. Finally, we thank everyone involved in the revision process for helping us improve the manuscript. This project was funded by the “Promotions-Plattform Geisenheim – Gießen –Marburg” by the Hessian Ministry for Science and Art.
Funding
This work was funded by the Hessian Ministry for Science and the Arts.
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
L.C. wrote the manuscript text and conducted the fieldwork and lab work. E.M. and S.L. supervised the writing process and contributed to the interpretation of the results. E.M., S.L. and I.L. helped with the experimental design. K.E. supervised the DNA extraction process. S.L. supervised the genotyping process. E.M. supervised the research project. All the authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors have no relevant financial or non-financial conflicts of interest to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Capria, L., Liepelt, S., Eimert, K. et al. Neutral genetic diversity follows a latitudinal gradient in the endangered plant Arnica montana L.: a range-wide study. Conserv Genet 25, 87–100 (2024). https://doi.org/10.1007/s10592-023-01559-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-023-01559-6